

Abstract
The anti-cancer effects of emodin, including inhibition of proliferation, invasion, metastasis and angiogenesis, were confirmed by various previous studies. However, the specific mechanisms were not clear. In this study, we investigated emodin’s anti-angiogenesis effect and focused on the mechanisms in human osteosarcoma (OS). OS cells were implanted to nude mice to form OS xenografts. Immunofluorescence assay was used to assess vWF expression in tumor tissue. MTT assay was employed to screen proper emodin concentrations unrelated with proliferation inhibition. siRNA technique was utilized to silence SIRT1 expression in OS cells. Expression levels of SIRT1 and VEGF were investigated by real-time PCR and western blotting. H4-k16Ac expression which indicated the deacetylation activity of SIRT1 was also detected by western blotting. As in results, HMGB1 treatment exacerbated OS angiogenesis both in vivo and in vitro. Emodin administration attenuated angiogenesis in both OS and HMGB1 treated OS in vivo and in vitro. After emodin treatment, the expression level and deacetylation activity of SIRT1 were dramatically enhanced. HMGB1-induced angiogenesis was more striking in SIRT1 silenced OS cells. SIRT1 silencing also impaired the anti-angiogenesis effect of emodin in OS cells. In conclusion: SIRT expression and deacetylation activity elevation are involved in emodin’s anti-angiogenesis effect in human OS.
Keywords: Osteosarcoma, angiogenesis, SIRT1, HMGB1, emodin
Introduction
In adolescents and children, osteosarcoma (OS) is considered the most frequent malignant bone tumor which is characterized by osteoid tissue generation or immature bone formation [1]. The incidence of OS was reported 5/million. As the anti-tumor treatment has been developing rapidly in recent decades, the 5-year survival rate of OS is currently 60%-70%, the prognosis of OS is still poor because of its aggressive malignancy [2]. Most patients with OS were diagnosed until obvious clinical manifestations such as bone fractures and local pain were observed, thus the OS was often found at advanced stage [3]. The indications for curative surgical treatment are limited because of metastasis [4]. Therapeutic effects of other conventional therapies including chemotherapy or radiotherapy are also undermined due to metastasis, chemoresistance, side effects and dyscrasia. Thus, effective novel agents inhibiting metastasis without serious general side effects are favorable and promising in OS clinical treatment.
It is generally accepted the notion that the progression and development of malignant tumor are largely associated with angiogenesis [5]. In physiological conditions, the novel vessel formation is restricted. In pathological conditions, such as acute inflammation and wound healing, the angiogenesis is also localized. However, in malignant tumors, the angiogenesis is usually uncontrolled in order to fit the blood supply to the boosting tumor growth [6,7]. Blood vessel extension, irregularity and circuitry are often found. The tumor angiogenesis is also found highly associated with tumor invasion and metastasis [8,9]. Thus, drugs or agents with anti-angiogenesis activity are promising for malignant tumor treatment and prevention.
Emodin, also referred as 1,3,8-trihydroxy-6-methylanthraquinone, is plant original, which is derived from rhizome of Rheum palmatum L [10]. In traditional medicine in eastern and southern Asia, from ancient times, Rheum palmatum L. has been used as an effective agent in treatment of peptic ulcer [11], indigestion, hemorrhoid, and several infectious diseases [12]. In recent decades, studies found anti-cancer activities of emodin, characterized by inhibition of tumor growth [13], invasion [14] and metastasis [15]. Also, emodin was reported to suppress tumor angiogenesis by blocking vascular endothelial growth factor (VEGF) signaling in cancer cells [16]. However, further interpretations are still needed to elucidate the exact mechanisms.
The sirtuin (SIRT) family (SIRT1-SIRT7) was reported associated with regulation of multiple pathophysiological events and has been drawing more and more attention recently [17,18]. SIRT1 is the most studied SIRT member and is known for its association with cancer [19]. It was suggested that the activation of SIRT1 played a promoting role in cancer development and progression [20]. Several well-known cancer suppressors, such as p53 and HIC1, were supposed the down-stream molecules in SIRT1 signaling [21]. A recent study pointed out that the angiogenesis was suppressed after SIRT1 activation which down-regulated VEGF transcription by inhibiting HMGB1 induced hypoxia- induced factor (HIF)-1 regulated angiogenesis [22].
In the present study, we investigated the possible involvement of SIRT1 signaling pathway in anti-angiogenesis effect of emodin in OS. We hypnotized that: (1) Emodin could inhibit VEGF expression in OS; (2) Emodin attenuates angiogenesis in OS by regulating SIRT1/HMGB1 signaling pathway. We believe that results in this study would not only broaden our knowledge of mechanisms of angiogenesis in OS, but also provide ground for possible clinical application of emodin in OS treatment in the future.
