

Abstract
High mobility group box-B1 (HMGB1) is upregulated in tumors, inflammations, and other injuries. However, its extracellular role and signaling in wound healing remains unclear. In the present study, we examined the HMGB1 levels in hematoma samples in fractured bones and in human macrophagy U937 cells under hypoxia with enzyme-linked immunosorbent assay (ELISA) and western blotting. Then we investigated the activation of the extracellular signal-regulated kinases (ERK) and c-Jun N-terminal kinases (JNK) signaling western blotting in osteoblast MG-63 cells under hypoxia, with or without HMGB1 treatment. And then we assessed the effects of extracellular HMGB1 on cell proliferation of MG-63 cells with CCK-8 assay. It was demonstrated that HMGB1 expression was significantly up regulated in hematoma samples in fractured bones and in U937 cells under hypoxia. MG-63 cells under hypoxia showed an increased HMGB1 in the cytoplasm rather than in nuclei. And the extracellular HMGB1 ameliorated the hypoxia-induced viability reduction and promoted the proliferation of MG-63 cells. Moreover, the MG-63 cells incubated with HMGB1 had increased ERK1/2 phosphorylation, whereas such effect was blocked by the TLR-4 knockout with SIRNA-TLR-4 transfection. In conclusion, we found the up regulation HMGB1 in the hematoma of fractured bones and in macrophage U937 cells under hypoxia, and the hypoxia-up regulated HMGB1 promoted the proliferation of osteoblast MG-63 cells and activated the phosphorylation of ERK and JNK. And the proliferation promotion and the activation of ERK/JNK signaling was TLR-4-dependent.
Keywords: Hypoxia, HMGB1, osteoblast cells, proliferation, ERK/JNK signaling
Introduction
Bone fracture is a prevalent medical condition, in which the urgent alteration of the bone microenvironment causes a reduced blood supply and subsequent hypoxia. And the healing process of bone fracture starts with the hematoma and inflammation surrounding the injured bone and ends with the re-structure of the broken bone [1,2]. However, the healing process is promoted or hindered by several factors mainly via affecting the growth and differentiation of osteoblasts and the mineralization of the collagen matrix [3]. Serious hypoxia is one of the most prominent outcomes following fractures, significantly influencing their healing process [4]. Hypoxia promotes the expression of such transcriptional factors and cytokines [5] as Hypoxia-inducible factors (HIFs), vascular endothelial growth factor (VEGF) [6], bone morphogenetic protein 2 (BMP-2) [7,8].
Studies have shown a significant interaction between the local and systemic inflammatory response after severe trauma in small animal models. However, the involvement of the inflammatory responses in mechanisms of bone fracture healing is still poorly comprehended. Various types of immune cells and cytokines have been indicated to be involved in the wound healing or bone regeneration [9-12]. Studies have indicated the infiltration of immune cells into already existing fracture hematomas on the early inflammatory phase of fracture healing [13,14]. And such cytokines as interleukin (IL)-8 [15], transforming growth factor beta (TGF-β) [16] and IL-6 [17] are confirmed to be upregulated by hypoxia in an association with the bone fracture.
High-mobility group protein B1 was initially described as a cytokine-like factor in models of sepsis [18], and now is well-known to be a key mediator of inflammation in multiple injury models, such as hemorrhagic lung injury [19], hepatic ischemia-reperfusion [20], hemorrhagic shock [21]. HMGB1 interacts with Toll-like receptor (TLR)-4 [22-24] and initiates initial inflammatory response to injury. However, the role of HMGB1 in the inflammatory response following bone fractures is unknown.
The purpose of this study was to examine the promotion to HMGB1 in the hematoma specimens and in macrophages under hypoxia. Then we investigated the regulation of HMGB1 on the proliferation of osteoblast cells, and on the activation of TLR-4 and RAGE under normoxia or hypoxia. We also examined the phosphorylation of extracellular signal-regulated kinases (ERK) and c-Jun N-terminal kinases (JNK) in the HMGB1-treated osteoblast cells under normoxia or hypoxia. In addition, the xx-specific SIRNA was transfected into the HMGB1-treated osteoblast cells under hypoxia and re-examined the cell proliferation and the ERK/JNK phosphorylation. Our study recognized the key regulatory role of hypoxia-induced HMGB1 on the proliferation of osteoblast cells via regulating the ERK/JNK phosphorylation, TLR-4-dependently.
Materials and methods
Hematoma samples, cell culture and treatment
Total of 31 patients with open femur fracture were enrolled from March 2014 to March 2015 in the Department of Orthopedics and Traumatology, Nanfang Hospital, Southern Medical University, and were involved in this study. The accumulated hematoma samples in the wound and the fresh bleeding sample during surgery were collected for the HMGB1 assay. Written consent was obtained from each patient before the study, which was also approved by the Ethics Committee of Nanfang Hospital, Southern Medical University.
U937 cells were cultured and maintained in RPMI-1640 medium (Hyclone, Logan, UT, USA) which was supplemented with 10% or 2% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA), with 50 μg/ml penicillin and with 50 μg/ml streptomycin (CSPC Pharmaceutical Group Limited, Shijiazhuang, China). Cultures can be established by centrifugation with subsequent resuspension of 1-2 × 105 viable cells/mL. Fresh medium was updated every 3 to 4 days (depending on cell density). For hypoxia treatment, cells were placed in a hypoxia incubator infused with a gas mixture of 5% CO2 and nitrogen to obtain 2% oxygen concentration. For the HMGB1 assay, U937 cells were incubated under normoxia or under hypoxia for 8, 12, 24 or 48 hours, then the supernatant of U937 cells and cells in each group were collected for HMGB1 assay.
