

Abstract
The aflatoxins were discovered in toxic peanut meal causing “turkey X” disease, which killed large numbers of turkey poults, ducklings and chicks in the UK in the early 1960s. Extracts of toxic feed induced the symptoms in experimental animals, and purified metabolites with properties identical to aflatoxins B1 and G1 (AFB1 and AFG1) were isolated from Aspergillus flavus cultures. Structure elucidation of aflatoxin B1 was accomplished and confirmed by total synthesis in 1963. AFB1 is a potent liver carcinogen in rodents, non-human primates, fish and birds, operating through a genotoxic mechanism involving metabolic activation to an epoxide, formation of DNA adducts and, in humans, modification of the p53 gene. Aflatoxins are unique among environmental carcinogens, in that elucidation of their mechanisms of action combined with molecular epidemiology provides a foundation for quantitative risk assessment; extensive evidence confirms that contamination of the food supply by AFB1 puts an exposed population at increased risk of developing hepatocellular carcinoma (HCC). Molecular biomarkers to quantify aflatoxin exposure in individuals were essential to link aflatoxin exposure with liver cancer risk. Biomarkers were validated in populations with high HCC incidence in China and The Gambia, West Africa; urinary AFB1–N7-Guanine excretion was linearly related to aflatoxin intake, and levels of aflatoxin– serum albumin adducts also reflected aflatoxin intake. Two major cohort studies employing aflatoxin biomarkers identified their causative role in HCC etiology. Results of a study in Shanghai men strongly support a causal relationship between HCC risk and the presence of biomarkers for aflatoxin and HBV infection, and also show that the two risk factors act synergistically. Subsequent cohort studies in Taiwan confirm these results. IARC classified aflatoxin as a Group 1 human carcinogen in 1993, based on sufficient evidence in humans and experimental animals indicating the carcinogenicity of naturally occurring mixtures of aflatoxins, aflatoxin B1, G1 and M1. Aflatoxin biomarkers have also been used to show that primary prevention to reduce aflatoxin exposure can be achieved by low-technology approaches at the subsistence farm level in sub-Saharan Africa. Also, in residents of Qidong, China, oral dosing with chlorophyllin, a chlorophyll derivative, prior to each meal led to significant reduction in aflatoxin–DNA biomarker excretion, supporting the feasibility of preventive measures to reduce HCC risk in populations experiencing unavoidable aflatoxin exposure. The systematic, comprehensive approach used to create the total aflatoxin database justifies optimism for potential success of preventive interventions to ameliorate cancer risk attributable to aflatoxin exposure. This strategy could serve as a template for the development, validation and application of molecular and biochemical markers for other carcinogens and cancers as well as other chronic diseases resulting from environmental exposures.
Keywords: chromatography, LC/MS, aflatoxins
Discovery of aflatoxin and main features of its toxicology and epidemiology
Aflatoxin occupies a unique position among environmental toxins and carcinogens, in that extensive elucidation of its toxicology combined with molecular epidemiology have provided a foundation for quantitative risk assessments based upon an understanding of its mechanisms of action. A time-line depicting highlights of aflatoxin research is shown in Figure 1 and the intent of this paper is to review and comment on several of these seminal lines of investigation. The aflatoxins were discovered 50 years ago as the causative agent of the “turkey X” disease epidemic, which resulted in the death of thousands of turkey poults, ducklings and chicks fed a toxic peanut meal (Blount 1961). Subsequent laboratory studies using extracts of groundnut cultures of Aspergillus flavus confirmed the existence of a toxic principle capable of inducing acute liver toxicity (Lancaster et al. 1961; Sargeant et al. 1961). Structural elucidation of aflatoxin B1 was accomplished and confirmed by its total synthesis in 1963 (Asao et al. 1963). AFB1 and AFB2 were named due to their strong blue fluorescence under ultraviolet light, whereas AFG1 and AFG2 fluoresced greenish-yellow. These properties facilitated the rapid development in the early 1960s of screening methods for grains and commodities that enabled subsequent promulgation of regulations to minimize contamination of human foods.
Figure 1.
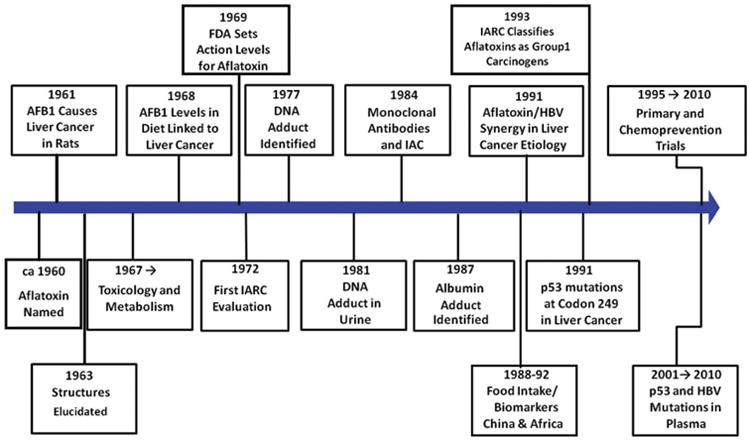
Time-lines in aflatoxin research.
Extensive research conducted since the aflatoxins were discovered has produced an impressive body of evidence supporting the conclusion that contamination of the food supply by aflatoxin B1 puts the exposed population at increased risk of developing HCC. Evidence showing that aflatoxin is a genotoxic liver carcinogen in experimental animals and poses a risk for humans can be summarized as follows. AFB1 is mutagenic to micro-organisms and to cultured animal and human cells in the presence of an appropriate activating system. Metabolic activation pathways leading to genotoxicity are identical in animals and humans, with the N7-guanine adduct being the major product in the DNA damage profile. Importantly, DNA adduct level relates quantitatively to tumor incidence and chemoprevention regimens that reduce DNA adduct formation also reduce or inhibit tumor induction. The carcinogenic potency of AFB1 has been well established in many species of animals, including rodents, nonhuman primates, fish and birds (Busby and Wogan 1984; Eaton and Groopman 1994; Wild and Turner 2002). The liver is the primary target organ affected and the toxin induces a high incidence of hepatocellular carcinoma; however, under certain circumstances, depending on animal species and strain, dose, route of administration and dietary factors, significant numbers of tumors have been found at other sites, including kidney and colon. Indeed, no animal species has been found to be completely resistant to aflatoxin-induced carcinogenesis. Wide cross-species potency, including sensitivity of primates, provided justification for the hypothesis that this agent contributes to risk of human liver cancer.
