

ABSTRACT
High levels of circulating immunocomplexes (ICs) are found in patients with either infectious or sterile inflammation. We report that patients with either Plasmodium falciparum or Plasmodium vivax malaria have increased levels of circulating anti-DNA antibodies and ICs containing parasite DNA. Upon stimulation with malaria-induced ICs, monocytes express an NF-κB transcriptional signature. The main source of IC-induced proinflammatory cytokines (i.e., tumor necrosis factor alpha [TNF-α] and interleukin-1β [IL-1β])in peripheral blood mononuclear cells from acute malaria patients was found to be a CD14+ CD16 (FcγRIIIA)+ CD64 (FcγRI)high CD32 (FcγRIIB)low monocyte subset. Monocytes from convalescent patients were predominantly of the classical phenotype (CD14+ CD16−) that produces high levels of IL-10 and lower levels of TNF-α and IL-1β in response to ICs. Finally, we report a novel role for the proinflammatory activity of ICs by demonstrating their ability to induce inflammasome assembly and caspase-1 activation in human monocytes. These findings illuminate our understanding of the pathogenic role of ICs and monocyte subsets and may be relevant for future development of immunity-based interventions with broad applications to systemic inflammatory diseases.
IMPORTANCE
Every year, there are approximately 200 million cases of Plasmodium falciparum and P. vivax malaria, resulting in nearly 1 million deaths, most of which are children. Decades of research on malaria pathogenesis have established that the clinical manifestations are often a consequence of the systemic inflammation elicited by the parasite. Recent studies indicate that parasite DNA is a main proinflammatory component during infection with different Plasmodium species. This finding resembles the mechanism of disease in systemic lupus erythematosus, where host DNA plays a central role in stimulating an inflammatory process and self-damaging reactions. In this study, we disclose the mechanism by which ICs containing Plasmodium DNA activate innate immune cells and consequently stimulate systemic inflammation during acute episodes of malaria. Our results further suggest that Toll-like receptors and inflammasomes have a central role in malaria pathogenesis and provide new insights toward developing novel therapeutic interventions for this devastating disease.
INTRODUCTION
Despite different etiologies and clinical manifestations, there are many parallels between malaria and systemic lupus erythematosus (SLE). In both diseases, nucleic acids are thought to be responsible for activating innate immune sensors and promoting systemic inflammation (1–4). Activation of nucleic-acid-sensing Toll-like receptors (NAS-TLRs) may be either pathogenic or protective in both SLE (5–8) and malaria (9–12). Likewise, tumor necrosis factor alpha (TNF-α), a cytokine induced by TLR activation, can either mediate resistance or enhance the pathogenesis of either disease (13–16). Intriguingly, for many decades effective antimalarial drugs have been used to treat SLE patients. These drugs accumulate in lysosomes, where they raise the pH, and are thought to mitigate the symptoms of SLE by preventing activation of endosomal TLRs (17).
How nucleic acids gain access to intracellular innate immune receptors is an important question in understanding the pathogenesis of SLE and malaria (7). In SLE, immunocomplexes (ICs) containing pathogenic anti-DNA/RNA antibodies are thought to be important carriers of human nucleic acids to the intracellular compartments of B cells and phagocytes, where they can activate the endosomal TLRs and possibly transit to the cytoplasm, where other DNA sensors can be engaged (4, 7, 8, 18, 19). Importantly, high levels of ICs are also found in both human and rodent malaria (20–22). However, the importance of DNA-containing ICs in activation of innate immune cells and pathogenesis of malaria is unknown.
The IgG Fc receptors have an important role in internalization of ICs by innate immune cells. Once bound to the Fc portion of IgG, Fc receptors can inhibit (e.g., FcγRIIB) or activate (e.g., FcγRIIIA and FcγRI) monocyte functions (23). Indeed, a loss-of-function polymorphism in the gene encoding the deactivating FcγRIIB protects against malaria but enhances susceptibility to SLE (24–28).
