

Abstract
The Let-7 microRNA (miRNA) family is frequently downregulated in multiple human tumors, including hepatocellular carcinoma (HCC). Our previous report demonstrated that hepatitis B virus X protein (HBx) suppressed the expression of let-7 in HepG2 hepatoma cells. However, the underlying mechanisms were not elucidated. Lin28B is known to negatively regulate the maturation of let-7, and this prompted us to determine whether HBx acts through Lin28B to suppress let-7. Real-time reverse-transcription polymerase chain reaction (qRT-PCR) was performed to examine let-7 expression before and after treatment with c-Myc-and Lin28B-specific siRNAs in HepG2 cells stably/transiently transfected with HBx. mRNA and protein analyses were employed to determine the correlation of HBx, c-Myc and Lin28B in HCC tissues and cells. Cell cycle and proliferation assays were performed to delineate the consequences of Lin28B repression in HepG2 cells expressing HBx. Lin28B was overexpressed in HBx-transfected cells and HBV-infected liver tissues. HBx-c-Myc-Lin28B axis mediated the repression of let-7 in HepG2 cells. Reduced expression of Lin28B inhibited the growth and cell cycle progression of HepG2 cells by derepressing let-7 and repressing c-Myc. There was not only a preliminary HBx-c-Myc-Lin28B-let-7 pathway but also another possible double-negative feedback loop between c-Myc/Lin28B and let-7 in HepG2 cells transfected with HBx, which together induced the deregulation of let-7. Lin28B has the potential to be a novel molecular target in the treatment of HBV+ HCC.
Keywords: HBx, hepatocellular carcinoma, Lin28B, let-7
Introduction
Non-coding RNAs (ncRNAs), such as siRNA, miRNA and long ncRNA, have been confirmed to participate in diverse eukaryotic cellular processes, including proliferation, differentiation, apoptosis and death [1]. miRNAs are 18-24 nucleotide RNAs that post-transcriptionally silence the expression of target mRNAs by degrading them or inhibiting their translation. First, the miRNA gene is transcribed as pri-miRNA, which is processed sequentially by DROSHA/DGCR8 (pre-miRNA) and Dicer1 to yield mature miRNAs. Increasing evidence has confirmed that the deregulation of miRNAs is related to many human diseases, including carcinogenesis, through the repression of target gene expression. miRNAs may play oncogenic or tumor suppressor roles according to their expression and function in tumors. The downregulation of the let-7 family is involved in the initiation and progression of many human tumors, including HCC [2], resulting in the re-expression of oncogenes, such as Ras, HMAGA2, c-Myc, STAT3, Bcl-xL, caspase 3 and Lin28B [3].
The dysregulation of miRNA genes in malignancy attributes to genome variations, epigenetic modifications and alterations in the biogenesis process. miRNAs are not only regulators but are also the targets of protein-coding genes, such as RNA-binding proteins. miRNAs are frequently involved in almost all tumor-signaling pathways. This is well illustrated by the direct induction of miR-15a/16-1, miR-34a, miR-23a, miR-26a, miR-143 and miR-145 by p53 in response to DNA damage [4]. These activated miRNAs induce cell cycle arrest and apoptosis, both of which are mechanisms of p53 protection of cells against transformation. c-Myc directly induces the expression of the oncogenic miR-17-92 cluster and represses the expression of the tumor-suppressor genes miR-34a, let-7 and miR-15a/16-1 [5]. In contrast, let-7 silences the expression of c-Myc and Lin28B, both of which are its own upstream regulator [6]. miRNAs and transcription factors are currently believed to co-regulate gene expression in the epigenome.
As a transactivating oncoprotein encoded by hepatitis B virus (HBV), HBx is associated with the initiation and progression of HCC [7]. Though HBx cannot directly bind to DNA, it alters host gene expression by activating cellular signal pathways (e.g., NF-κB, src, ras, AP-1, AP-2, PI3K/Akt, Jak/STAT, c-Myc, Smad, and Wnt) and by binding nuclear TFs (e.g., CREB, ATF-2, Oct-1, and TBP), these two modes of action contribute to malignant transition in hepatocytes [8]. Recently, it was reported that HBx transcript directly triggered the down-regulation of miR-15a/miR-16-1 via the microRNA targeting sequences in the viral RNA [9].