Human populations can be exposed to aflatoxins by consumption of commodities supporting growth of Aspergillus flavus and/or A. paraciticus during growth, harvest or storage. Grains and other foodstuffs sometimes found to contain aflatoxins include corn, peanuts, milo, sorghum, copra and rice (Busby and Wogan 1984). While contamination by the molds may be universal within a given geographical area, concentrations of aflatoxins in final products can vary from less than 1μg/kg (1ppb) to greater than 12,000 μg/kg (12 ppm). Indeed in a recent outbreak of aflatoxin-induced death of people in Kenya, the daily exposure to AFB1 was estimated to be 50mg per person (Probst et al. 2007).
One of the major controversies in risk assessment is the question of linearity of the dose–response curve for experimentally-induced cancers. Recent studies of aflatoxin carcinogenicity in the rainbow trout took advantage of low cost, low background of spontaneous tumors and high sensitivity, enabled the design of a study to provide an effective dose at 1% incidence (ED01); such a study would be impractical to conduct in rodents. In a total of more than 42,000 trout, AFB1 elicited a linear dose–response for liver cancer at the ED01 (Williams et al. 2009).
Aflatoxin–DNA adducts and other aflatoxin metabolites as biomarkers
Data demonstrating the high carcinogenic and toxic potency of AFB1 provided an impetus to characterize the metabolism and DNA adduct formation reactions of AFB1 to understand the underlying molecular mechanisms through which it induces cancer. Measurement of aflatoxin–DNA and –protein adducts were of major interest because they are direct products of (or surrogate markers for) damage to critical cellular macromolecular targets. The chemical structures of the major aflatoxin macromolecular DNA and protein adducts were elucidated (Essigmann et al. 1977; Sabbioni et al. 1987), and critical pathways emanating from the aflatoxin–epoxides are shown in Figure 2. The finding that the major aflatoxin–nucleic acid adduct, AFB1–N7-Gua, was excreted in urine of exposed rats (Bennett et al. 1981) spurred interest in using this metabolite as a possible biomarker of exposure and risk. The serum aflatoxin–albumin adduct was also examined as a biomarker of exposure. Owing to the longer half-life in vivo of the serum albumin adduct compared with the DNA adduct excreted in urine, the protein adduct integrates exposure over longer time periods. Despite kinetic differences, it was later shown in experimental animal models that formation of aflatoxin–DNA adducts in liver, excretion of the urinary aflatoxin–nucleic acid adduct and formation of the serum albumin adduct are highly correlated (Groopman, De Matos, et al. 1992), and exposure data produced by application of these two biomarkers are also directly comparable.
Figure 2.
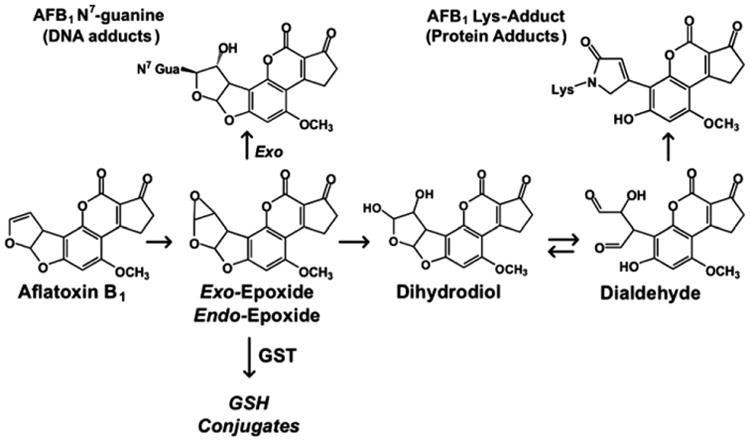
Chemical structures of the major aflatoxin macromolecular DNA and protein adducts (Sabbioni et al. 1987; Essigmann et al. 1977), and critical pathways emanating from the aflatoxin–epoxides.
Using combined immunological and analytical chemical methods, aflatoxin metabolites were measured in biological samples (Groopman et al. 1984, 1985). With this approach, a linear relationship was found between AFB1 dose in rats and excretion of the AFB1– N7-Gua adduct in urine over the initial 24 h period of exposure (Groopman, Hasler, et al. 1992). Subsequent studies in rodents that assessed the formation of aflatoxin macromolecular adducts after chronic administration also provided support for the use of DNA and protein adducts as molecular measures of exposure (Kensler et al. 1986; Egner et al. 1995). Recent studies using isotope-dilution mass spectrometry with liquid chromatography separation have demonstrated an increase in sensitivity of at least 1000-fold over technologies used for the analysis of aflatoxin bio-markers 15 years earlier (Egner et al. 2006; Scholl, Mc Coy, et al. 2006; Scholl, Turner, et al. 2006). Furthermore, repeated analysis of serum collected in 1983 from aflatoxin-exposed people has demonstrated that the aflatoxin–lysine adduct in albumin is stable for at least 25 years under a range of temperature storage conditions (Scholl and Groopman 2008).
Experimental animal models played a very important role in development of analytical methods for measuring aflatoxin biomarkers that were applicable for identifying effective chemoprevention strategies against aflatoxin carcinogenesis. The hypothesis tested was that reduction of aflatoxin–DNA adduct levels by chemopreventive agents would be mechanistically related to, and therefore predictive of, cancer preventive efficacy. Studies using multiple doses of aflatoxin and the chemopreventive agent ethoxyquin examined effects on DNA adduct formation and removal and hepatic tumorigenesis in rats. Treatment with ethoxyquin reduced both area and volume of liver occupied by pre-neoplastic foci by >95%. This same protocol also dramatically reduced binding of AFB1 to hepatic DNA, from 90% initially, to 70% at the end of a 2-week dosing period. This experiment was then repeated with several different chemopreventive agents and in all cases aflatoxin-derived DNA and protein adducts were reduced; however, even under optimal conditions, the reduction in the macromolecular adducts always under-represented the magnitude of reduction in tumor burden (Roebuck et al. 1991; Bolton et al. 1993).