In this study, we report that ICs containing Plasmodium DNA activate intracellular DNA sensors. Our data indicate a previously undescribed role of the proinflammatory activity of ICs by demonstrating their ability to induce inflammasome assembly, caspase-1 activation, and interleukin-1β (IL-1β) secretion, primarily via CD14+ CD16 (FcγRIIIA)+ CD64 (FcγRI)high CD32 (FcγRIIB)low monocytes. Our findings have important implications for understanding the role of ICs and monocyte subsets in malaria pathogenesis and, more broadly, for understanding other infectious and autoimmune diseases.
RESULTS
Increased levels of cytokines and circulating ICs in sera of malaria patients.
The levels of IL-6, IL-8, and IL-10 in the plasma of malaria patients used in this study (see Fig. S1 in the supplemental material) are consistent with our prior data (29, 30). To evaluate the immunostimulatory properties of sera from malaria patients, we incubated peripheral blood mononuclear cells (PBMCs) from healthy donors with RPMI medium containing 20% sera from either Plasmodium vivax- or Plasmodium falciparum-infected subjects. We found that only sera from malaria patients, not those from healthy controls, triggered TNF-α production (Fig. 1A). The TNF-α levels were undetectable in similarly diluted (20%) patient sera (data not shown), ensuring that it was released from monocytes and not presented a priori in the tissue culture medium.
FIG 1 .

High levels of ICs in sera from P. falciparum- and P. vivax-infected patients. (A) Monocytes from healthy donors were incubated with individual sera from P. vivax (n = 3) and P. falciparum (n = 8) malaria patients at a 1:5 (20%) dilution in tissue culture medium or stimulated with 100 ng/ml of LPS, and TNF-α production was evaluated 24 h later by ELISA. (B) IC levels in sera from malaria patients and healthy individuals, as well as in purified IC preparations, as positive controls. (C) ICs in the sera of P. vivax-infected subjects (n = 22) before and 30 to 45 days after treatment as well as in healthy donors (n = 4). Different colors indicate different levels of parasitemia. IC levels shown in panels B and C were measured by the MicroVue CIC-C1q enzyme immunoassay kit. Horizontal lines indicate the average for each group. P values were determined by Student’s t test (A and B) and paired t test (C).
We hypothesized that circulating ICs were responsible for stimulating monocytes. Indeed, we detected high levels of ICs in sera of the same patients with acute untreated malaria (Fig. 1B). The levels of ICs were also measured in sera from P. vivax-infected patients before and after chemotherapy and compared with healthy donors as baseline controls. We found high levels of ICs in sera from patients during acute P. vivax episodes (mean, ~16.1 µg-equivalents [µg-eq]/ml) compared to healthy donors (mean, ~2.3 µg-eq/ml). The IC levels in the sera of treated patients were intermediate (mean, ~12.1 µg-eq/ml) and differed significantly from patient serum pretreatment and healthy controls (Fig. 1C). We found no correlation between the level of ICs and parasitemia.
Circulating ICs from malaria patients contain Plasmodium DNA.
We next measured the levels of IgM and IgG specific for double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA) in the sera of P. vivax- and P. falciparum-infected patients. As in SLE patients, the levels of anti-dsDNA IgG and IgM were significantly increased in the sera of acutely infected patients over those in sera from healthy donors (Fig. 2). Anti-dsDNA IgG persisted at least 30 to 40 days after treatment and parasitological cure in either P. vivax- or P. falciparum-infected individuals. In contrast, anti-ssDNA IgG and IgM levels in malaria patients did not differ from the corresponding levels measured in healthy donors (data not shown).
FIG 2 .