Materials and methods
Cell lines, culturing and treatment
Human osteosarcoma cell lines SOSP-9607, MG63 and SAOS-2 which were purchased from China Center for Type Culture Collection (CCTCC, China) were used in this study. SOS-9607 and MG63 cells were maintained in RPMI 1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA), 2.0 mM L-glutamine (Sigma-Aldrich, USA) and 1% antibiotic solution (Sigma-Aldrich, USA, containing 150 μmol/L streptomycin and 100 U/ml penicillin). SAOS-2 cells were maintained in DMEM medium (Gibco, USA) supplemented with 10% FBS, 2.0 mM L-glutamine and 1% antibiotic solution. Cells were cultured in humidified condition with 5% CO2 and 95% fresh air at 37°C. In some cases, cells were pre-treated with exogenous HMGB1 protein (HMG Biotech, Italy) at 40 ng/ml and emodin at serial diluted concentrations (0, 15, 30, 45, 60 μmol/L).
In vivo animal study
Male BALB/c nude mice were purchased from Animal experimental center of Fourth Military Medical University. Cell suspension containing 1×106 SOSP-9607/MG63/SAOS-2 cells were injected into left dorsa region of mice subcutaneously. Mice were treated by HMGB1 protein by single intramuscular injection (800 ng/mouse) or/and emodin (300 μg/Kg bodyweight) by 14-day continuous intraperitoneal injections. 4 weeks after injection, xenograft tumor tissue was harvested and prepared for Western blotting, real-time PCR and immunofluorescence assays.
Cell proliferation determination
Proliferation of SOSP-9607, MG63 and SAOS-2 cells was determined by colorimetric 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyltetrazolium bromide (MTT) assay. Same amount of cells (1×104/mL) were seeded into wells in a 96-well plate and then cultured for 24 hours. After being washed by PBS, 150 μL dimethylsulfoxide (DMSO, Sigma-Aldrich) was used to incubate cells in each well. A plate reader (Bio-Rad) was utilized to detect the absorbance at 540 nm (A540). The proliferation inhibition rate was calculated by “[1-A540 (experimental well)/A540 (control well)] ×100%”.
Small interfering RNA (siRNA) transfection
Sequence of siRNA against human SIRT1 gene was designed by TaKaRa (Tokyo, Japan) and synthesized accordingly. Specifically, the sequence was: sense 5’-CCAAGCAGCUAAGAGUAAUTT-3’, antisense 5’-AUUACUCUUAGCUGCUUGGTT-3’. SOSP-9607, MG63 and SAOS-2 cells were incubated for 24 hours and then the above siRNA and vehicle control (GenePharma, Shanghai) was transfected into cells by using PepMuteTM siRNA Transfection Reagent (SignaGen) per se the manufacturer’s instructions.
Real-time RT-PCR
The total RNA was extracted from cells by using TRIZOL Reagent (Invitrogen) according to the manufacturer’s instructions. SuperScript III Reverse Transcriptase (Invitrogen) was used to perform the reverse transcription and cDNA synthesis. Finally, by using All-in-one qPCR kit (Gene Copoeia), the quantitative real-time PCR was carried out. Primers were provided by TaKaRa, specifically, for SIRT1 was: sense 5’-CAGCAAGGCGAGCATAAA-3’, antisense 5’-TTCAGAACCACCAAAGCG-3’; for GAPDH (internal reference) was: sense 5’-TTGCCATCAATGACCCCTTCA-3’, antisense 5’-CGCCCCACTTGATTTTGGA-3’.
Western blotting
By using RIPA lysis buffer system (Beyotime) and Protein Extraction kit (Beyotime), total protein from tissue and cultured cells was extracted. A BCA protein assay kit (Solarbio) was used to detect protein concentration. 50 μg protein sample was loaded and then separated by vertical electrophoresis in SDS-polyacrylamide gels (8%-10%) and transferred to PVDF membranes (Millipore) electronically. Antibodies against SIRT1 (Abcam), acetylated histone 4 lysine 16 residue (H4-k16Ac, Invitrogen), VEGF (Abcam) and GAPDH (internal reference, Abcam) were used to incubate the membranes. SuperSignal West Pico kit (Peirce) was used to detect the immunoblots which were the analyzed by ImageJ2x software.