Aflatoxin biomarkers in human epidemiology
Liver cancer, including hepatocellular carcinoma (HCC) and cholangiocarcinoma, accounts for 5.7% of all reported cancer cases and is the sixth most common cancer diagnosed worldwide (Parkin et al. 2005). The incidence of liver cancer varies widely, and the burden of this nearly always fatal disease is much higher in economically less-developed countries of Asia and sub-Saharan Africa (Groopman et al. 2008). Overall, more than 650,000 new cases and over 200,000 deaths occur annually in the People's Republic of China alone (Kew 2002; Wang et al. 2002). In contrast with most common cancers in the economically developed world, where over 90% of liver cancer cases are diagnosed after the age of 45 years, in high-risk regions onset begins in both men and women by 20 years of age and peaks between 40–49 years of age in men and between 50–59 years in women (Vatanasapt et al. 1995; Parkin et al. 2005; Chen et al. 2006).
Molecular biomarkers capable of quantifying aflatoxin exposure of individuals in a population, equivalent to those for determining HBV status, were essential to link aflatoxin exposure to liver cancer risk through molecular epidemiology. Early work conducted in populations with high HCC incidence in China and The Gambia, West Africa, examined relationships between dietary aflatoxin intake and the levels of urinary aflatoxin biomarkers (Groopman et al. 1985, Groopman, Hall, et al. 1992; Groopman, Zhu, et al. 1992). These were the first dose–response investigations to be conducted in humans. Urinary AFB1–N7-Gua and AFM1 showed linear dose-dependent relationships between aflatoxin intake and excretion. Gan et al. (1988) and Wild et al. (1992) also monitored levels of aflatoxins serum albumin adducts and observed a highly significant association between aflatoxin intake and adduct level. Interestingly, these studies also indicated that the kinetics of formation and excretion of AFB1–N7-Gua in urine were similar in rats and humans, thereby adding to our insights into the mechanisms of aflatoxin carcinogenesis.
Data obtained from cohort studies have the greatest power to determine a true relationship between exposure and disease outcome because they begin with a cohort of healthy individuals, collect biomarker samples and then follow the cohort until significant numbers of cases occur. A nested study within the cohort can then be designed to match cases and controls. An advantage of this method is that the controls are truly matched to the cases since both were recruited at the same time and with the same health status. A major disadvantage, however, is the time needed in follow-up (often years) to accrue cases in sufficient numbers to enable valid statistical analysis. This disadvantage can be overcome in part by enrolling large numbers of people (often tens of thousands) to ensure case accrual at a reasonable rate.
To date two major cohort studies employing aflatoxin biomarkers have demonstrated the role of this carcinogen in the etiology of HCC. The first study, comprising more than 18,000 men residing in Shanghai, China, examined HBV infection and aflatoxin exposure as independent and interactive risk factors for HCC. The nested case-control data revealed a statistically significant increase in the relative risk (RR) of 3.4 for those HCC cases where urinary aflatoxin biomarkers, but no HBV markers were detected. In men who were HBsAg-positive (i.e. had been HBV infected) but not aflatoxin exposed, the RR was 7. However, men exhibiting both urinary aflatoxin biomarkers and positive HBsAg status, the RR was 59 (Ross et al. 1992; Qian et al. 1994). These results strongly support a causal relationship between the presence of the chemical and viral-specific biomarkers and the HCC risk, and also show that the two risk factors act synergistically.
Subsequent cohort studies in Taiwan substantially confirmed the results from the Shanghai investigation. Wang et al. (1996) examined HCC cases and controls nested within a cohort and found that in HBV-infected people there was an adjusted odds ratio of 2.8 for detectable compared with non-detectable aflatoxin– albumin adducts and 5.5 for high compared with low levels of aflatoxin metabolites in urine. In a follow-up study, there was a dose–response relationship between urinary AFM1 levels and risk of HCC in chronic HBV carriers. Similar to the Shanghai data, the HCC risk associated with AFB1 exposure was more striking among the HBV carriers with detectable AFB1–N7-Gua in urine.
A recent case-control study nested within a community-based cohort was conducted in Taiwan to assess the interaction of HBV and aflatoxin in the development of HCC (Wu et al. 2009). The adjusted ORs (95% CIs) were 1.54 (1.01–2.36) and 1.76 (1.18– 2.58), respectively, for those with AFB1–albumin adducts and urinary AFB1 metabolite levels above the mean compared with those with levels below the mean. Further, the ORs (95% CI) were 10.38 (5.73–18.82) and 15.13 (7.83–29.25) for HBV carriers having levels of AFB1-albumin adducts and urinary AFB1 metabolites above the mean, respectively. These data continue to bolster the combined AFB1 exposure and HBV infection effects in the risk of liver cancer.
Collectively, available epidemiologic and experimental data fulfill criteria defined by Hill (1965) to differentiate causation from statistical association resulting from environmental chemical exposures, and thus establish aflatoxin as a causative agent for HCC in humans. The criteria include strength of association between dietary aflatoxin levels and HCC incidence, consistency across multiple studies, temporality between exposure and clinical disease, biological gradient (dose–response) in experimental models, biological plausibility based on mechanisms of action in experimental models and humans, and experimental modulation of disease biomarkers by chemoprevention. Aflatoxin is among few environmental carcinogens for which documentation is sufficient to meet these criteria.