Due to the pleiotropic functions of HBx, we previously sought to identify its effect on the host cellular miRNA expression [10]. Consistent with the report of Wang’s team [2], we found that HBx negatively regulates the expression of the let-7 family in vitro and in vivo. However, the underlying mechanisms of let-7 deregulation were not elucidated. Consistent with microarray results, we further validated the repression of let-7 in HepG2 cell lines that were stably or transiently transfected with HBx, using qRT-PCR. We demonstrated that HBx induced the upregulation of c-Myc in HepG2 cells [10], which is capable of transactivating Lin28B. As anticipated, Lin28B was overexpressed in HepG2 cells that expressed HBx and in HBV+ HCC tissues. Accordingly, the loss of c-Myc/Lin28B function impaired the HBx-induced downregulation of let-7. Finally, Lin28B-specific siRNA inhibited the proliferation and cell cycle progression of HepG2 cells transfected with HBx. Moreover, we also found that there was not only a preliminary HBx-c-Myc-Lin28B-let-7 pathway but also another possible feedback loop between c-Myc/Lin28B and let-7 in HepG2 cells. These findings provide insight into mechanisms of HBx-induced miRNA deregulation and highlight the potential of targeting Lin28B as a treatment for HBV+ HCC.
Materials and methods
Cell culture and tissue
Human hepatoma cell line HepG2 was purchased from ATCC (American Type Culture Collection, Manassas, VA) and cultured in DMEM (Dulbecco’s modification of Eagle’s medium) (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Hyclone, South Logan, Utah). The HepG2 cells that were stably transfected with HBx or control vector included HepG2-vc, HepG2-hbx, HepG2-2.1 and HepG2.2.15 were established and cultured as described [10]. All cell lines were maintained in a 5% CO2, humidified atmosphere.
Clinical samples used for immunohistochemistry (IHC) were obtained from a human HCC tissue array (LV8013; US Biomax), which contained 19 HCCs, 1 intrahepatic cholangiocarcinoma, 10 cirrhotic liver tissues and 10 normal liver tissues. Detailed information, including differentiation status, TNM grading and HBV/HCV status, were also provided for each sample.
Oligoribonucleotides
All RNA oligoribonucleotides, including c-Myc, Lin28B-specific siRNA, and negative control small RNAs (NC), were purchased from Genepharma (Shanghai, China). The HBx-, c-Myc- and Lin28B-expressing plasmids were constructed as described [10]. The sequences of oligoribonucleotides were provided in Table S1.
Cell transfection
The transfection of RNA oligoribonucleotides was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Unless otherwise indicated, 50 nM of RNA oligoribonucleotides was used in each transfection.
Real-time reverse-transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted with TRIzol reagent (Invitrogen). cDNA for qRT-PCR was synthesized with PrimeScript RT reagent Kit With gDNA Eraser (Takara Biotechnology, DaLian, China). The qRT-PCR assays were performed using the SYBR PrimeScript RT-PCR kit (Takara Biotechnology) for c-Myc, Lin28B and GAPDH. The assay kits for U6 and let-7 were purchased from Genepharma.
Immunoblots
Cell cytosolic protein fractions were prepared using RIPA buffer (Beyotime Biotechnology, Haimen, China). The antibodies used in western blotting and IHC were for Lin28B (ab71415, Abcam) and GAPDH (G9295; Sigma-Aldrich, USA). Western blots and IHC were performed according to standard procedures.
Cell viability
Cell proliferation was analyzed with the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumamoto, Japan). HepG2-hbx cells were treated with Lin28B or c-Myc siRNA, and the growth curve was determined by measuring the absorbance at 450 λ at 24 h, 48 h, 72 h and 96 h post-transfection.