MG-63 cells were also grown in RPMI-1640 medium supplemented with 10% FBS, 50 μg/mL penicillin and 50 μg/mL streptomycin. The two types of cells were incubated at 37°C, with 5% CO2 in a humid incubator. For hypoxia treatment, cells were placed in a hypoxia incubator infused with a gas mixture of 5% CO2 and nitrogen to obtain 2% oxygen concentration. For the HMGB1 treatment, MG-63 cells with 85-95% confluence were incubated with RPMI-1640 (2% FBS) supplemented with HMGB1 for 0, 0.2 or 1 μg/mL for 24, 48 or 72 hours under normoxia or under hypoxia. To knockout the TLR-4 in MG-63 cells, SIRNA-TLR-4 and control SIRNA were synthesized by Gene Pharma Technology (Shanghai, China) and were transfected into MG-63 cells with a concentration of 30 or 60 nM by lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Elisa for HMGB1
HMGB1 in the hematoma or fresh bleeding samples, or in the supernatant of macrophage U937 cells were examined with the enzyme-linked immunosorbent assay (ELISA) kit (Westang Bio, Shanghai, China) under the guidance of the product’s manual. The microplate was added with 100 μL standard samples or with the serially-diluted samples, and was incubated at 37°C for two hours. Then samples were aspirated, and the plate was washed for four times with 100 μL phosphate buffered saline with Tween 20 (PBST) in each well. Then the plate was added with 100 μL antibody against HMGB1 in each well, and was incubated 37°C for one hour. Post four-time washing, the plate was added with 100 μL secondary antibody conjugated with horseradish peroxidase and was incubated for 30 minutes at 37°C. Post the inoculation with 100 μL substrate at dark for 15 minutes, the specific binding optical density of each well was determined immediately at 450 nm.
MRNA preparation and quantitative analysis
MRNA samples from MG-63 cells were prepared with the TRizol reagent (Life Technologies, Grand Island, NY, USA), and were quantified by real-time quantitative PCR (RT-qPCR). SYBR Green PCR Kits (Takara, Tokyo, Japan) was utilized to quantify the mRNA level of ERK, JNK, TLR-4, or Tubulin in MG-63 cells. The QPCR was performed on the ABI PRISM 7300 detection system. The primers for each molecule were synthesized by Shanghai Sangon Company (Sangon, Shanghai, China). All data were presented as the fold change over the internal control Tubulin and were calculated with the ∆∆Ct method [25].
Western blotting assay
Cytosolic or nuclear protein samples were isolated with the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, Waltham, MA, USA), and were supplemented with a Protease Inhibitor Cocktail (Abcam, Cambridge, UK). Protein samples were separated by the 10% or 12% SDS-PAGE gel, and were transferred to nitrocellulose membrane (Millipore, Bedford, MA, USA). Then the membrane was blocked with 2% BSA (dissolved in PBST) overnight at 4°C to cover the non-specific binding on the membrane, and the HMGB1, ERK with or without the phosphorylated Thr202/Thr204, JNK with or without the phosphorylated Thr183, TLR-4 or Tubulin was detected with rabbit polyclonal IGG against each marker (all from Cell Signaling Technology Inc. (Danvers, MA, USA) or Tubulin (Sinobio, Beijing, China). A peroxidase-conjugated secondary antibody against rabbit IGG (Jackson ImmunoResearch, West Grove, PA, USA) and the electrochemoluminescence (ECL) detection system (Amersham, Uppsala, Sweden) were used to present the specific binding. The level of HMGB1 or TLR-4 was presented as a relative gray value to Tubulin, whereas the level of p-ERK or p-JNK was presented as the relative gray value to ERK or to JNK.
Cell proliferation assay with CCK-8
Proliferation of MG-63 cells under normoxia or hypoxia, with or without the HMGB1 treatment, with the transfection with siRNA-TLR-4 or SIRNA-Con was performed with CCK-8 assay (DOJINDO, Kumamoto, Japan). Briefly, MG-63 cells in each group were incubated with CCK-8. The 490 nm absorbance of cells was detected after visual color occurrence.
Statistical analysis
Data was presented as mean ± SEM. And statistical difference was analyzed with SPSS18.0 software (IBM SPSS, Armonk, NY, USA). The difference between two groups was analyzed by Student’s t test. A p value less than 0.05 was considered to be significant.
Results
HMGB1 was up regulated in the hematoma of fractured bones and in macrophage U937 cells under hypoxia
To recognize the role of HMGB1 in bone fracture, we firstly examined the level of HMGB1 in the hematoma specimens (n=31) which were produced during the bone fracture, with the fresh bleeding samples (n=31) as control. It was demonstrated in Figure 1A that the average HMGB1 level in the hematoma samples from bone fracture was 31.730 ± 3.197 ng/mL, significantly higher than the level of 9.845 ± 0.967 ng/mL in the fresh bleeding samples from bone fracture (P < 0.0001). Then we investigated the regulation of HMGB1 by hypoxia in the human macrophage, which is the main type of HMGB1 producers in blood. As shown in Figure 1B, from 12 to 48 hours post treatment, the HMGB1 level in the supernatant of macrophage U937 cells was significantly up regulated by the hypoxia treatment (P < 0.01 or P < 0.001), with the U937 cells under normoxia as control. In addition, we analyzed the nucleus and cytosol distribution of HMGB1 in U937 cells under hypoxia or normoxia. The western blotting (Figure 1C) results indicated that the HMGB1 in nucleus was not markedly different between the normoxia and hypoxia groups. However, the cytosolic HMGB1 was promoted by hypoxia, the relative cytosol/nucleus level of HMGB1 was significantly up regulated by hypoxia at 24 or 48 hours post treatment (P < 0.01 respectively). Thus, we confirmed the up regulation of HMGB1 in the hematoma of fractured bones and in macrophage U937 cells under hypoxia.