The aflatoxin p53 mutation linkage
The relationship between aflatoxin exposure and development of HCC was further highlighted by molecular biological studies of the p53 tumor suppressor gene, the gene most commonly mutated in many human cancers (Harris 1993; Greenblatt et al. 1994; Olivier et al. 2002). Many studies of p53 mutations in HCC occurring in populations exposed to high levels of dietary aflatoxin have found high frequencies of guanine to thymine transversions, with a striking clustering of mutations in the third base of codon 249 (Bressac et al. 1991; Hsu et al. 1991).
Previous mechanistic studies showed that AFB1 induced almost exclusively guanine to thymine transversions in bacteria (Foster et al. 1983) and that aflatoxin–8,9-epoxide bound preferentially to codon 249 of p53 in a plasmid in vitro (Puisieux et al. 1991). Furthermore, Aguilar et al. (1993) examined mutagenesis of the p53 gene in human HepG2 cells and hepatocytes exposed to AFB1 and showed preferential induction of guanine to thymine transversion in the third position of codon 249. A further study has mapped AFB1 adduct formation to codon 249 (Denissenko et al. 1998).
While the detection of specific codon 249 p53 mutations in liver tumors has provided insight into the etiology of this disease, application of this knowledge to the early detection of HCC offers great promise for prevention (Sidransky and Hollstein 1996). It is now well-recognized that DNA derived from cells undergoing necrosis or apoptosis shed fragments of their DNA into the circulation, and these fragments can be detected and quantified in serum or plasma by a number of different molecular strategies (Gormally et al. 2007). Kirk et al. (2000) first reported the detection of codon 249 p53 mutations in plasma of HCC patients from The Gambia. They also found that a small number of cirrhosis patients harbored this mutation. Consistent with the strong relationship between cirrhosis and HCC development, Jackson et al. (2001) found that specific p53 mutations detected by SOMA analysis of serum were comparable to those identified by sequencing of DNA from 25 HCC samples. Jackson et al. (2003) further explored the temporality of detection of this mutation in plasma before and after clinical diagnosis of HCC in the same patient. More recent studies have provided additional evidence that the number of mutant p53 copies in plasma DNA also tracks with disease development (Kirk et al. 2005). Thus, this aflatoxin-specific mutation in a key gene linked to cancer development has the potential to serve as a biological effect biomarker for aflatoxin exposure.
Intervention trials in high-risk populations
Protective interventions against aflatoxin exposures in developing countries can take many forms. It is axiomatic that as economic development increases, affordability of a more diverse and more highly processed diet also increases; this usually also leads to concomitant reduction in aflatoxin exposures (Groopman et al. 2008). Given that economic development generally proceeds at a very slow rate, intervention strategies would be useful in reducing risks resulting from aflatoxin contamination of dietary staples. These strategies can take two forms, viz. primary and secondary prevention. In primary prevention, the goal is to reduce the frequency and levels of aflatoxin exposure by interventions such as planting pest-resistant varieties of staple crops, attempting to lower mold growth in harvested crops, and improving storage methods to enhance rapid drying following harvest. Active research is in progress to develop mold-resistant strains of grains and genetic manipulation of molds to lower aflatoxin production (Yu et al. 2005; Menkir et al. 2006). For secondary prevention measures, one avenue is to block the uptake of unavoidably ingested aflatoxins by use of trapping agents and another is to modulate the metabolism of ingested aflatoxins to enhance detoxification processes; thereby reducing internal dose and subsequent risk.
The use of aflatoxin biomarkers to monitor efficacy of primary prevention has been recently reported (Turner et al. 2005). This work was based on earlier research that showed extensive aflatoxin exposure in Guinea, West Africa, via consumption of contaminated groundnuts as a dietary staple (Diallo et al. 1995; Sylla et al. 1999); it also revealed that post-harvest storage was associated with increased aflatoxin exposure. A community-based intervention study was conducted, in which educational materials detailing post-harvest measures to limit aflatoxin contamination of the groundnut crop were provided by local agricultural support workers (Turner et al. 2005). Altered trajectories of aflatoxin–albumin adduct levels measured in 600 subjects over a 5-month post-harvest period were used to assess efficacy of the intervention. In control villages, the aflatoxin–albumin adduct level in serum increased, whereas in villages employing the intervention, adduct levels after 5 months of storage were similar to those immediately post-harvest. Mean levels at this time were 60% lower in intervention than in control villages. The effectiveness of this intervention suggests that significant reduction in aflatoxin exposure can be achieved through implementation of low-technology approaches at the subsistence farm level in sub-Saharan Africa, particularly in a setting of limited dietary complexity.
Another strategy for risk reduction used chlorophyllin, a water-soluble derivative of chlorophyll that had been shown to be an effective agent for reducing liver tumor incidence in aflatoxin-treated rainbow trout, a species highly sensitive to aflatoxin-induced HCC (Breinholt et al. 1995). In a randomized, double-blind, placebo-controlled chemoprevention trial conducted in people residing in Qidong, China, chlorophyllin was tested to determine if it could alter the disposition of aflatoxin (Egner et al. 2001). One hundred and eighty healthy adults were randomly assigned to ingest 100 mg of chlorophyllin or a placebo three times a day prior to each meal for 4 months. Aflatoxin–N7-Gua could be detected in 105 of 169 urine samples; chlorophyllin consumption at each meal led to an overall 55% reduction in median urinary levels of this aflatoxin biomarker compared with those in subjects taking placebo.
Chemoprevention is a key strategy for the secondary prevention of cancer, as illustrated by the use of aflatoxin biomarkers as intermediate endpoints in a Phase IIa trial employing oltipraz in Qidong, China (Kensler et al. 1998; Wang et al. 1999). Urinary AFM1 levels were reduced by 51% compared with the placebo group in persons receiving a 500 mg weekly dose of the drug. Median levels of AFB1–mercapturic acid (a glutathione conjugate derivative) were elevated six-fold in the 125-mg group, but were unchanged in the 500-mg group. Increased AFB1–mercapturic acid reflects induction of aflatoxin conjugation through the actions of GSTs. The apparent lack of induction in the 500-mg group probably reflects masking caused by diminished aflatoxin–8, 9-epoxide formation for conjugation through the inhibition of CYPlA2 seen in this group. This initial study demonstrated for the first time that aflatoxin biomarkers could be modulated in humans in a manner that would predict decreased HCC risk.