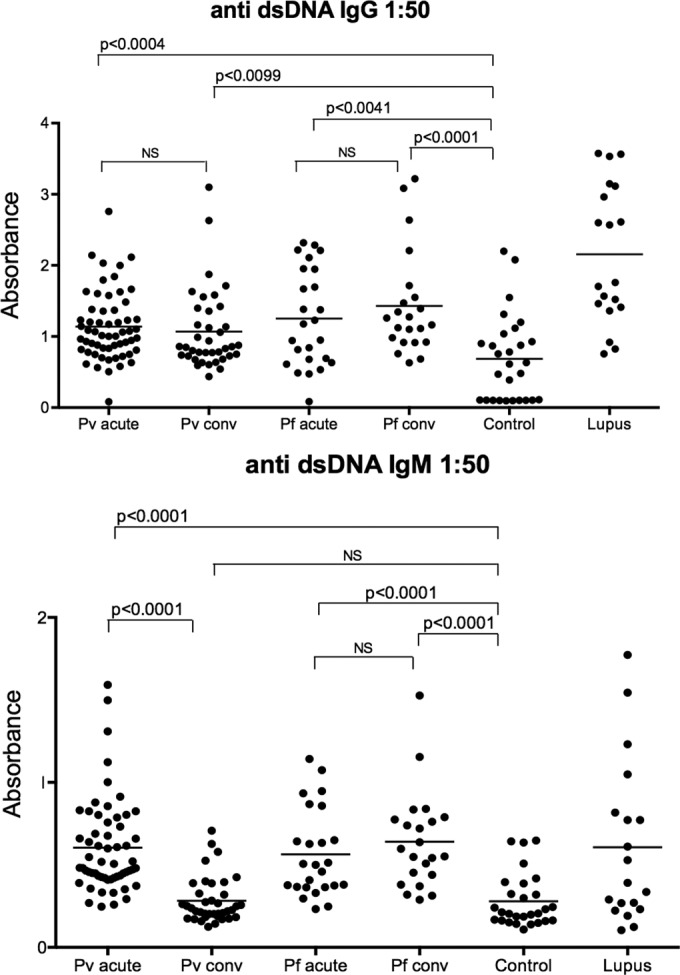
High levels of circulating anti-dsDNA in patients undergoing acute episodes of P. vivax and P. falciparum malaria. Levels of circulating anti-dsDNA IgG (top) and IgM (bottom) measured by ELISA in sera from patients with acute episodes of P. vivax (n = 58) and P. falciparum (n = 25) malaria and convalescent (conv) individuals 30 to 45 days after antimalarial therapy. Sera from uninfected healthy donors (control; n = 28) and lupus patients (n = 19) were used as negative and positive controls, respectively. Significant differences are indicated with P values using the Mann-Whitney U test. NS, nonsignificant.
The levels of human and parasite DNA present in ICs and total plasma from malaria patients were determined by quantitative PCR using CCND1- and SSUrRNA-specific primers. CCND1 is a single-copy gene optimized to quantify human DNA (http://www.rtprimerdb.org, primer ID 3605). SSUrRNA is a multicopy gene from P. vivax, which allows amplification and quantitation of DNA in a highly sensitive and Plasmodium species-specific manner. We observed increased levels of both human and parasite DNA in the plasma of P. vivax malaria patients before treatment. Circulating human DNA persisted, although at lower levels, whereas parasite DNA was not detectable in plasma following antimalarial chemotherapy (Fig. 3A). Similarly, parasite DNA was detected in purified ICs from P. vivax malaria patients before and (at much lower levels) after treatment. Human DNA was detected at very low levels in IC preparations from the same patients (Fig. 3B). Benzonase treatment degraded DNA and confirmed the presence of DNA in ICs from malaria patients (Fig. 3C).
FIG 3 .