Cell cycle analysis
Twenty-four hours after the siRNA and NC transfections, the HepG2-hbx cells were treated with nocodazole (100 ng/ml, M1404, Sigma) for 20 h. Floating and adherent cells were harvested, combined, washed once in phosphate-buffered saline (PBS), and fixed in 70% ethanol overnight. The DNA content was then examined using the Cell Cycle Detection Kit (KGA512, KeyGen Biotechnology, Nanjing, China) and analyzed using BD FACSCalibur flow cytometer.
Statistical analysis
Data were analyzed with SPSS 16.0, and the significance level was set at P < 0.05. The results of quantitative-PCR were compared using a Student’s t-test.
Results
Let-7 was downregulated in HepG2 cells expressing HBx
Our previous microarray assay demonstrated that let-7 was downregulated in HBx-expressing HCC cells [10], which was consistent with Wang’s report [2]. To extend our study, we confirmed significant repression of let-7 in HepG2 cells stably and transiently transfected with HBx using qRT-PCR (Figure 1).
Figure 1.
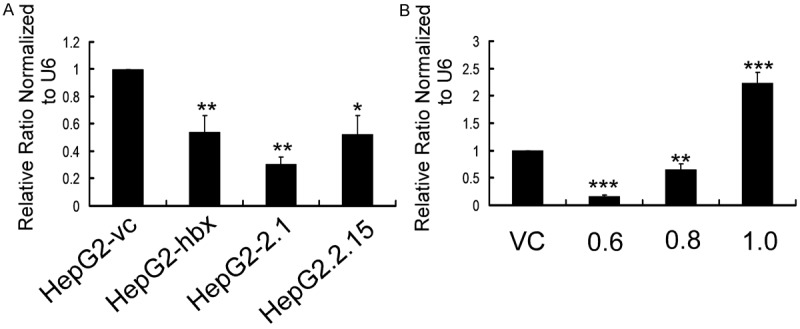
Let-7 was downregulated in HepG2 cells that were stably and transiently transfected with HBx plasmid. A. Let-7 expression was measured using qRT-PCR and normalized by U6 expression in HepG2 cells that stably expressed HBx or control vector. B. The expression of let-7 normalized to U6 was detected by qRT-PCR in HepG2 cells transiently transfected with HBx-expressing plasmid or control vehicle. HepG2 cells were transfected with 0.6, 0.8 and 1 µg PCDNA3.1-hbx or 0.6 µg PCDNA3.1 as a control. Cells were collected for analysis 48 h after each transfection. Data are presented as the mean ± SE fold change. (*P < 0.05, **P < 0.01, ***P < 0.001; Student’s t-test; n=3).
Interestingly, we found that the levels of let-7 were inversely correlated with the levels of HBx at pro-apoptotic dosage (1.0 µg) during transient transfection (Figure 1B). Numerous apoptotic cells appeared when we increased the dose of plasmid in HepG2 cells (data not shown). This was consistent with our previous experiments with the miR-16 family [10], which also suggested a non-coding RNA mechanism of the pro-/anti-apoptosis function of HBx [11,12]. However, the exact role of let-7 in the HBx-associated apoptosis of hepatocytes remains to be clarified.
Lin28B was over-expressed in HepG2 cells expressing HBx and in HBV-infected tissues
To elucidate the underlying mechanisms of HBx-induced let-7, we noted the report that Lin28B promoted tumorigenesis by blocking the biogenesis of pri-/pre-let-7 [13]. Lin28B was first cloned from and shown to be over-expressed in HCC samples and cells [14]. As a result, we hypothesized that Lin28B was a downstream target of HBx and responsible for the suppression of let-7. qRT-PCR and western blot results showed that the mRNA and protein expression of Lin28B were significantly upregulated in HepG2 cells expressing HBx (Figure 2A and 2B). Furthermore, IHC assay demonstrated that Lin28B expression was induced in HBV-infected HCC tissues and cirrhotic livers compared with normal liver (Figure 2C). Taken together, our results verified that there was a positive correlation between the expression of HBx/HBV and Lin28B in HCC tissues and cells.