Although the oltipraz clinical trial demonstrated the proof of principle for increasing pathways leading to aflatoxin detoxication in humans, the practicality of using a drug-based preventive method in the economically developing world is limited. Not only is there a potential for adverse health effects from any long-term exposure to a drug, but also the expense of this type of intervention may make it cost-prohibitive for these populations. Many foods contain high levels of such enzyme inducers (Talalay and Fahey 2001; Fahey and Kensler 2007). Sulforaphane, found in cruciferous vegetables, is a potent activator leading to increased expression of carcinogen detoxifying enzymes that has been extensively examined for its chemopreventive properties (Fahey et al. 2002; Dinkova-Kostova et al. 2007). A beverage consisting of hot water infusions of 3-day old broccoli sprouts, containing defined concentrations of glucoraphanin, the stable glucosinolate precursor of sulforaphane, was recently evaluated for its ability to alter the disposition of aflatoxin. In this study, 200 healthy adults residing in Qidong, China, drank such infusions containing either 400 or < 3 μmol glucoraphanin nightly for 2 weeks. Urinary levels of aflatoxin–N7-guanine were similar between the two intervention treatment groups. However, urinary levels of dithiocarbamate metabolites showed striking inter-individual differences in sulforaphane bioavailability. When this variability was taken into account, a significant inverse association was evident between urinary excretion of dithiocarbamates and aflatoxin– N7-guanine adducts in individuals receiving broccoli sprout glucosinolates (Kensler et al. 2005). This preliminary study illustrates the potential usefulness of an inexpensive, easily implemented food-based method for secondary prevention in a population at high HCC risk by virtue of aflatoxin exposure.
Summary
The past 50 years of research into the toxicology and epidemiology of aflatoxin represents, perhaps, the most extensively documented study in the field of environmental carcinogenesis. On the basis of the totality of evidence, IARC classified aflatoxin as a Group 1 human carcinogen in 1993 (Baan et al. 2009). The IARC evaluation concluded that there is sufficient evidence in humans for the carcinogenicity of aflatoxins as a cause of hepatocellular carcinoma. Additional evidence cited in support of this conclusion included sufficient evidence in experimental animals for the carcinogenicity of naturally occurring mixtures of aflatoxins, aflatoxin B1, G1 and M1. Furthermore, it was pointed out that the carcinogenicity of aflatoxins operates by a genotoxic mechanism of action that involves metabolic activation to a genotoxic epoxide metabolite, formation of DNA adducts and modification of the p53 gene. In human HCCs from areas of high exposure to aflatoxins, up to 50% of tumors have been shown to harbor a specific mutation in the p53 tumor suppressor gene.
A striking discovery produced through the molecular epidemiology of aflatoxin as a risk factor for HCC was the interaction with hepatitis B virus infection, which acts synergistically to vastly increase attributable to each factor alone. This discovery has important potential implications for future research strategies in environmental carcinogenesis. It is presently unclear whether the HBV/aflatoxin/liver cancer relationship is unique, or whether it may represent the “tip of an iceberg” of unrecognized syngergistic interactions between chemical toxicants, infectious agents and chronic inflammation that might also affect risks for other cancers and possibly other environmentally linked diseases. This question clearly merits extensive further research.
The systematic, comprehensive approach used to create the total aflatoxin database justifies optimism with respect to the potential success of preventive interventions to ameliorate cancer risk attributable to aflatoxin exposure, and could serve as a template for the development, validation and application of molecular and biochemical markers for other carcinogens and cancers as well as other chronic diseases resulting from environmental exposures. Key elements to define health risks from environmental agents and to devise strategies to mitigate their effects were identified by the aflatoxin experience. These include experimental animal models mimicking the human disease, observational epidemiology associating exposure with disease incidence, development of mechanism-based molecular biomarkers in animal models, biomarker validation in animals by dose–response and modulation studies, biomarker validation by transitional studies in exposed humans, and association of biomarkers with risk in prospective studies in exposed humans.
Acknowledgments
This work was supported in part by grants P01 ES006052 and P30 ES003819 from the USPHS.
References
- Aguilar F, Hussain SP, Cerutti P. Aflatoxin B1 induces the transversion of G→T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc Natl Acad Sci USA. 1993;90(18):8586–8590. doi: 10.1073/pnas.90.18.8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asao T, Buchi G, Abdel-Kader M, Chang S, Wick E, Wogan GN. Aflatoxins B and G. J Am Chem Soc. 1963;85:1706–1707. doi: 10.1021/ja01082a031. [DOI] [PubMed] [Google Scholar]