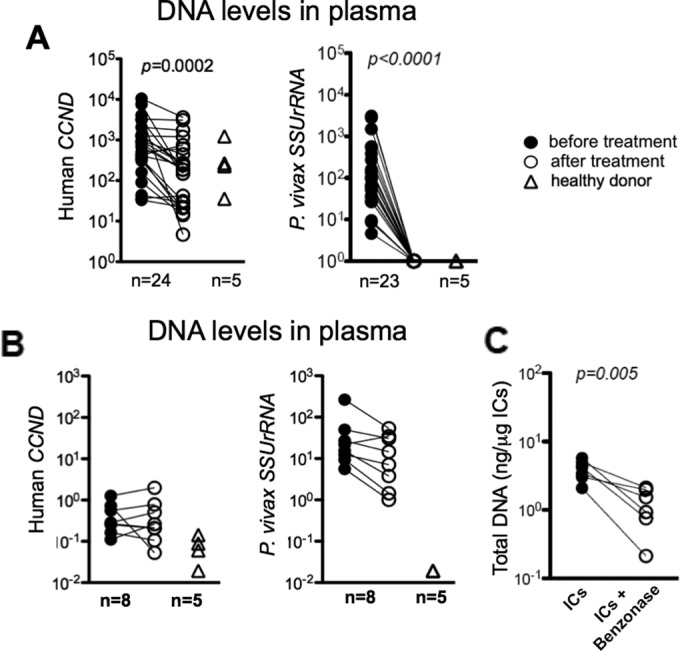
High levels of human and parasite DNA in plasma and ICs of P. vivax malaria patients. (A and B) Quantitative PCR to determine levels of human CYCLIN D1 (CCND) and P. vivax SSUrRNA (18S from rRNA) genes in plasma from P. vivax patients (n = 24) before and 30 to 45 days after treatment as well as in healthy donors (n = 5) (A) and DNA extracted from purified ICs from P. vivax malaria patients (n = 8) before and 30 to 45 days after treatment as well as healthy donors (n = 5) (B). (C) Quantification of total DNA extracted from purified ICs from P. vivax malaria patients (n = 6) before and after Benzonase treatment. Significant differences are indicated with P values using a paired t test.
An NF-κB transcriptional signature in monocytes stimulated with ICs.
We next profiled mRNA expression of highly enriched monocytes (over 99% purity) collected from four healthy donors (Fig. 4). A custom code set for NanoString analysis was designed for 98 genes related to inflammatory responses. Genes were divided into six groups: NOD-like receptor signaling pathway; TLR signaling pathway; NF-κB signaling pathway; cytokines and cytokine receptors; adhesion molecules, chemokines, and chemokine receptors; and interferon (IFN)-stimulated genes. Of these, 27 genes had augmented expression upon IC stimulation (fold change, >1.8; P value, <0.05) (see Table S1 in the supplemental material). Of the 27 differentially expressed genes, five were from the NF-κB family, and most of the genes with enhanced expression were proinflammatory cytokines and chemokines known to be induced by NF-κB (Fig. 4).
FIG 4 .
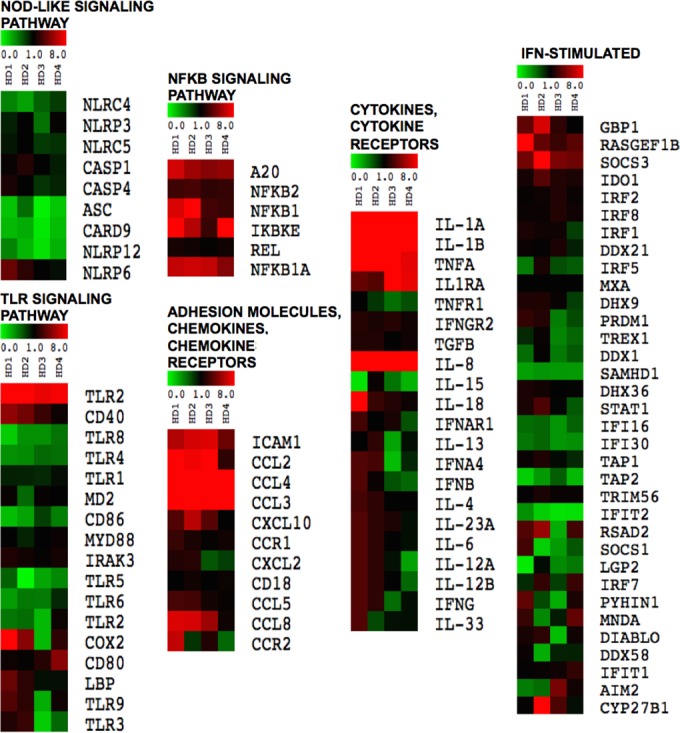
NF-κB transcriptional signature in monocytes stimulated with ICs. Purified monocytes from four different healthy donors were either unstimulated or stimulated with ICs (60 µg/ml) for 6 h and analyzed for gene expression using the NanoString technology. Data were normalized using housekeeping genes, and fold increase in gene expression was calculated in reference to unstimulated samples from the same patient. Different signaling pathways are depicted in the heat maps and show differentially expressed genes (rows) for each healthy donor (columns) that have diminished (green), unaltered (black), and enhanced (red) expression. Table S1 in the supplemental material provides the P values of genes which showed significantly enhanced expression as determined by the unpaired t test.