Figure 2.
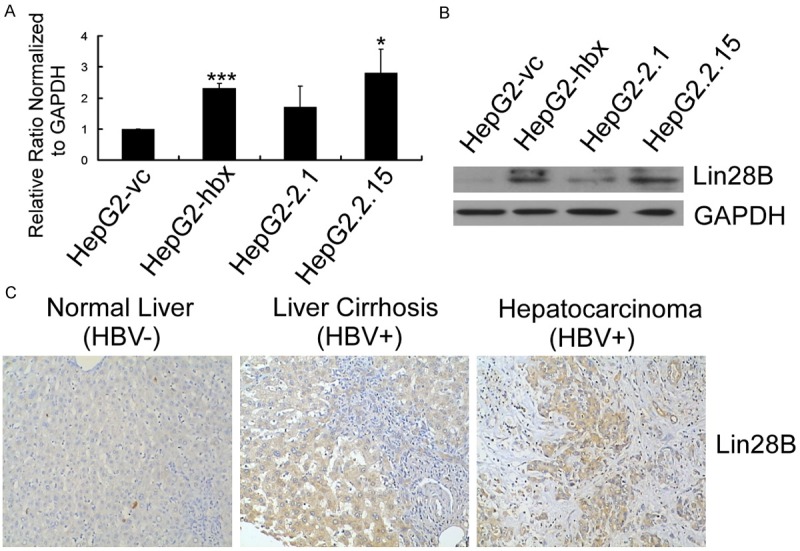
Lin28B was overexpressed in HepG2 cells expressing HBx and HBV+ HCC tissues. (A) qRT-PCR and (B) representative western blot results demonstrated that HBx upregulated Lin28B transcription and translation in HepG2 cells. Relative Lin28B expression was normalized to GAPDH. The qRT-PCR data are presented as the mean ± SE fold change compared with HepG2-vc (*P < 0.05, **P < 0.01, ***P < 0.001; n=3, Student’s t-test). (C) The expression of Lin28B protein was detected by IHC. Representative results are presented.
HBx-c-Myc-Lin28B pathway induced let-7 suppression in HepG2 cells
We previously demonstrated that HBx induced the upregulation of c-Myc in HepG2 cells [10], whereas Lin28B transactivation was necessary for c-Myc to suppress let-7 [15]. We therefore hypothesized there might be a HBx-c-Myc-Lin28B pathway accounting for the suppression of let-7 in HepG2 cells. We transfected HepG2-hbx cells with c-Myc/Lin28B-specific siRNAs and found that both siRNAs significantly reactivated the expression of let-7 (Figure 3C). This restoration of expression could also be reproduced in HepG2.2.15 cells (data not shown). These results suggest that HBx downregulates let-7 expression, at least partly, through the induction of Lin28B and/or c-Myc in HepG2 cells.
Figure 3.