Figure 1.
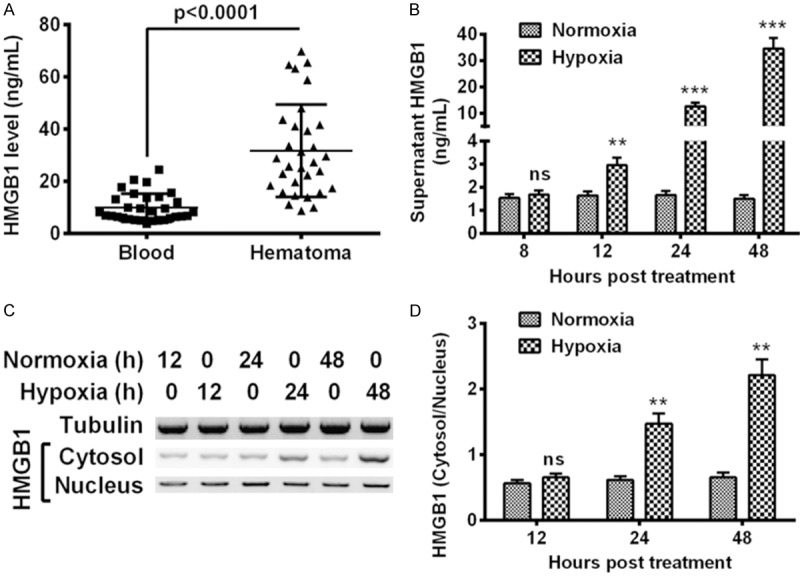
High mobility group box-B1 (HMGB1) levels in the hematoma of fractured bones and in macrophage U937 cells under hypoxia. A: Enzyme-linked immunosorbent assay (ELISA) for HMGB1 in the hematoma specimens or in the fresh bleeding samples from fractured bones; B: HMGB1 level in the supernatant of macrophage U937 cells under normoxia or hypoxia for 8, 12, 24 or 48 hours; C: Western blot analysis of HMGB1 in cytosol and in nucleus in U937 cells under normoxia or hypoxia for 12, 24 or 48 hours, with tubulin as internal control; D: Relative level of HMGB1 in cytosol to in nucleus in U937 cells. The data represent three independent experiments, and shown as Mean ± SE. *P < 0.05, **P < 0.01, ***P < 0.001. ns: no significance.
HMGB1 promotes the proliferation of osteoblast MG-63 cells and up regulates TLR-4
We then investigated the regulation of the hypoxia-promoted HMGB1 on the proliferation of MG-63 cells. The growth curve of MG-63 cells which were treated with 0, 0.2 or 1 μg/mL HMGB1 was assessed. It was demonstrated that the MG-63 cells which were treated with 0.2 μg/mL HMGB1 grew to higher levels at 24, 48 or 72 hour post treatment (P < 0.05 or P < 0.01) under normoxia. And the treatment with 1 μg/mL HMGB1 promoted even a higher level of cell proliferation than the treatment with 0.2 μg/mL HMGB1 (P < 0.05 respectively). Such promotion to the cell proliferation was also confirmed in the MG-63 cells under hypoxia. Figure 2B indicated that activity reduction of MG-63 cells was markedly inhibited by the treatment with 0.2 μg/mL HMGB1 at 24, 48 or 72 hour post treatment (P < 0.05 or P < 0.01).
Figure 2.
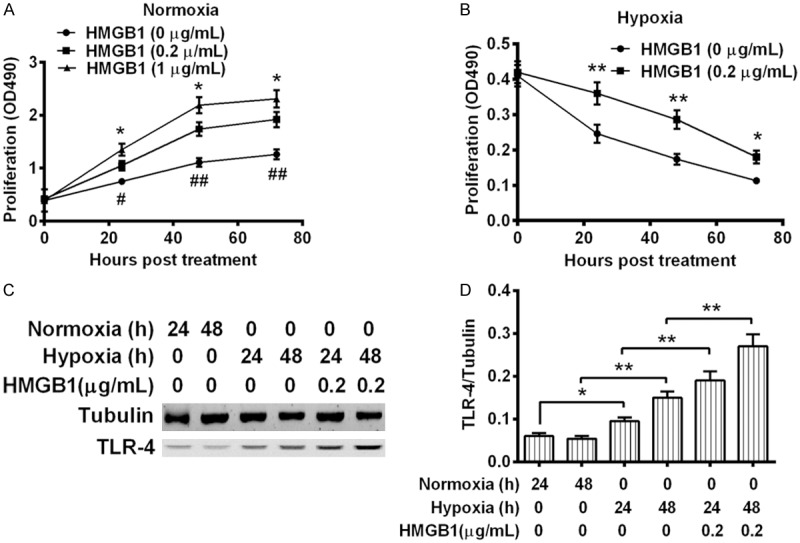
Growth curves of and TLR-4 expression in the osteoblast MG-63 cells treated with HMGB1 under normoxia or hypoxia. (A and B) Growth curve of HMGB1-treated osteoblast MG-63 cells under normoxia (A) or under hypoxia (B) MG-63 cells with more than 85% confluence were treated with 0, 0.2 or 1 μg/mL HMGB1 for 24, 48 or 72 hours, under normoxia or under hypoxia, and then were examined with CCK-8 assay. (C) Western blot analysis of Toll-like receptor (TLR)-4 in U937 cells which were treated with or without 0.2 μg/mL HMGB1, under normoxia or under hypoxia for 24 or 48 hours, with tubulin as internal control; (D) Relative level of TLR-4 to tubulin in U937 cells. Results were repeated in triplicate and were shown as Mean ± SE. * or #P < 0.05, ** or ##P < 0.01.