Von Willebrand factor (vWF) immunofluorescence assay
Harvested tumor tissue was fixed, dehydrated and then embedded in paraffin. 5-μm thick slice were made and incubated with anti-vWF antibody (Abcam) for 1 hour at 37°C. After washing and incubation with second antibody (Santa Cruz) for 30 minutes at 37°C, the fluorescence was detected and images were captured by a fluorescence microscope (Nikon).
Statistical considerations
The data in this study was presented as (mean ± SD). Differences between two groups were assessed performed with two sample two-tailed t test. Differences among multiple groups were assessed by one-way analysis of variance (ANOVA). P < 0.05 was considered statistically significant. The statistical analysis was performed by software SPSS (version 16.0, SPSS).
Results
Emodin significantly attenuated HMGB1-associated VEGF-induced angiogenesis in vivo
As shown in Figure 1, after nude mice were implanted with human osteosarcoma cells (SOSP-9607, MG63 and SAOS-2), exogenous HMGB1 administration was found playing a positive role in promoting role in tumor angiogenesis which was evidenced by increased VEGF and vWF expression. However, after administrated by emodin, both of VEGF and vWF expression were suppressed in xenografted tumor tissue harvested from nude mice.
Figure 1.

Emodin’s anti-angiogenesis effect in human OS graft tumor in vivo. Upper part of this figure demonstrated the immunoblots of VEGF in human OS graft tumors (SOSP-9607, MG-63 and SAOS-2) harvested from nude mice. Lower part of this figure showed captured images of immunofluorescent staining of vWF in graft tumors. Ctrl: tumor bearing nude mice, Ctrl+Emodin: tumor bearing nude mice treated with emodin; Ctrl+HMGB: tumor bearing nude mice treated with HMGB; tumor bearing nude mice received co-administration of emodin and HMGB. aDifferences were significant when compared with Ctrl; cdifferences were significantly from Ctrl+Emodin.
Proliferation of cultured human osteosarcoma cells was inhibited by emodin
Emodin incubation at serial diluted concentrations inhibited the proliferation of SOSP-9607, MG63 and SAOS-2 cells in a concentration-dependent manner (Figure 2). Emodin showed significant inhibitory effect on proliferation at 15 μmol/L for SOSP-9607 and MG63 cells; at 20 μmol/L for SAOS-2 cells manner (Figure 2). Thus, concentration below 15 μmol/L and 20 μmol/L were selected for anti-angiogenesis evaluation in SOSP-9607/MG63 and SAOS-2 cells respectively for the subsequent experiments.
Figure 2.
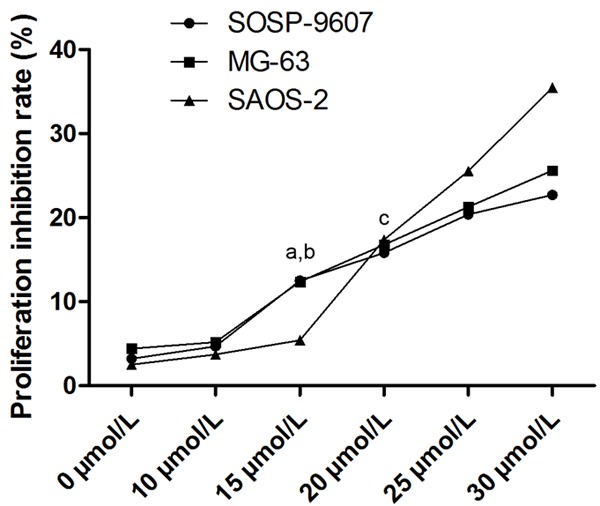
Effects of emodin incubation on proliferation of human OS cells. This chart showed proliferation inhibitory effect of emodin on SOSP-9607, MG-63 and SAOS-2 cells at serial concentrations ranging from 0 to 30 μmol/L. aDifferences were significantly from 10 μmol/L in SOSP-9607 cells; bdifferences were significantly from 10 μmol/L in MG-63 cells; cdifferences were significantly from 15 μmol/L in SAOS-2 cells.
Emodin dramatically reduced HMGB1 induced VEGF production in cultured human osteosarcoma cells
In cultured SOSP-9607, MG63 and SAOS-2 human osteosarcoma cells, emodin treatment significantly decreased VEGF expression in these cells in a concentration-dependent manner (Figure 3). Exogenous HMGB1 administration was found significantly induced cellular VEGF production which is considered fundamental in tumor angiogenesis. After emodin incubation, however, also in a concentration- dependent manner, HMGB1-induced VEGF production was found reduced dramatically.
Figure 3.