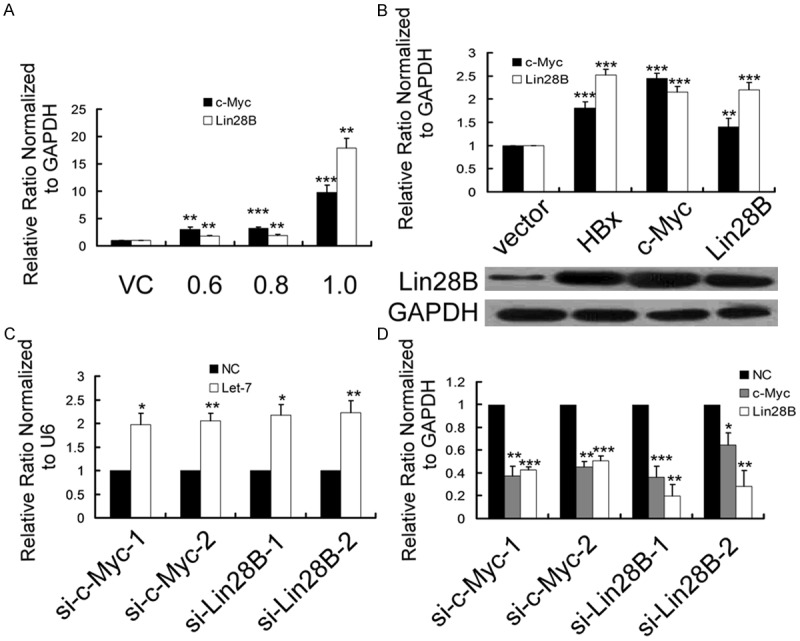
HBx suppressed the expression of let-7 by activating c-Myc and Lin28B in HepG2 cells. A. qRT-PCR results showed that HBx upregulated the transcription of c-Myc and Lin28B in a dose-dependent manner in HepG2 cells. Relative c-Myc and Lin28B expression was normalized to GAPDH. HepG2 cells were transfected with 0.6, 0.8 and 1 µg PCDNA3.1-hbx or 0.6 µg PCDNA3.1 as a control. Cells were collected for analysis 48 h after each transfection. B. qRT-PCR and representative western blot results showed that Lin28B and c-Myc were activated by ectopically expressed HBx, c-Myc and Lin28B in HepG2 cells. Relative expression of c-Myc and Lin28B was normalized to GAPDH. C, D. The relative expression of c-Myc, Lin28B and let-7 was analyzed by qRT-PCR at 48 h after the transfection of si-c-Myc/si-Lin28B into HepG2-hbx cells compared with the negative control (NC) siRNA transfectants, normalized to GAPDH and U6, respectively. The data are presented as the mean fold change ± SE. The qRT-PCR data are presented as the mean fold change ± SE compared with VC (n=3, Student’s t-test). (*P < 0.05, **P < 0.01, ***P < 0.001; n=3, Student’s t-test).
Based on these findings, we carried out several other analyses to further clarify the HBx-c-Myc-Lin28B pathway. Both c-Myc and Lin28B were transcriptionally activated in a dose-dependent manner in HepG2 cells transiently transfected with HBx (Figure 3A). We noticed that the mRNA of c-Myc and Lin28B were aberrantly upregulated > 10-fold when HBx plasmid reached a pro-apoptotic dose (1.0 µg). Nonetheless, whether the upregulation of let-7 (Figure 1B) was a negative feedback for the aberrant activation of c-Myc and Lin28B or resulted from other pro-apoptotic mechanisms remains to be explored. Surprisingly, both c-Myc and Lin28B themselves are the targets of let-7 [16]. These results strongly suggest that there may be a double-negative feedback loop between c-Myc/Lin28B and let-7. Next, we found that c-Myc and Lin28B transient transfection, respectively, could effectively activate the mRNA and protein expression of Lin28B in HepG2 cells (Figure 3B). In addition, ectopically expressed Lin28B induced high expression of c-Myc (Figure 3B). This might be attributed to Lin28B-induced let-7 repression, which abrogated the post-transcriptional silencing of c-Myc by let-7. Conversely, qRT-PCR assays demonstrated that either c-Myc- or Lin28B-specific siRNAs significantly downregulated the expression of Lin28B and c-Myc in HepG2-hbx cells (Figure 3C). In contrast, let-7 was derepressed by these siRNAs (Figure 3D).
In summary, our results suggest not only a feed-forward HBx-c-Myc-Lin28B-let-7 pathway but also another complex double-negative feedback loop between let-7 and Lin28B/c-Myc (Figure 4). However, more studies are needed to clarify this pathway. In particular the significance of let-7 deregulation in dose-dependent transfection may aid in understanding the HBx-induced malignant transition (survival or apoptosis) of hepatocytes during the acute/chronic HBV infection.
Figure 4.