The inflammatory response exerted by HMGB1 is usually initiated by the interaction of HMGB1 with TLR-4 [22-24]. To recognize the mechanism underlining the proliferation promotion by HMGB1 in osteoblast MG-63 cells, we then examined the level of TLR-4 in MG-63 cells under normoxia or hypoxia, with or without the HMGB1 treatment. The western blot analysis (Figure 2C) indicated that hypoxia markedly upregulated TLR-4 in MG-63 cells under hypoxia rather than under normoxia (P < 0.05 or P < 0.01, Figure 2D), particularly in the presence of 0.2 μg/mL HMGB1 (P < 0.01 for column 5 vs. column 3, or for column 6 vs. column 4, Figure 2D). Therefore, HMGB1 promoted the proliferation of osteoblast MG-63 cells and up regulated TLR-4.
HMGB1 induces the phosphorylation of ERK and JNK in MG-63 cells
HMGB1 initiates inflammatory reactions through the activation of ERK [26] and JNK [27] signaling. To clarify the mechanisms by which the hypoxia-induced HMGB1 promotes the proliferation of MG-63 cells, we then determined the expression and the phosphorylation of ERK and JNK in the HMGB1-treated MG-63 cells under normoxia or hypoxia. It was indicated in Figure 3A that the mRNA level of ERK was significantly unregulated by hypoxia at 12 hour post treatment (P < 0.05). Particularly, such upregulation was aggravated by additional HMGB1 treatment (0.2 μg/mL) (P < 0.05). And such aggravation lasted to the 24 hour post treatment (P < 0.05). And the mRNA level of JNK was also upregulated by the HMGB1 treatment under hypoxia in MG-63 cells under hypoxia at 12 hour post treatment (P < 0.05, Figure 3B). Then we examined the phosphorylation of both signaling markers in the HMGB1-treated MG-63 cells under normoxia or hypoxia with western blot analysis (Figure 3C). Results demonstrated that the phosphorylation levels of both markers were markedly promoted by the hypoxia treatment (P < 0.05 or P < 0.01, Figure 3D), and the hypoxia-promoted phosphorylation was also aggravated by the HMGB1 treatment (P < 0.01 respectively, Figure 3D). Taken together, the hypoxia upregulated the phosphorylation of both ERK and JNK, and the upregulation was aggravated by HMGB1.
Figure 3.
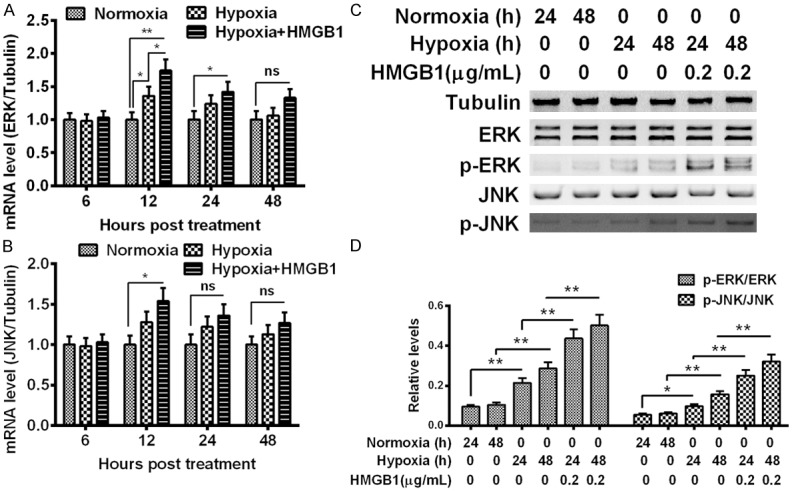
Promotion of ERK and JNK in the MG-63 cells treated with HMGB1 under normoxia or hypoxia. (A and B) mRNA levels of extracellular signal-regulated kinases (ERK) (A) and c-Jun N-terminal kinases (JNK) (B) in MG-63 cells which were cultured under normoxia or hypoxia, and were treated with 0.2 μg/mL HMGB1 for 6, 12, 24 or 48 hours, under normoxia or under hypoxia, and then were examined with MTT assay. (C) Western blot analysis of ERK or phosphorylated ERK, of JNK or phosphorylated JNK in MG-63 cells under normoxia or hypoxia, treated with or without with 0.2 μg/mL HMGB1, for 24 or 48 hours, with tubulin as internal control; (D) Relative phosphorylated ERK or phosphorylated JNK to total ERK or total JNK in the HMGB1-treated MG-63 cells under normoxia or hypoxia. The data represent three independent experiments, and shown as Mean ± SE. *P < 0.05, **P < 0.01, ns: no significance.