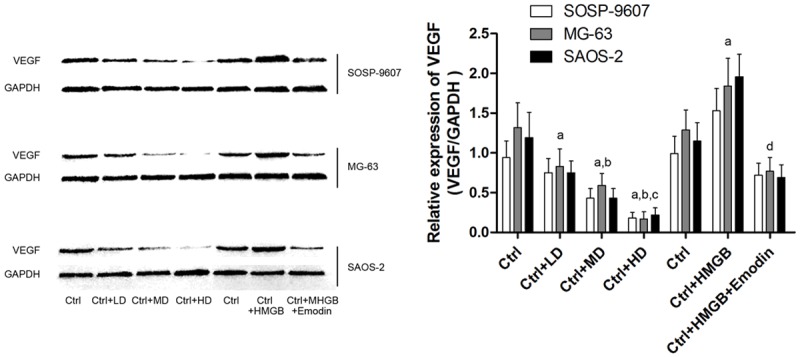
Effects of emodin and HMGB incubation on VEGF expression in human OS cells. Left part of this figure demonstrated the immunoblots of VEGF in human OS cells including SOSP-9607, MG-63 and SAOS-2 cells respectively. Ctrl: human OS cells; Ctrl+LD: human OS cells incubated with 2.5 μmol/L emodin; Ctrl+MD: human OS cells incubated with 5 μmol/L emodin; Ctrl+HD: human OS cells incubated with 10 μmol/L emodin. aDifferences were significantly from Ctrl; bdifferences were significantly from Ctrl+LD; cdifferences were significantly from Ctrl+MD; ddifferences were significantly from Ctrl+HMGB.
Emodin elevated SIRT1 expression level and SIRT1 mediated deacetylase activity in cultured human osteosarcoma cells
SIRT1 was considered to execute the role as a deacetylase on multiple nuclear transfactors. Previously, H4-k16 was thought one of the substrates of SIRT1. Expression of acetylase from of H4-k16 (H4-k16Ac) is considered an indicator of deacetylase activity of SIRT1. In this study, we found both of SIRT1 expression level and deacetylase activity were elevated after emodin treated OS cells (Figure 4). We also found that in HMGB1 incubated human osteosarcoma cells, emodin significantly increased both SIRT1 expression level and deacetylase activity (Figure 4). In all, emodin increased SIRT1 expression and deacetylase activity in human OS cells, which was not affected by HMGB1 incubation.
Figure 4.

Effects of emodin and HMGB incubation on SIRT1 expression and deacetylase activity in human OS cells. Left part of this figure demonstrated the immunoblots of SIRT1 and H4-k16AC in human OS cells including SOSP-9607, MG-63 and SAOS-2 cells respectively. Upper part of the right part of this figure showed the mRNA levels in human OS cells. Ctrl: human OS cells; Ctrl+LD: human OS cells incubated with 2.5 μmol/L emodin; Ctrl+MD: human OS cells incubated with 5 μmol/L emodin; Ctrl+HD: human OS cells incubated with 10 μmol/L emodin. aDifferences were significantly from Ctrl; bdifferences were significantly from Ctrl+LD; cdifferences were significantly from Ctrl+MD; ddifferences were significantly from Ctrl+HMGB.
SIRT1 silencing impaired emodin’s anti-VEGF production activity in cultured human osteosarcoma cells
After SIRT1 was silenced by siRNA, compared with counterpart transfected with siRNA vehicle control, we found that HMGB1 incubation dramatically increased VEGF expression in human OS cells (Figure 5). Furthermore, compared with vehicle siRNA control, SIRT1 silencing significantly impaired emodin’s ability of reducing VEGF expression which was believed crucial in malignant tumor angiogenesis in human OS cells (Figure 5).
Figure 5.

Effects of HMGB-incubated SIRT1-/-human OS cells on VEGF expression. Upper part of this figure demonstrated the immunoblots of VEGF and SIRT1 in HMGB-incubated SIRT1/human OS cells. Ctrl: human OS cells; siRNA: human OS cells incubated with SIRT1 siRNA; Ctrl+Emodin: human OS cells incubated with emodin; siRNA+Emodin: SIRT1/human OS cells incubated with emodin. ADifferences were significantly from Ctrl, Bdifferences were significantly from siRNA; Cdifferences were significantly from Ctrl+Emodin. aDifferences were significantly from Ctrl; bdifferences were significantly from HMGB.