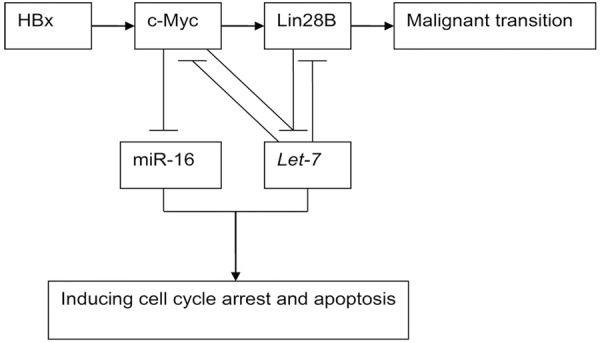
Double-negative feedback loop between c-Myc/Lin28B and let-7.
Reduced expression of Lin28B inhibited the proliferation and cell cycle progression of HepG2-hbx cells
The dysregulation of the Lin28B-let-7-Lin28B loop is involved in the initiation and progression of hepatocarcinogenesis. We sought to determine the therapeutic potential of targeting Lin28B in HBV+ HCC in vitro. CCK-8 analysis showed that si-Lin28B and si-c-Myc significantly inhibited the growth of HepG2-hbx cells at 48 h, 72 h and 96 h post-transfection compared with NC-transfected cells (Figure 5A). Fluorescence-activated cell sorting (FACS) assays demonstrated that si-Lin28B transfection arrested HepG2-hbx cells at the G1 phase (Figure 5B).
Figure 5.
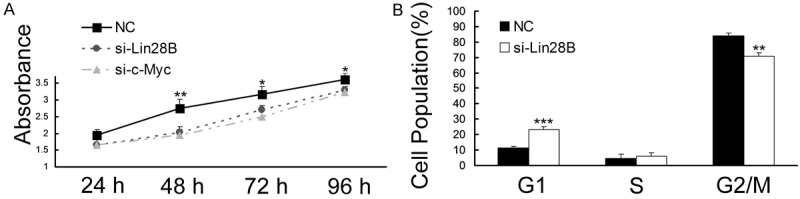
si-Lin28B inhibited proliferation and cell cycle progression in HepG2 cells. A. The growth curves of HepG2-hbx cells transfected with si-Lin28B or NC were generated using the absorbance from the CCK-8 analysis at the indicated time points. The reduced expression of Lin28B significantly inhibited the growth of HepG2-hbx cells at 48, 72 and 96 h post-transfection. B. HepG2-hbx cells were treated with nocodazole (100 ng/ml) 24 h after the transfection of si-Lin28B or NC, and cell cycle distribution was analyzed 20 h later. The data are presented as the mean fold change ± SE. (**P < 0.01, ***P < 0.001; n=3, Student’s t-test).
Discussion
HBx, as a co-transactivator that promotes hepatocarcinogenesis mainly by altering the expression of protein-coding genes and physically binding to TFs. At present, miRNAs are most extensively documented in the role of regulating gene expression together with TFs. These observations prompted us to investigate the downstream miRNAs of HBx. We previously demonstrated that HBx caused widespread suppression of miRNAs, including let-7 [10], the repression of which contributed to hepatocarcinogenesis by derepressing the expression of HMGA2, p16 (INK4A), Bcl-xL, STAT3, caspase-3, c-Myc and Lin28B [2,17-20]. Moreover, it has also been reported that HBx suppressed the expression of the let-7 family in HepG2 cells, and there was inversely correlated expression between HBx and the let-7 family in HBV+ HCC or in adjacent tissues [2]. However, the detailed mechanisms of HBx-induced let-7 repression were not elucidated. We validated the repression of let-7 in HepG2 cells expressing HBx using qRT-PCR and delineated a preliminary HBx-c-Myc-Lin28B-let-7 pathway in HepG2 cells in vitro.