TLR-4 knockout inhibits the HMGB1-promoted proliferation of MG-63 cells
We next elucidated the role of the main cell membrane HMGB1 receptors TLR4, in HMGB1-induced proliferation of MG-63 cells. TLR-4-specific SIRNA was utilized to knockout the TLR-4 expression in the MG-63 cells, and results demonstrated in Figure 4A that siRNA-TLR-4 with 30 or 60 nM reduced the relative TLR-4 mRNA level from 1.00 ± 0.12 or 1.00 ± 0.13 to 0.53 ± 0.058 or to 0.32 ± 0.037 (P < 0.01 respectively). And the siRNA-TLR-4 transfection also significantly blocked the hypoxia-induced TLR-4 in MG-63 cells in protein level at either 24 or 48 hour post transfection (P < 0.01 respectively, Figure 4B). Then we re-curved the growth of MG-63 cells under hypoxia or normoxia, post the transfection with 60 NM SIRNA-TLR-4 or SIRNA-Con. As shown in Figure 4C, the SIRNA-TLR-4 significantly reduced the proliferation level of MG-63 cells under normoxia at 24, 48 or 72 hour post transfection, compared with the SIRNA-Con (P < 0.05 or P < 0.01). Moreover, the knockout of TLR-4 with siRNA-TLR-4 markedly reduced the cell viability than the siRNA-Con transfection (P < 0.05 or P < 0.01, Figure 4D). Thus, we confirmed the TLR-4-dependence of the HMGB1-induced MG-63 cell proliferation under normoxia, or of the HMGB1-mediated cellular viability amelioration of MG-63 cells under hypoxia.
Figure 4.
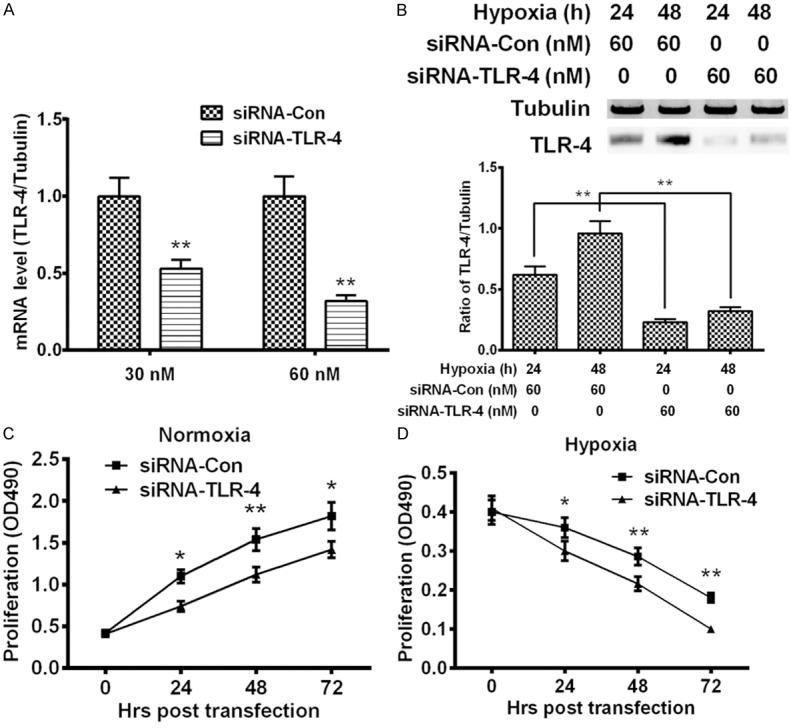
Growth curves of HMGB1-treated MG-63 cells post the TLR-4 knockout under normoxia or hypoxia. (A) The relative level of Toll-like receptor (TLR)-4 in MG-63 cells which were transfected with 30 or 60 NM TLR-4-specific SIRNA (SIRNA-TLR-4) or with control SIRNA (SIRNA-Con) for 12 hours; (B) Western blotting of TLR-4 in the siRNA-TLR-4- or siRNA-Con-transfected MG-63 cells under hypoxia for 24 hours; (C and D) Growth curve of siRNA-TLR-4- or SIRNA-Con-transfected MG-63 under normoxia (C) or under hypoxia (D). MG-63 cells with more than 85% confluence were transfected with 30 or 60 NM SIRNA-TLR-4 or with siRNA-Con were incubated under normoxia or under hypoxia for 24, 48 or 72 hours, and then were examined with CCK-8 assay. All experiments were performed respectively in triplicate. *P < 0.05, **P < 0.01.
TLR-4 knockout reduces the HMGB1-induced phosphorylation of ERK and JNK in MG-63 cells
We also re-evaluated the phosphorylation levels of ERK and JNK in the hypoxia-treated MG-63 cells post the knockout of TLR-4. The level of ERK and JNK with or without phosphorylation were analyzed with western blotting (Figure 5A) in the hypoxia-treated MG-63 cells which were transfected with 60 NM SIRNA-TLR-4 or with siRNA-Con. Results demonstrated that compared to the SIRNA-Con, siRNA-TLR-4 significantly blocked the hypoxia-induced phosphorylation of ERK at either 24 or 48 hour post transfection (P < 0.01 respectively, Figure 5B). And the hypoxia-induced phosphorylation level of JNK was also markedly inhibited by SIRNA-TLR-4 with 60 NM (P < 0.01 respectively, Figure 5C). Therefore, the hypoxia- and following promoted HMGB1-induced phosphorylation of ERK and JNK was TLR-4-dependent.