ICs derived from malaria patients induce cytokine production by human monocytes.
We then examined if ICs from malaria patients could induce cytokine production by PBMCs from healthy donors. We incubated PBMCs from healthy donors with different concentrations of purified ICs and measured TNF-α, IL-1β, and IL-10 levels in culture supernatants 24 h after stimulation. We observed that 60 µg/ml of purified ICs induced near-maximal cytokine production by PBMCs (see Fig. S2A in the supplemental material) and chose this concentration for the remaining experiments.
We next evaluated whether DNA is an important component for the immunostimulatory activity of ICs. To address this question, we used E6446, a compound that binds DNA and RNA in the lysosomal compartment and blocks activation of TLR7 as well as TLR9 (10, 31) and potentially other DNA/RNA sensors. PBMCs stimulated with lipopolysaccharide (LPS) and E5564, a TLR4 antagonist (32), were used as controls. Pretreatment of PBMCs with the DNA/RNA inactivator E6446 (2 µM) for 3 h followed by stimulation with ICs for 24 h resulted in 40 to 50% inhibition of IL-1β but no inhibition of TNF-α or IL-10. At a higher concentration, E6446 (20 µM) inhibited 70% of TNF-α and 90% of IL-1β as well as IL-10 production (see Fig. S2B in the supplemental material). Control experiments using the TLR4 antagonist E5564 (2 µM) resulted in ~100% inhibition of LPS-induced cytokines but had no effect on IC-induced cytokine production by PBMCs. Collectively, these data suggest that parasite DNA fragments formed during malaria are main components of ICs and are involved in induction of cytokines by human PBMCs.
We previously demonstrated that PBMCs from malaria patients express various gamma interferon (IFN-γ)-inducible genes and become hyperresponsive to TLR agonists (30). Relative to unprimed cells, priming with IFN-γ enhanced IL-1β and TNF-α and decreased IL-10 production by PBMCs from healthy donors stimulated with malaria-induced ICs (Fig. 5A). We also evaluated the main cell source of cytokines in PBMCs stimulated with ICs. For these experiments, we used total PBMCs, PBMCs depleted of monocytes, and a highly enriched monocyte population (CD14+ cells). Our results show that >95% of the cells in the purified population are CD14+ CD16− (Fig. 5B, bottom left panel). A minor population of monocytes expressed low levels of CD14 and high levels of CD16. Monocytes were the main source of TNF-α, IL-1β, and IL-10 (Fig. 5B). Importantly, IFN-γ priming upregulated expression of CD64 and downregulated expression of CD32, while expression of CD16, CD11b, and CD35 was unchanged. As CD32 is a deactivating FcγR, these results further suggest that IFN-γ priming shifts the balance of FcγR expression toward a proinflammatory one (Fig. 5C; also see Fig. S3 in the supplemental material).
FIG 5 .