Several stemness-related genes, including Lin28, which are referred to as oncofetal genes, are thought to promote tumorigenesis upon re-expression in somatic cells. Lin28 was initially identified in C. elegans, where it was shown to be responsible for the timing of development [21]. Physiologically, Lin28 is activated mainly in various stem cell populations and silenced in terminally differentiated somatic tissues. Recently, Lin28 was selected together with OCT4, NANOG and SOX2 to reprogram human somatic fibroblasts to pluripotency as a replacement for c-Myc. Lin28B, as a homologue of Lin28, was first cloned from and shown to be overexpressed in human hepatocellular carcinoma cells and clinical samples [14]. Lin28B facilitates cellular transformation in vitro, and overexpression is associated with advanced disease across multiple tumor types, including HCC, by blocking the biogenesis of let-7 [22]. Recently, we also reported that Lin28B promotes colon cancer migration and recurrence [23]. We found that HBx induced the overexpression of Lin28B in hepatoma cells and tissues (Figure 3). These results may partly explain why HBV+ HCCs consistently show much more aggressive tumor growth characteristics. Lin28B and let-7 antagonize each other, maintaining a balancing equilibrium between these factors in tumorigenesis [6]. A rebalancing of the Lin28/let-7 regulatory loop could yield ALDH1+ cancer stem cells and drive liver cancer in murine models arising through a “reprogramming-like” mechanism [24,25]. Our results illustrate the significance of HBx-induced re-expression of Lin28B in HBV-associated hepatocarcinogenesis.
The relationship between HBx and apoptosis is controversial. It has been reported that HBx could inhibit hepatocyte apoptosis either by inducing the NF-κB family [26], by inhibiting TGF-β-induced apoptosis [27], by recruiting the HBXIP/survivin complex [28] or by antagonizing the apoptotic function of p53 [12]. However, there is a p53-independent pro-apoptotic effect of HBx in vivo and in vitro [11]. The authors of this paper stated that HBx required an additional death stimulus to promote apoptosis when stably expressed at low levels in cell cultures, and they tested whether transient overexpression of HBx was sufficient to induce apoptosis. The results showed that transient overexpression of HBx induces apoptosis in HepG2 and MMHD3 cells. After the transfection of HepG2 cells with different amounts of pCMV-HBx, the percentage of apoptotic cells varied in a dose-dependent manner. Interestingly, we also found similar dose-dependent results when we transiently transfected pcDNA3.1-hbx plasmid into HepG2, SK-HEP-1 and Huh7 cells [10]. Importantly, we observed that miR-16 and let-7 exhibited inversely correlated expression with HBx when when HBx reached a pro-apoptotic dose in HepG2 cells (Figure 1B), both of miR-16 and let-7 are capable of inducing apoptosis. Meanwhile, the mRNA of c-Myc and Lin28B were upregulated > 10-fold with this dose of HBx (Figure 3A). Taken together, we propose that there is a c-Myc/Lin28B-let-7-c-Myc/Lin28B double-negative feedback loop in HepG2 cells.
In conclusion, our data suggests critical downstream roles for Lin28B in HBx-induced deregulation of host cellular let-7 expression and increased proliferation of HepG2 cells. Targeting Lin28B should be considered as a prospective treatment for HBV+ HCC patients.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81302161) and the Health and Family Planning Commission of Sichuan Province (150215). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Lu Y, Toh ST, Sung WK, Tan P, Chow P, Chung AY, Jooi LL, Lee CG. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3. J Hepatol. 2010;53:57–66. doi: 10.1016/j.jhep.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 3.Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70–80. doi: 10.3747/co.v17i1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 5.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ji J, Wang XW. A Yin-Yang balancing act of the lin28/let-7 link in tumorigenesis. J Hepatol. 2010;53:974–975. doi: 10.1016/j.jhep.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 8.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–170. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Jiang L, Ji X, Yang B, Zhang Y, Fu XD. Hepatitis B viral RNA directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16-1 in hepatocytes. J Biol Chem. 2013;288:18484–18493. doi: 10.1074/jbc.M113.458158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu G, Yu F, Xiao Z, Xu K, Xu J, Tang W, Wang J, Song E. Hepatitis B virus X protein downregulates expression of the miR-16 family in malignant hepatocytes in vitro. Br J Cancer. 2011;105:146–153. doi: 10.1038/bjc.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terradillos O, Pollicino T, Lecoeur H, Tripodi M, Gougeon ML, Tiollais P, Buendia MA. p53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene. 1998;17:2115–2123. doi: 10.1038/sj.onc.1202432. [DOI] [PubMed] [Google Scholar]