Figure 5.
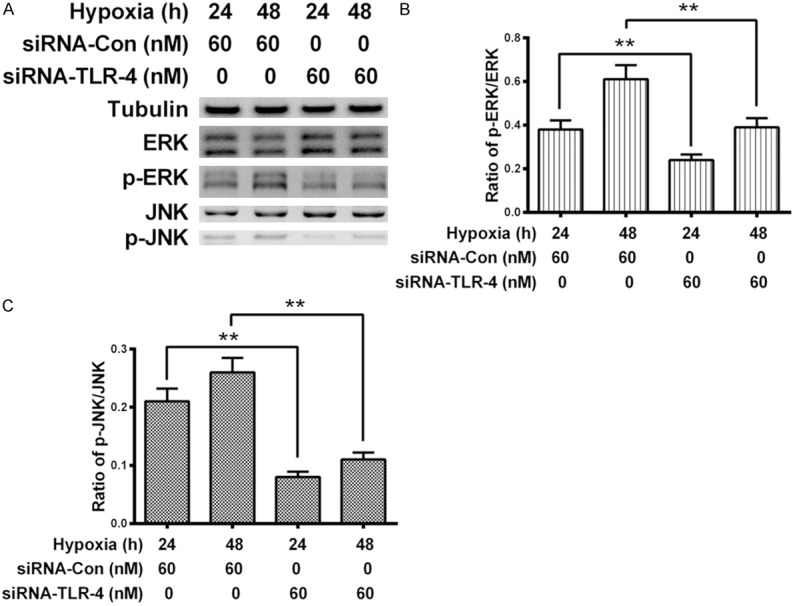
Promotion of ERK and JNK in the MG-63 cells post the TLR-4 knockout under normoxia or hypoxia. (A) Western blot analysis of ERK or phosphorylated ERK, of JNK or phosphorylated JNK in the siRNA-TLR-4- or SIRNA-Con-transfected (60 NM) MG-63 cells under hypoxia for 24 or 48 hours; (B and C) Relative level of phosphorylated ERK to total ERK (B) or relative phosphorylated JNK or total JNK (C) in the SIRNA-TLR-4- or SIRNA-Con-transfected MG-63 cells under hypoxia. Each result was averaged for three independent experiments, and was presented as Mean ± SE. *P < 0.05, **P < 0.01.
Discussion
As a proinflammatory cytokine, HMGB1 can be stimulated to release into extracellular milieu and to function in response to injury, infection, and inflammation [26]. Various factors, such as necrosis [28], apoptosis [29], oxidative stress [30] and hypoxia [31] are recognized to promote the HMGB1 release. Hypoxia has been reported to induce HMGB1 in hepatocytes [32], in cardiomyocytes [33] and other types of cells. In particularly, high level of HMGB1 is secreted from macrophages in response to the stimulation with lipopolysaccharides or cytokines [34,35]. Recently, it was indicated in a pig trauma model that HMGB1 was promoted in the fracture hematoma [36]. In the present study, we recognized the significant upregulation of HMGB1 in the human hematoma specimens which were produced during the bone fracture. And further investigations confirmed the up regulated release and translocation from nucleus to cytosol of HMGB1 by hypoxia in the human macrophages, which is the main type of HMGB1 producers in blood.
Recently, HMGB1 has been indicated to promote the proliferation of such type’s cells as smooth muscle cells [37], mesangial cells [38], fibroblasts [39], and glioma cells [40]. And it is active in bone tissues [41,42]. However, its role in bone injury or fracture healing is rarely reported. In the current study, we confirmed the promotion to the proliferation of osteoblast MG-63 cells either under mormoxia or under hypoxia. Moreover, the HMGB1-mediated inflammatory response is usually via the interaction of HMGB1 with TLR-4 [22-24]. And our study confirmed that the hypoxia-induced HMGB1 upregulated in MG-63 cells the TLR-4 level, knockout of which with SIRNA markedly blocked the HMGB1-mediated proliferation promotion in MG-63 cells.
In addition, the ERK [26] and JNK [27] signaling was reported to be implicated in the HMGB1-initiated inflammatory reactions. We also investigated the activation of both ERK and JNK signaling in the HMGB1-treated MG-63 cells under normoxia or hypoxia. And our results confirmed the upregulation of the expression and the phosphorylation of ERK and JNK by hypoxia and the HMGB1 treatment. And the TLR-4 knockout reduced the HMGB1-induced phosphorylation of ERK and JNK in MG-63 cells. Therefore, we confirmed the hypoxia- and following promoted HMGB1-induced phosphorylation of ERK and JNK was TLR-4-dependent. However, there are several open questions about our study. Firstly, though our results demonstrated a positive regulation of HMGB1 on the osteoblast cells in vitro, the in vivo effect of it still needs to be further investigated. Secondly, the fracture healing is a result of multiple physiological processes, such as growth and differentiation of osteoblasts, the mineralization of the collagen matrix [3], and the revascularization. The exact role of HMGB1 in such processes is also unclear. Last but not the least, we found respectively the promotion of MG-63 cell proliferation and of ERK/JNK signaling, however, the detailed regulatory pathways linking the activation of ERK/JNK signaling with the proliferation of MG-63 cells is also needed to investigate.