Discussion
In this study, both of in vivo and in vitro investigations were implemented to explore the possible mechanisms of anti-angiogenesis activity of emodin. In the in vivo study, OS cells were planted to nude mice to form xenograft tumor. We found that HMGB1 administration aggravated the tumor associated angiogenesis which was then significantly attenuated by emodin administration. In the in vitro part, three kinds of human OS cell lines, namely SOSP-9607, MG63 and SAOS-2 cells were used. HMGB1 incubation induced over-expression of VEGF which was attenuated by emodin treatment. Moreover, the anti-angiogenesis activity of emodin was found associated with its promoting effect on SIRT1 expression and de acetylase activity in HMGB1 incubated OS cells. This association was then testified by the impairment of the inhibitory effect of emodin on VEGF over-expression in HMGB1-incubated OS cells.
OS is originated from mesenchymal stem cell (MSC) and considered as the most common cancer of bone, especially in adolescents [23]. It was reported that the overall 5-year survival rate of OS was 60%-70% [24]. The prognosis of OS is pessimistic due to its poor response to chemotherapy and high rate of relapse and metastasis. Except for cell proliferation and invasion, angiogenesis is also considered as one of the remarkable characteristics of malignant tumor including OS [25]. In addition, to some extent, it was believed that angiogenesis was the biological basis for cancer cell proliferation and invasion [26]. In this process, endothelial cells were recruited and stimulated to proliferate. These cells would migrate through basement membrane and extracellular matrix (ECM) and eventually form novel tubular blood vessels [27]. Therefore, on one hand, more blood supply would be introduced to support cancer cell proliferation; on the other hand, cancer cells would have more chance of invasion and metastasis because of more contact with blood vessels.
Many molecules were described involved in the process of cancer-associated angiogenesis, which were called angiogenic molecules, such as basic fibroblast growth factor (bFGF) [28], placental growth factor (PLGF) [29], epidermal growth factor (EGF) [30], VEGF [31] and so on. In previously studies, VEGF was reported positively correlated with tumor angiogenesis [32]. Ubiquitously distributed in cell nucli, HMGB1 is exerting massive biological functions. Previous studies have reviled the association of HMGB1 in the occurrence and development of malignant tumors. Over-expression of HMGB1 was found indicating the development and prognosis in several human cancers [33,34]. HMGB1 could interact with multiple down-stream molecules such as NF-κB [35], mitogen-activated protein kinase (MAPK) [36] and receptors for advanced glycation end products (RAGE) [37] to activate signaling pathways which play parts in tumor growth, invasion, metastasis and angiogenesis. In the present study, we found that in nude mice bearing human OS xenograft tumor, HMGB1 administration significantly increased angiogenesis in tumor tissue, which was indicated by elevated expression of VEGF and vWF. In the in vitro study, similar observation was obtained that HMGB1 treatment induced VEGF production in OS cells.
Angiogenesis inhibition could be a promising therapeutic strategy for malignant tumor prevention and treatment. Reagents with this anti-angiogenic effect are of great clinical significance. Emodin is one of the natural anthraquinones extracted from Rheum palmatum L. Literature records reporting emodin’s anti-cancer activity could be found as early as 1970s. Following studies found emodin could suppress cancer proliferation, invasion and metastasis in a variety of cancer cells including gastric cancer, cervical cancer, hepatic cancer and so on [38-40]. A recent study reported that emodin inhibited angiogenesis in pancreatic cancer by regulating NF-κB-associated angiogenic factors [41]. It was reported in another study that emodin suppressed VEGF-induced angiogenesis in human [42]. In this study, we found that in HMGB1 administrated nude mice, emodin treatment significantly attenuated tumor angiogenesis in tumor tissue. We further confirmed that emodin also dramatically down-regulated VEGF in HMGB1 incubated human OS cells.
Additionally, in our current study, possible mechanisms were also investigated. Some previous studies pointed out that VEGF gene transcription was regulated by its upper stream nuclear transfector-HIF-1 [43]. The HIF-1 activity is largely dependent on the acetylation degree of its alpha subunit, namely HIF-1α. It was reported that HMGB1 induced tube formation by elevating acetylation of HIF-1α, while SIRT1 was the main molecule inducing HIF-1α deacetylation directly [44]. Thus, SIRT1 is the possible regulator in HMGB1-induced angiogenesis in OS and target of emodin. In the present study, we tried to testify this presupposition. In HMGB1-incubated OS cells, emodin significantly elevated SIRT1 expression level and its deacetylation activity. Thus, HMGB1-induced HIF-1α acetylation is attenuated by emodin-regulated SIRT1-induced HIF-1α deacetylation. As a result, HIF-1α-induced VEGF production was decreased by emodin administration. Furthermore, siRNA technique was also employed to silence SIRT1 expression in OS cells. We found that in SIRT1 silenced OS cells, HMGB1 treatment increased cellular VEGF production compared with wild type. Moreover, we also found that, in OS cells, SIRT1 silencing impaired emodin’s inhibiting effect against VEGF production. These results indicated that SIRT1 was the molecular target of emodin when exerting its anti-angiogenesis effect in OS.