- 12.Wang XW, Gibson MK, Vermeulen W, Yeh H, Forrester K, Sturzbecher HW, Hoeijmakers JH, Harris CC. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995;55:6012–6016. [PubMed] [Google Scholar]
- 13.Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell. 2010;140:445–449. doi: 10.1016/j.cell.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, Chen Y, Ito H, Watanabe A, Ge X, Kodama T, Aburatani H. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene. 2006;384:51–61. doi: 10.1016/j.gene.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, Jung J, Gao P, Dang CV, Beer MA, Thomas-Tikhonenko A, Mendell JT. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci U S A. 2009;106:3384–3389. doi: 10.1073/pnas.0808300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong X, Li N, Liang S, Huang Q, Coukos G, Zhang L. Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J Biol Chem. 2010;285:41961–71. doi: 10.1074/jbc.M110.169607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsang WP, Kwok TT. Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis. 2008;13:1215–1222. doi: 10.1007/s10495-008-0256-z. [DOI] [PubMed] [Google Scholar]
- 18.Wang YC, Chen YL, Yuan RH, Pan HW, Yang WC, Hsu HC, Jeng YM. Lin-28B expression promotes transformation and invasion in human hepatocellular carcinoma. Carcinogenesis. 2010;31:1516–1522. doi: 10.1093/carcin/bgq107. [DOI] [PubMed] [Google Scholar]
- 19.Lan FF, Wang H, Chen YC, Chan CY, Ng SS, Li K, Xie D, He ML, Lin MC, Kung HF. Hsa-let-7g inhibits proliferation of hepatocellular carcinoma cells by downregulation of c-Myc and upregulation of p16(INK4A) Int J Cancer. 2011;128:319–331. doi: 10.1002/ijc.25336. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A, Tatsumi T, Ishida H, Noda T, Nagano H, Doki Y, Mori M, Hayashi N. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. 2010;52:698–704. doi: 10.1016/j.jhep.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 21.Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/s0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- 22.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, O’Sullivan M, Lu J, Phillips LA, Lockhart VL, Shah SP, Tanwar PS, Mermel CH, Beroukhim R, Azam M, Teixeira J, Meyerson M, Hughes TP, Llovet JM, Radich J, Mullighan CG, Golub TR, Sorensen PH, Daley GQ. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang M, Wu G, Hou X, Hou N, Liang L, Jia G, Shuai P, Luo B, Wang K, Li G. LIN28B Promotes Colon Cancer Migration and Recurrence. PLoS One. 2014;9:e109169. doi: 10.1371/journal.pone.0109169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen LH, Robinton DA, Seligson MT, Wu L, Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, Yang EH, Powers JT, Shinoda G, Shah SP, Hammer RE, Daley GQ, Zhu H. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell. 2014;26:248–261. doi: 10.1016/j.ccr.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Lin X, Zhong X, Kaur S, Li N, Liang S, Lassus H, Wang L, Katsaros D, Montone K, Zhao X, Zhang Y, Butzow R, Coukos G, Zhang L. Double-negative feedback loop between reprogramming factor LIN28 and microRNA let-7 regulates aldehyde dehydrogenase 1-positive cancer stem cells. Cancer Res. 2010;70:9463–9472. doi: 10.1158/0008-5472.CAN-10-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clippinger AJ, Gearhart TL, Bouchard MJ. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF-kappaB and the mitochondrial permeability transition pore. J Virol. 2009;83:4718–4731. doi: 10.1128/JVI.02590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL. Hepatitis B virus X protein inhibits transforming growth factor-beta-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J Biol Chem. 2000;275:25858–25864. doi: 10.1074/jbc.M003578200. [DOI] [PubMed] [Google Scholar]
- 28.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, Reed JC. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–2740. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.