In conclusion, we found the upregulation HMGB1 in the hematoma of fractured bones and in macrophage U937 cells under hypoxia, and the hypoxia-upregulated HMGB1 promoted the proliferation of osteoblast MG-63 cells and activated the phosphorylation of ERK and JNK. And the proliferation promotion and the activation of ERK/JNK signaling was TLR-4-dependent.
Acknowledgements
Present study was supported by the grant from Nanfang Hospital, Southern Medical University.
Disclosure of conflict of interest
None.
References
- 1.McKibbin B. The biology of fracture healing in long bones. J Bone Joint Surg Br. 1978;60-B:150–162. doi: 10.1302/0301-620X.60B2.350882. [DOI] [PubMed] [Google Scholar]
- 2.Remedios A. Bone and bone healing. Vet Clin North Am Small Anim Pract. 1999;29:1029–1044. doi: 10.1016/s0195-5616(99)50101-0. [DOI] [PubMed] [Google Scholar]
- 3.Harada S, Rodan GA. Control of osteoblast function and regulation of bone mass. Nature. 2003;423:349–355. doi: 10.1038/nature01660. [DOI] [PubMed] [Google Scholar]
- 4.Heppenstall RB, Goodwin CW, Brighton CT. Fracture healing in the presence of chronic hypoxia. J Bone Joint Surg Am. 1976;58:1153–1156. [PubMed] [Google Scholar]
- 5.Warren SM, Steinbrech DS, Mehrara BJ, Saadeh PB, Greenwald JA, Spector JA, Bouletreau PJ, Longaker MT. Hypoxia regulates osteoblast gene expression. J Surg Res. 2001;99:147–155. doi: 10.1006/jsre.2001.6128. [DOI] [PubMed] [Google Scholar]
- 6.Akeno N, Czyzyk-Krzeska MF, Gross TS, Clemens TL. Hypoxia induces vascular endothelial growth factor gene transcription in human osteoblast-like cells through the hypoxia-inducible factor-2alpha. Endocrinology. 2001;142:959–962. doi: 10.1210/endo.142.2.8112. [DOI] [PubMed] [Google Scholar]
- 7.Bouletreau PJ, Warren SM, Spector JA, Peled ZM, Gerrets RP, Greenwald JA, Longaker MT. Hypoxia and VEGF up-regulate BMP-2 mRNA and protein expression in microvascular endothelial cells: implications for fracture healing. Plast Reconstr Surg. 2002;109:2384–2397. doi: 10.1097/00006534-200206000-00033. [DOI] [PubMed] [Google Scholar]
- 8.Wang HS, Han JS. Research progress on combat trauma treatment in cold regions. Mil Med Res. 2014;1:8. doi: 10.1186/2054-9369-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naik AA, Xie C, Zuscik MJ, Kingsley P, Schwarz EM, Awad H, Guldberg R, Drissi H, Puzas JE, Boyce B, Zhang X, O’Keefe RJ. Reduced COX-2 expression in aged mice is associated with impaired fracture healing. J Bone Miner Res. 2009;24:251–264. doi: 10.1359/jbmr.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerstenfeld LC, Thiede M, Seibert K, Mielke C, Phippard D, Svagr B, Cullinane D, Einhorn TA. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 2003;21:670–675. doi: 10.1016/S0736-0266(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O’Keefe RJ. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. 2002;109:1405–1415. doi: 10.1172/JCI15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolar P, Schmidt-Bleek K, Schell H, Gaber T, Toben D, Schmidmaier G, Perka C, Buttgereit F, Duda GN. The early fracture hematoma and its potential role in fracture healing. Tissue Eng Part B Rev. 2010;16:427–434. doi: 10.1089/ten.TEB.2009.0687. [DOI] [PubMed] [Google Scholar]
- 13.Andrew JG, Andrew SM, Freemont AJ, Marsh DR. Inflammatory cells in normal human fracture healing. Acta Orthop Scand. 1994;65:462–466. doi: 10.3109/17453679408995493. [DOI] [PubMed] [Google Scholar]
- 14.Hauser CJ, Zhou X, Joshi P, Cuchens MA, Kregor P, Devidas M, Kennedy RJ, Poole GV, Hughes JL. The immune microenvironment of human fracture/soft-tissue hematomas and its relationship to systemic immunity. J Trauma. 1997;42:895–903. doi: 10.1097/00005373-199705000-00021. [DOI] [PubMed] [Google Scholar]
- 15.Hoff P, Maschmeyer P, Gaber T, Schutze T, Raue T, Schmidt-Bleek K, Dziurla R, Schellmann S, Lohanatha FL, Rohner E, Ode A, Burmester GR, Duda GN, Perka C, Buttgereit F. Human immune cells’ behavior and survival under bioenergetically restricted conditions in an in vitro fracture hematoma model. Cell Mol Immunol. 2013;10:151–158. doi: 10.1038/cmi.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westhauser F, Zimmermann G, Moghaddam S, Bruckner T, Schmidmaier G, Biglari B, Moghaddam A. Reaming in treatment of nonunions in long bones: cytokine expression course as a tool for evaluation of non-union therapy. Arch Orthop Trauma Surg. 2015;135:1107–16. doi: 10.1007/s00402-015-2253-3. [DOI] [PubMed] [Google Scholar]