In conclusion, our results demonstrate that HMGB1 administration exacerbates tumor angiogenesis by inducing VEGF in human OS. Emodin alleviates tumor angiogenesis by reducing cellular VEGF production in human OS. Furthermore, SIRT expression and deacetylation activity elevation are involved in emodin’s anti-angiogenesis effect in human OS.
Disclosure of conflict of interest
None.
References
- 1.Saeter G. Osteosarcoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2007;18(Suppl 2):ii77–78. doi: 10.1093/annonc/mdm047. [DOI] [PubMed] [Google Scholar]
- 2.Fagioli F, Biasin E, Mereuta OM, Muraro M, Luksch R, Ferrari S, Aglietta M, Madon E. Poor prognosis osteosarcoma: new therapeutic approach. Bone Marrow Transplant. 2008;41(Suppl 2):S131–134. doi: 10.1038/bmt.2008.71. [DOI] [PubMed] [Google Scholar]
- 3.Hamscho N, Grunwald F. Prognosis of primary osteosarcoma. J Nucl Med. 2003;44:996–997. author reply 997. [PubMed] [Google Scholar]
- 4.Nathan SS, Gorlick R, Bukata S, Chou A, Morris CD, Boland PJ, Huvos AG, Meyers PA, Healey JH. Treatment algorithm for locally recurrent osteosarcoma based on local diseasefree interval and the presence of lung metastasis. Cancer. 2006;107:1607–1616. doi: 10.1002/cncr.22197. [DOI] [PubMed] [Google Scholar]
- 5.Russo G, Mischi M, Scheepens W, De la Rosette JJ, Wijkstra H. Angiogenesis in prostate cancer: onset, progression and imaging. BJU Int. 2012;110:E794–808. doi: 10.1111/j.1464-410X.2012.11444.x. [DOI] [PubMed] [Google Scholar]
- 6.Ruf W, Yokota N, Schaffner F. Tissue factor in cancer progression and angiogenesis. Thromb Res. 2010;125(Suppl 2):S36–38. doi: 10.1016/S0049-3848(10)70010-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yano S, Goto H, Yamamoto A, Kanematsu T, Sone S. Angiogenesis in the progression of lung cancer. Intern Med. 2003;42:305–309. doi: 10.2169/internalmedicine.42.305. [DOI] [PubMed] [Google Scholar]
- 8.Zheng HC, Li YL, Sun JM, Yang XF, Li XH, Jiang WG, Zhang YC, Xin Y. Growth, invasion, metastasis, differentiation, angiogenesis and apoptosis of gastric cancer regulated by expression of PTEN encoding products. World J Gastroenterol. 2003;9:1662–1666. doi: 10.3748/wjg.v9.i8.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boedefeld WM 2nd, Bland KI, Heslin MJ. Recent insights into angiogenesis, apoptosis, invasion, and metastasis in colorectal carcinoma. Ann Surg Oncol. 2003;10:839–851. doi: 10.1245/aso.2003.02.021. [DOI] [PubMed] [Google Scholar]
- 10.Pooja T, Karunagaran D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur J Pharmacol. 2014;742:55–64. doi: 10.1016/j.ejphar.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 11.Chung JG, Wang HH, Wu LT, Chang SS, Chang WC. Inhibitory actions of emodin on arylamine N-acetyltransferase activity in strains of Helicobacter pylori from peptic ulcer patients. Food Chem Toxicol. 1997;35:1001–1007. doi: 10.1016/s0278-6915(97)87269-9. [DOI] [PubMed] [Google Scholar]
- 12.Kim JR, Oh DR, Cha MH, Pyo BS, Rhee JH, Choy HE, Oh WK, Kim YR. Protective effect of polygoni cuspidati radix and emodin on Vibrio vulnificus cytotoxicity and infection. J Microbiol. 2008;46:737–743. doi: 10.1007/s12275-008-0232-x. [DOI] [PubMed] [Google Scholar]
- 13.Masaldan S, Iyer VV. Exploration of effects of emodin in selected cancer cell lines: enhanced growth inhibition by ascorbic acid and regulation of LRP1 and AR under hypoxia-like conditions. J Appl Toxicol. 2014;34:95–104. doi: 10.1002/jat.2838. [DOI] [PubMed] [Google Scholar]