- Baan R, Grosse Y, Straif K, Secretan B, El Ghissassi F, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, et al. A review of human carcinogens– Part F: chemical agents and related occupations. Lancet Oncol. 2009;10(12):1143–1144. doi: 10.1016/s1470-2045(09)70358-4. [DOI] [PubMed] [Google Scholar]
- Bennett RA, Essigmann JM, Wogan GN. Excretion of an aflatoxin-guanine adduct in the urine of aflatoxin B1-treated rats. Cancer Res. 1981;41(2):650–654. [PubMed] [Google Scholar]
- Blount WP. Turkey “X” disease. J Br Turkey Fed. 1961;9(2):55–58. [Google Scholar]
- Bolton MG, Munoz A, Jacobson LP, Groopman JD, Maxuitenko YY, Roebuck BD, Kensler TW. Transient intervention with oltipraz protects against aflatoxin-induced hepatic tumorigenesis. Cancer Res. 1993;53(15):3499–3504. [PubMed] [Google Scholar]
- Breinholt V, Hendricks J, Pereira C, Arbogast D, Bailey G. Dietary chlorophyllin is a potent inhibitor of aflatoxin B1 hepatocarcinogenesis in rainbow trout. Cancer Res. 1995;55:57–62. [PubMed] [Google Scholar]
- Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350(6317):429–431. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- Busby WF, Wogan GN. Aflatoxins. In: Searle CE, editor. Chemical Carcinogens. Washington, DC: American Chemical Society; 1984. [Google Scholar]
- Busby WF, Wogan GN. Aflatoxins. In: Searle CD, editor. Chemical Carcinogens. Washington (DC): American Chemical Society; 1984. [Google Scholar]
- Chen JG, Zhu J, Parkin DM, Zhang YH, Lu JH, Zhu YR, Chen TY. Trends in the incidence of cancer in Qidong, China, 1978–2002. Int J Cancer. 2006;119(6):1447–1454. doi: 10.1002/ijc.21952. [DOI] [PubMed] [Google Scholar]
- Denissenko MF, Koudriakova TB, Smith L, O'Connor TR, Riggs AD, Pfeifer GP. The p53 codon 249 mutational hotspot in hepatocellular carcinoma is not related to selective formation or persistence of aflatoxin B1 adducts. Oncogene. 1998;17(23):3007–3014. doi: 10.1038/sj.onc.1202214. [DOI] [PubMed] [Google Scholar]
- Diallo MS, Sylla A, Sidibe K, Sylla BS, Trepo CR, Wild CP. Prevalence of exposure to aflatoxin and hepatitis B and C viruses in Guinea, West Africa. Nat Toxins. 1995;3(1):6–9. doi: 10.1002/nt.2620030103. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Fahey JW, Wade KL, Jenkins SN, Shapiro TA, Fuchs EJ, Kerns ML, Talalay P. Induction of the phase 2 response in mouse and human skin by sulforaphane-containing broccoli sprout extracts. Cancer Epidemiol Biomarkers Prev. 2007;16(4):847–851. doi: 10.1158/1055-9965.EPI-06-0934. [DOI] [PubMed] [Google Scholar]
- Eaton DL, Groopman JD. The Toxicology of Aflatoxins: Human Health, Veterinary, and Agricultural Significance. San Diego: Academic Press; 1994. [Google Scholar]
- Egner PA, Gange SJ, Dolan PM, Groopman JD, Munoz A, Kensler TW. Levels of aflatoxin-albumin biomarkers in rat plasma are modulated by both long-term and transient interventions with oltipraz. Carcinogenesis. 1995;16(8):1769–1773. doi: 10.1093/carcin/16.8.1769. [DOI] [PubMed] [Google Scholar]
- Egner PA, Wang JB, Zhu YR, Zhang BC, Wu Y, Zhang QN, Qian GS, Kuang SY, Gange SJ, Jacobson LP, et al. Chlorophyllin intervention reduces aflatoxin–DNA adducts in individuals at high risk for liver cancer. Proc Natl Acad Sci USA. 2001;98(25):14601–14606. doi: 10.1073/pnas.251536898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner PA, Groopman JD, Wang JS, Kensler TW, Friesen MD. Quantification of aflatoxin-B1–N7-Guanine in human urine by high-performance liquid chromatography and isotope dilution tandem mass spectrometry. Chem Res Toxicol. 2006;19(9):1191–1195. doi: 10.1021/tx060108d. [DOI] [PubMed] [Google Scholar]
- Essigmann JM, Croy RG, Nadzan AM, Busby WF, Jr, Reinhold VN, Buchi G, Wogan GN. Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc Natl Acad Sci USA. 1977;74(5):1870–1874. doi: 10.1073/pnas.74.5.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Kensler TW. Role of dietary supplements/nutraceuticals in chemoprevention through induction of cytoprotective enzymes. Chem Res Toxicol. 2007;20(4):572–576. doi: 10.1021/tx7000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, Talalay P, Lozniewski A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci USA. 2002;99(11):7610–7615. doi: 10.1073/pnas.112203099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Eisenstadt E, Miller JH. Base substitution mutations induced by metabolically activated aflatoxin B1. Proc Natl Acad Sci USA. 1983;80(9):2695–2698. doi: 10.1073/pnas.80.9.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan LS, Skipper PL, Peng XC, Groopman JD, Chen JS, Wogan GN, Tannenbaum SR. Serum albumin adducts in the molecular epidemiology of aflatoxin carcinogenesis: correlation with aflatoxin B1 intake and urinary excretion of aflatoxin M1. Carcinogenesis. 1988;9(7):1323–1325. doi: 10.1093/carcin/9.7.1323. [DOI] [PubMed] [Google Scholar]
- Gormally E, Caboux E, Vineis P, Hainaut P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutat Res. 2007;635(2-3):105–117. doi: 10.1016/j.mrrev.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54(18):4855–4878. [PubMed] [Google Scholar]