- 17.Herlin M, McGuigan FE, Luthman H, Akesson K. Polymorphisms in inflammation associated genes ALOX15 and IL-6 are associated with bone properties in young women and fracture in elderly. Bone. 2015;79:105–109. doi: 10.1016/j.bone.2015.05.035. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 19.Kim JY, Park JS, Strassheim D, Douglas I, Diaz DVF, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama I, Ishizaka A, Abraham E. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288:L958–L965. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 20.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang R, Harada T, Mollen KP, Prince JM, Levy RM, Englert JA, Gallowitsch-Puerta M, Yang L, Yang H, Tracey KJ, Harbrecht BG, Billiar TR, Fink MP. Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol Med. 2006;12:105–114. doi: 10.2119/2006-00010.Yang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 23.Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–C924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- 24.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.van Beijnum JR, Buurman WA, Griffioen AW. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1) Angiogenesis. 2008;11:91–99. doi: 10.1007/s10456-008-9093-5. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Mi Y, Yang H, Hu A, Zhang Q, Shang C. The activation of HMGB1 as a progression factor on inflammation response in normal human bronchial epithelial cells through RAGE/JNK/NF-kappaB pathway. Mol Cell Biochem. 2013;380:249–257. doi: 10.1007/s11010-013-1680-0. [DOI] [PubMed] [Google Scholar]
- 28.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 29.Gauley J, Pisetsky DS. The translocation of HMGB1 during cell activation and cell death. Autoimmunity. 2009;42:299–301. doi: 10.1080/08916930902831522. [DOI] [PubMed] [Google Scholar]
- 30.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamada T, Torikai M, Kuwazuru A, Tanaka M, Horai N, Fukuda T, Yamada S, Nagayama S, Hashiguchi K, Sunahara N, Fukuzaki K, Nagata R, Komiya S, Maruyama I, Fukuda T, Abeyama K. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum. 2008;58:2675–2685. doi: 10.1002/art.23729. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, Lotze M, Tang D, Tsung A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63:114–121. doi: 10.1016/j.jhep.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Hu X, Xie J, Xu W, Jiang H. Beta-1-adrenergic receptors mediate Nrf2-HO-1-HMGB1 axis regulation to attenuate hypoxia/reoxygenation-induced cardiomyocytes injury in vitro. Cell Physiol Biochem. 2015;35:767–777. doi: 10.1159/000369736. [DOI] [PubMed] [Google Scholar]
- 34.Palmblad K, Schierbeck H, Sundberg E, Horne AC, Harris HE, Henter JI, Antoine DJ, Andersson U. High systemic levels of the cytokineinducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol Med. 2014;20:538–547. doi: 10.2119/molmed.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Wu CX, Sun H, Liu Q, Guo H, Gong JP. LPS induces HMGB1 relocation and release by activating the NF-kappaB-CBP signal transduction pathway in the murine macrophage-like cell line RAW264.7. J Surg Res. 2012;175:88–100. doi: 10.1016/j.jss.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 36.Horst K, Eschbach D, Pfeifer R, Hubenthal S, Sassen M, Steinfeldt T, Wulf H, Ruchholtz S, Pape HC, Hildebrand F. Local inflammation in fracture hematoma: results from a combined trauma model in pigs. Mediators Inflamm. 2015;2015:126060. doi: 10.1155/2015/126060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang HL, Peng LP, Chen WJ, Tang SH, Sun BZ, Wang CL, Huang R, Xu ZJ, Lei WF. HMGB1 enhances smooth muscle cell proliferation and migration in pulmonary artery remodeling. Int J Clin Exp Pathol. 2014;7:3836–3844. [PMC free article] [PubMed] [Google Scholar]
- 38.Feng XJ, Liu SX, Wu C, Kang PP, Liu QJ, Hao J, Li HB, Li F, Zhang YJ, Fu XH, Zhang SB, Zuo LF. The PTEN/PI3K/Akt signaling pathway mediates HMGB1-induced cell proliferation by regulating the NF-kappaB/cyclin D1 pathway in mouse mesangial cells. Am J Physiol Cell Physiol. 2014;306:C1119–C1128. doi: 10.1152/ajpcell.00385.2013. [DOI] [PubMed] [Google Scholar]
- 39.Chitanuwat A, Laosrisin N, Dhanesuan N. Role of HMGB1 in proliferation and migration of human gingival and periodontal ligament fibroblasts. J Oral Sci. 2013;55:45–50. doi: 10.2334/josnusd.55.45. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Liu C, Hou R. Knockdown of HMGB1 improves apoptosis and suppresses proliferation and invasion of glioma cells. Chin J Cancer Res. 2014;26:658–668. doi: 10.3978/j.issn.1000-9604.2014.12.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charoonpatrapong K, Shah R, Robling AG, Alvarez M, Clapp DW, Chen S, Kopp RP, Pavalko FM, Yu J, Bidwell JP. HMGB1 expression and release by bone cells. J Cell Physiol. 2006;207:480–490. doi: 10.1002/jcp.20577. [DOI] [PubMed] [Google Scholar]
- 42.Yang J, Shah R, Robling AG, Templeton E, Yang H, Tracey KJ, Bidwell JP. HMGB1 is a boneactive cytokine. J Cell Physiol. 2008;214:730–739. doi: 10.1002/jcp.21268. [DOI] [PubMed] [Google Scholar]