- 14.Han YM, Lee SK, Jeong DG, Ryu SE, Han DC, Kim DK, Kwon BM. Emodin inhibits migration and invasion of DLD-1 (PRL-3) cells via inhibition of PRL-3 phosphatase activity. Bioorg Med Chem Lett. 2012;22:323–326. doi: 10.1016/j.bmcl.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Jia X, Yu F, Wang J, Iwanowycz S, Saaoud F, Wang Y, Hu J, Wang Q, Fan D. Emodin suppresses pulmonary metastasis of breast cancer accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res Treat. 2014;148:291–302. doi: 10.1007/s10549-014-3164-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren H, Zhu C, Li Z, Yang W, Song E. Emodinloaded magnesium silicate hollow nanocarriers for anti-angiogenesis treatment through inhibiting VEGF. Int J Mol Sci. 2014;15:16936–16948. doi: 10.3390/ijms150916936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan RS, Fonseca-Kelly Z, Callinan C, Zuo L, Sachdeva MM, Shindler KS. SIRT1 activating compounds reduce oxidative stress and prevent cell death in neuronal cells. Front Cell Neurosci. 2012;6:63. doi: 10.3389/fncel.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Gao P, Zhang H, Chen H, Zheng W, Lv X, Xu T, Wei Y, Liu D, Liang C. SIRT1 inhibits angiotensin II-induced vascular smooth muscle cell hypertrophy. Acta Biochim Biophys Sin (Shanghai) 2011;43:103–109. doi: 10.1093/abbs/gmq104. [DOI] [PubMed] [Google Scholar]
- 19.Calvanese V, Fraga MF. SirT1 brings stemness closer to cancer and aging. Aging (Albany NY) 2011;3:162–167. doi: 10.18632/aging.100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi J, Luo J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta. 2010;1804:1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tseng RC, Lee CC, Hsu HS, Tzao C, Wang YC. Distinct HIC1-SIRT1-p53 loop deregulation in lung squamous carcinoma and adenocarcinoma patients. Neoplasia. 2009;11:763–770. doi: 10.1593/neo.09470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HY, Park SY, Lee SW, Lee HR, Lee WS, Rhim BY, Hong KW, Kim CD. Inhibition of HMGB1-induced angiogenesis by cilostazol via SIRT1 activation in synovial fibroblasts from rheumatoid arthritis. PLoS One. 2014;9:e104743. doi: 10.1371/journal.pone.0104743. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Janeway KA, Barkauskas DA, Krailo MD, Meyers PA, Schwartz CL, Ebb DH, Seibel NL, Grier HE, Gorlick R, Marina N. Outcome for adolescent and young adult patients with osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2012;118:4597–4605. doi: 10.1002/cncr.27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett JH, Thomas G, Evans AW, Speight PM. Osteosarcoma of the jaws: a 30-year retrospective review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:323–332. doi: 10.1067/moe.2000.108274. [DOI] [PubMed] [Google Scholar]
- 25.Mikulic D, Ilic I, Cepulic M, Orlic D, Giljevic JS, Fattorini I, Seiwerth S. Tumor angiogenesis and outcome in osteosarcoma. Pediatr Hematol Oncol. 2004;21:611–619. doi: 10.1080/08880010490501015. [DOI] [PubMed] [Google Scholar]
- 26.Mohammed ZM, McMillan DC, Edwards J, Mallon E, Doughty JC, Orange C, Going JJ. The relationship between lymphovascular invasion and angiogenesis, hormone receptors, cell proliferation and survival in patients with primary operable invasive ductal breast cancer. BMC Clin Pathol. 2013;13:31. doi: 10.1186/1472-6890-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheresh DA, Stupack DG. Regulation of angiogenesis: apoptotic cues from the ECM. Oncogene. 2008;27:6285–6298. doi: 10.1038/onc.2008.304. [DOI] [PubMed] [Google Scholar]
- 28.Walgenbach KJ, Gratas C, Shestak KC, Becker D. Ischaemia-induced expression of bFGF in normal skeletal muscle: a potential paracrine mechanism for mediating angiogenesis in ischaemic skeletal muscle. Nat Med. 1995;1:453–459. doi: 10.1038/nm0595-453. [DOI] [PubMed] [Google Scholar]
- 29.Albrecht ED, Robb VA, Pepe GJ. Regulation of placental vascular endothelial growth/permeability factor expression and angiogenesis by estrogen during early baboon pregnancy. J Clin Endocrinol Metab. 2004;89:5803–5809. doi: 10.1210/jc.2004-0479. [DOI] [PubMed] [Google Scholar]