- Groopman JD, Trudel LJ, Donahue PR, Marshak-Rothstein A, Wogan GN. High-affinity monoclonal antibodies for aflatoxins and their application to solid-phase immunoassays. Proc Natl Acad Sci USA. 1984;81(24):7728–7731. doi: 10.1073/pnas.81.24.7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groopman JD, Donahue PR, Zhu JQ, Chen JS, Wogan GN. Aflatoxin metabolism in humans: detection of metabolites and nucleic acid adducts in urine by affinity chromatography. Proc Natl Acad Sci USA. 1985;82(19):6492–6496. doi: 10.1073/pnas.82.19.6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groopman JD, DeMatos P, Egner PA, Love-Hunt A, Kensler TW. Molecular dosimetry of urinary aflatoxin–N7-guanine and serum aflatoxin–albumin adducts predicts chemoprotection by 1,2-dithiole-3-thione in rats. Carcinogenesis. 1992;13(1):101–106. doi: 10.1093/carcin/13.1.101. [DOI] [PubMed] [Google Scholar]
- Groopman JD, Hasler JA, Trudel LJ, Pikul A, Donahue PR, Wogan GN. Molecular dosimetry in rat urine of aflatoxin–N7-guanine and other aflatoxin metabolites by multiple monoclonal antibody affinity chromatography and immunoaffinity/high performance liquid chromatography. Cancer Res. 1992;52(2):267–274. [PubMed] [Google Scholar]
- Groopman JD, Hall AJ, Whittle H, Hudson GJ, Wogan GN, Montesano R, Wild CP. Molecular dosimetry of aflatoxin–N7-guanine in human urine obtained in The Gambia, West Africa. Cancer Epidemiol Biomarkers Prev. 1992;1(3):221–227. [PubMed] [Google Scholar]
- Groopman JD, Zhu JQ, Donahue PR, Pikul A, Zhang LS, Chen JS, Wogan GN. Molecular dosimetry of urinary aflatoxin–DNA adducts in people living in Guangxi Autonomous Region, People's Republic of China. Cancer Res. 1992;52(1):45–52. [PubMed] [Google Scholar]
- Groopman JD, Kensler TW, Wild CP. Protective interventions to prevent aflatoxin-induced carcinogenesis in developing countries. Annu Rev Public Health. 2008;29:187–203. doi: 10.1146/annurev.publhealth.29.020907.090859. [DOI] [PubMed] [Google Scholar]
- Harris CC. Multistep carcinogenesis. Jpn J Cancer Res. 1993;84(7) inside front cover. [PubMed] [Google Scholar]
- Hill AB. The environment and disease: Association or causation? Proc R Soc Med. 1965;58:295–300. [PMC free article] [PubMed] [Google Scholar]
- Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350(6317):427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- Jackson PE, Qian GS, Friesen MD, Zhu YR, Lu P, Wang JB, Wu Y, Kensler TW, Vogelstein B, Groopman JD. Specific p53 mutations detected in plasma and tumors of hepatocellular carcinoma patients by electrospray ionization mass spectrometry. Cancer Res. 2001;61(1):33–35. [PubMed] [Google Scholar]
- Jackson PE, Kuang SY, Wang JB, Strickland PT, Munoz A, Kensler TW, Qian GS, Groopman JD. Prospective detection of codon 249 mutations in plasma of hepatocellular carcinoma patients. Carcinogenesis. 2003;24(10):1657–1663. doi: 10.1093/carcin/bgg101. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Egner PA, Davidson NE, Roebuck BD, Pikul A, Groopman JD. Modulation of aflatoxin metabolism, aflatoxin-N7-guanine formation, and hepatic tumorigenesis in rats fed ethoxyquin: role of induction of glutathione S-transferases. Cancer Res. 1986;46(8):3924–3931. [PubMed] [Google Scholar]
- Kensler TW, He X, Otieno M, Egner PA, Jacobson LP, Chen B, Wang JS, Zhu Yr, Zhang BC, Wang JB, et al. Oltipraz chemoprevention trial in Qidong, People's Republic of China: Modulation of serum aflatoxin albumin adduct biomarkers. Cancer Epidemiol Biomarkers Prev. 1998;7:127–134. [PubMed] [Google Scholar]
- Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, Ye LX, Wang JB, Wu Y, Sun Y, et al. Broccoli sprout modulation of the urinary excretion of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in Qidong, People's Republic of china. J Nutr. 2005;135(12):3051S–3051S. doi: 10.1158/1055-9965.EPI-05-0368. [DOI] [PubMed] [Google Scholar]
- Kew MC. Epidemiology of hepatocellular carcinoma. Toxicology. 2002;181–182:35–38. doi: 10.1016/s0300-483x(02)00251-2. [DOI] [PubMed] [Google Scholar]
- Kirk GD, Camus-Randon AM, Mendy M, Goedert JJ, Merle P, Trepo C, Brechot C, Hainaut P, Montesano R. Ser-249 p53 mutations in plasma DNA of patients with hepatocellular carcinoma from The Gambia. J Natl Cancer Inst. 2000;92(2):148–153. doi: 10.1093/jnci/92.2.148. [DOI] [PubMed] [Google Scholar]
- Kirk GD, Lleonart ME, Villar S, Groopman JD, Mendy M, Hainaut P, Friesen M. Quantitative detection of 249ser TP53 mutations in plasma DNA and risk for hepatocellular carcinoma. J Hepatol. 2005;42:95–96. [Google Scholar]
- Lancaster MC, Jenkins FP, Philp JM. Toxicity associated with certain samples of groundnuts. Nature. 1961;192:1095–1096. [Google Scholar]
- Menkir A, Brown RL, Bandyopadhyay R, Chen ZY, Cleveland TE. A USA-Africa collaborative strategy for identifying, characterizing, and developing maize germplasm with resistance to aflatoxin contamination. Mycopathologia. 2006;162(3):225–232. doi: 10.1007/s11046-006-0056-3. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2002;2(1):a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Probst C, Njapau H, Cotty PJ. Outbreak of an acute aflatoxicosis in Kenya in 2004: identification of the causal agent. Appl Environ Microbiol. 2007;73(8):2762–2764. doi: 10.1128/AEM.02370-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puisieux A, Lim S, Groopman J, Ozturk M. Selective targeting of p53 gene mutational hotspots in human cancers by etiologically defined carcinogens. Cancer Res. 1991;51(22):6185–6189. [PubMed] [Google Scholar]
- Qian GS, Ross RK, Yu MC, Yuan JM, Gao YT, Henderson BE, Wogan GN, Groopman JD. A Follow-up-Study of Urinary Markers of Aflatoxin Exposure and Liver-Cancer Risk in Shanghai, People's Republic of China. Cancer Epidemiol Biomarkers Prev. 1994;3(1):3–10. [PubMed] [Google Scholar]