- 30.Dunn IF, Heese O, Black PM. Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neurooncol. 2000;50:121–137. doi: 10.1023/a:1006436624862. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Zan LP, Wang XD, Lu YJ, Ou TM, Lin J, Huang ZS, Gu LQ. Stabilization of VEGF G-quadruplex and inhibition of angiogenesis by quindoline derivatives. Biochim Biophys Acta. 2014;1840:2970–2977. doi: 10.1016/j.bbagen.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Cao Y, E G, Wang E, Pal K, Dutta SK, Bar-Sagi D, Mukhopadhyay D. VEGF exerts an angiogenesis-independent function in cancer cells to promote their malignant progression. Cancer Res. 2012;72:3912–3918. doi: 10.1158/0008-5472.CAN-11-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res. 2013;19:4046–4057. doi: 10.1158/1078-0432.CCR-13-0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abe A, Kuwata T, Yamauchi C, Higuchi Y, Ochiai A. High Mobility Group Box1 (HMGB1) released from cancer cells induces the expression of pro-inflammatory cytokines in peritoneal fibroblasts. Pathol Int. 2014;64:267–275. doi: 10.1111/pin.12167. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Kou YB, Zhu JS, Chen WX, Li S. Knockdown of HMGB1 inhibits growth and invasion of gastric cancer cells through the NFkappaB pathway in vitro and in vivo. Int J Oncol. 2014;44:1268–1276. doi: 10.3892/ijo.2014.2285. [DOI] [PubMed] [Google Scholar]
- 36.Wang L, Yu L, Zhang T, Leng Z, Guan Y, Wang X. HMGB1 enhances embryonic neural stem cell proliferation by activating the MAPK signaling pathway. Biotechnol Lett. 2014;36:1631–1639. doi: 10.1007/s10529-014-1525-2. [DOI] [PubMed] [Google Scholar]
- 37.Stoetzer OJ, Fersching DM, Salat C, Steinkohl O, Gabka CJ, Hamann U, Braun M, Feller AM, Heinemann V, Siegele B, Nagel D, Holdenrieder S. Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumour Biol. 2013;34:81–90. doi: 10.1007/s13277-012-0513-1. [DOI] [PubMed] [Google Scholar]
- 38.Guo J, Xiao B, Liu Q, Gong Z, Le Y. Suppression of C-myc expression associates with anti-proliferation of aloe-emodin on gastric cancer cells. Cancer Invest. 2008;26:369–374. doi: 10.1080/07357900701788130. [DOI] [PubMed] [Google Scholar]
- 39.Yu JQ, Bao W, Lei JC. Emodin regulates apoptotic pathway in human liver cancer cells. Phytother Res. 2013;27:251–257. doi: 10.1002/ptr.4703. [DOI] [PubMed] [Google Scholar]
- 40.Yaoxian W, Hui Y, Yunyan Z, Yanqin L, Xin G, Xiaoke W. Emodin induces apoptosis of human cervical cancer hela cells via intrinsic mitochondrial and extrinsic death receptor pathway. Cancer Cell Int. 2013;13:71. doi: 10.1186/1475-2867-13-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin SZ, Wei WT, Chen H, Chen KJ, Tong HF, Wang ZH, Ni ZL, Liu HB, Guo HC, Liu DL. Antitumor activity of emodin against pancreatic cancer depends on its dual role: promotion of apoptosis and suppression of angiogenesis. PLoS One. 2012;7:e42146. doi: 10.1371/journal.pone.0042146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meng G, Liu Y, Lou C, Yang H. Emodin suppresses lipopolysaccharide-induced pro-inflammatory responses and NF-kappaB activation by disrupting lipid rafts in CD14-negative endothelial cells. Br J Pharmacol. 2010;161:1628–1644. doi: 10.1111/j.1476-5381.2010.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamazaki Y, Egawa K, Nose K, Kunimoto S, Takeuchi T. HIF-1-dependent VEGF reporter gene assay by a stable transformant of CHO cells. Biol Pharm Bull. 2003;26:417–420. doi: 10.1248/bpb.26.417. [DOI] [PubMed] [Google Scholar]
- 44.Park SY, Lee SW, Kim HY, Lee WS, Hong KW, Kim CD. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1alpha activation. Eur J Immunol. 2014;24:201444908. doi: 10.1002/eji.201444908. [DOI] [PubMed] [Google Scholar]