- Roebuck BD, Liu YL, Rogers AE, Groopman JD, Kensler TW. Protection against aflatoxin B1-induced hepatocarcinogenesis in F344 rats by 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione (oltipraz): predictive role for short-term molecular dosimetry. Cancer Res. 1991;51(20):5501–5506. [PubMed] [Google Scholar]
- Ross RK, Yuan JM, Yu MC, Wogan GN, Qian GS, Tu JT, Groopman JD, Gao YT, Henderson BE. Urinary aflatoxin biomarkers and risk of hepatocellular carcinoma. Lancet. 1992;339(8799):943–946. doi: 10.1016/0140-6736(92)91528-g. [DOI] [PubMed] [Google Scholar]
- Sabbioni G, Skipper PL, Buchi G, Tannenbaum SR. Isolation and characterization of the major serum albumin adduct formed by aflatoxin B1 in vivo in rats. Carcinogenesis. 1987;8(6):819–824. doi: 10.1093/carcin/8.6.819. [DOI] [PubMed] [Google Scholar]
- Sargeant K, Sheridan A, O'Kelly J, Carnaghan RBA. Toxicity associated with certain samples of groundnuts. Nature. 1961;192:1096–1097. [Google Scholar]
- Scholl PF, Groopman JD. Long-term stability of human aflatoxin B1 albumin adducts assessed by isotope dilution mass spectrometry and high-performance liquid chromatography-fluorescence. Cancer Epidemiol Biomarkers Prev. 2008;17(6):1436–1439. doi: 10.1158/1055-9965.EPI-07-2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl PF, McCoy L, Kensler TW, Groopman JD. Quantitative analysis and chronic dosimetry of the aflatoxin B1 plasma albumin adduct Lys-AFB1 in rats by isotope dilution mass spectrometry. Chem Res Toxicol. 2006;19(1):44–49. doi: 10.1021/tx050251r. [DOI] [PubMed] [Google Scholar]
- Scholl PF, Turner PC, Sutcliffe AE, Sylla A, Diallo MS, Friesen MD, Groopman JD, Wild CP. Quantitative comparison of aflatoxin B1 serum albumin adducts in humans by isotope dilution mass spectrometry and ELISA. Cancer Epidemiol Biomarkers Prev. 2006;15(4):823–826. doi: 10.1158/1055-9965.EPI-05-0890. [DOI] [PubMed] [Google Scholar]
- Sidransky D, Hollstein M. Clinical implications of the p53 gene. Annu Rev Med. 1996;47:285–301. doi: 10.1146/annurev.med.47.1.285. [DOI] [PubMed] [Google Scholar]
- Sylla A, Diallo MS, Castegnaro J, Wild CP. Interactions between hepatitis B virus infection and exposure to aflatoxins in the development of hepatocellular carcinoma: a molecular epidemiological approach. Mutat Res. 1999;428(1-2):187–196. doi: 10.1016/s1383-5742(99)00046-0. [DOI] [PubMed] [Google Scholar]
- Talalay P, Fahey JW. Phytochemicals from cruciferous plants protect against cancer by modulating carcinogen metabolism. J Nutr. 2001;131(11 Suppl):3027S–3033S. doi: 10.1093/jn/131.11.3027S. [DOI] [PubMed] [Google Scholar]
- Turner PC, Sylla A, Gong YY, Diallo MS, Sutcliffe AE, Hall AJ, Wild CP. Reduction in exposure to carcinogenic aflatoxins by postharvest intervention measures in west Africa: a community-based intervention study. Lancet. 2005;365(9475):1950–1956. doi: 10.1016/S0140-6736(05)66661-5. [DOI] [PubMed] [Google Scholar]
- Vatanasapt V, Martin N, Sriplung H, Chindavijak K, Sontipong S, Sriamporn S, Parkin DM, Ferlay J. Cancer incidence in Thailand, 1988–1991. Cancer Epidemiol Biomarkers Prev. 1995;4:475–483. [PubMed] [Google Scholar]
- Wang JS, Shen X, He X, Zhu Yr, Zhang BC, Wang JB, Qian GS, Kuang SY, Zarba A, Egner PA, et al. Protective Alterations in Phase 1 and 2 Metabolism of Aflatoxin B1 by Oltipraz in Residents of Qidong, People's Republic of China. J Natl Cancer Inst. 1999;91(4):347–354. doi: 10.1093/jnci/91.4.347. [DOI] [PubMed] [Google Scholar]
- Wang LY, Hatch M, Chen CJ, Levin B, You SL, Lu SN, Wu MH, Wu WP, Wang LW, Wang Q, et al. Aflatoxin exposure and risk of hepatocellular carcinoma in Taiwan. Int J Cancer. 1996;67(5):620–625. doi: 10.1002/(SICI)1097-0215(19960904)67:5<620::AID-IJC5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, Brechot C, Harris CC. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002;181/182:43–47. doi: 10.1016/s0300-483x(02)00253-6. [DOI] [PubMed] [Google Scholar]
- Wild CP, Turner PC. The toxicology of aflatoxins as a basis for public health decisions. Mutagenesis. 2002;17(6):471–481. doi: 10.1093/mutage/17.6.471. [DOI] [PubMed] [Google Scholar]
- Wild CP, Hudson GJ, Sabbioni G, Chapot B, Hall AJ, Wogan GN, Whittle H, Montesano R, Groopman JD. Dietary intake of aflatoxins and the level of albumin-bound aflatoxin in peripheral blood in The Gambia, West Africa. Cancer Epidemiol Biomarkers Prev. 1992;1(3):229–234. [PubMed] [Google Scholar]
- Williams DE, Orner G, Willard KD, Tilton S, Hendricks JD, Pereira C, Benninghoff AD, Bailey GS. Rainbow trout (Oncorhynchus mykiss) and ultra-low dose cancer studies. Comp Biochem Physiol C Toxicol Pharmacol. 2009;149(2):175–181. doi: 10.1016/j.cbpc.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HC, Wang Q, Yang HI, Ahsan H, Tsai WY, Wang LY, Chen SY, Chen CJ, Santella RM, Wang LY, et al. Aflatoxin B1 exposure, hepatitis B virus infection, and hepatocellular carcinoma in Taiwan. Cancer Epidemiol Biomarkers Prev. 2009;18(3):846–853. doi: 10.1158/1055-9965.EPI-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Cleveland TE, Nierman WC, Bennett JW. Aspergillus flavus genomics: gateway to human and animal health, food safety, and crop resistance to diseases. Rev Iberoam Micol. 2005;22(4):194–202. doi: 10.1016/s1130-1406(05)70043-7. [DOI] [PubMed] [Google Scholar]