Monocytes are the main source of cytokines in PBMCs stimulated with ICs derived from malaria patients. (A) PBMCs isolated from healthy donors were either unprimed or primed with 100 ng IFN-γ and stimulated with 60 µg/ml of ICs from either P. vivax or P. falciparum malaria patients, and cytokine levels were measured 24 h later. (B) CD14+ monocytes were positively selected from PBMCs of three healthy donors. Total purified monocytes, monocyte-depleted PBMCs, and total PBMCs were either unprimed or primed with IFN-γ (100 ng/ml) and stimulated with 60 µg/ml of ICs from malaria patients. TNF-α, IL-1β, and IL-10 were measured by CBA 24 h after stimulation. Results are representative of one out of three experiments. (C) PBMCs were either unprimed or primed with 100 ng of IFN-γ overnight and stimulated with ICs for 24 h. FACS analysis was performed with gating on CD14+ cells and evaluating the levels of expression (MFI) of CD16, CD32, CD64, CD11b, or CD35. Results shown are representative of one out of three experiments. P values shown in panel A were determined by a paired t test.
ICs induce high levels of proinflammatory cytokines by CD14+ CD16+ CD32low monocytes from P. vivax malaria patients.
We next looked at the responsiveness of PBMCs from P. vivax malaria patients to ICs. As previously reported (30, 33) for various TLR agonists, we found that PBMCs from patients undergoing acute malaria episodes were highly responsive and produced high levels of IL-1β and TNF-α, but not IL-10, upon stimulation with ICs (Fig. 6A). As shown in Fig. 6B, and in Fig. S4 in the supplemental material (and previously reported), the frequency of CD14+ CD16 (FcγRIIIA)+ cells is increased in patients with acute P. vivax infection and drops to levels seen in healthy individuals 30 to 40 days posttreatment (34, 35). Furthermore, we defined CD14+ CD16+ cells as the primary source of cytokines among different monocytes from malaria patients (Fig. 6C). Hence, after treatment the profile of cytokines produced by PBMCs shifted back to an anti-inflammatory one, producing high IL-10 levels and low IL-1β and TNF-α levels (Fig. 6A), which coincided with the lower frequency of CD14+ CD16+ cells in convalescent patients. Importantly, while expression of the deactivating FcγRIIB (CD32) was unchanged, CD64 mean fluorescence intensity (MFI) was increased in CD14+ CD16− cells as well as CD14+ CD16+ cells from malaria patients (Fig. 6D). Thus, the ratios of CD16/CD32 and CD64/CD32 expression by monocytes were changed to proinflammatory ones during malaria. We also looked at expression of receptors that may interact with complement-coated ICs, i.e., CD35 (CR1) and CD11b (a component of CR3), and found no difference in expression levels when comparing patients before and after treatment and parasitological cure (see Fig. S5 in the supplemental material).
FIG 6 .

Purified ICs induce high levels of proinflammatory cytokines by CD14+ CD16+ monocytes from malaria patients. (A) PBMCs from P. vivax malaria patients (n = 6) before and 30 to 45 days after treatment were isolated and stimulated with 60 µg/ml of ICs for 24 h. The levels of TNF-α, IL-1β, and IL-10 were measured in supernatants by CBA. The P values were determined by a paired t test. (B) Flow cytometric analysis shows an increased frequency of CD14+ CD16+ cells in PBMCs from two P. vivax malaria patients. The frequency of CD14+ CD16+ cells decreased to levels similar to those for healthy donors at 30 to 40 days after treatment and parasitological cure. Results of three additional patients and healthy donors are shown in Fig. S4A in the supplemental material. (C) PBMCs from two different P. vivax malaria patients were isolated and stimulated with 60 µg/ml of ICs from three different patients for 10 h in culture containing brefeldin A and submitted to flow cytometric analysis to measure the expression of intracellular TNF-α and IL-1β in CD14+ CD16− as well as CD14+ CD16+ monocytes. The results are representative of two out of five patients. (D) Top panels show the gating strategy to identify the monocyte subsets: CD14+ CD16−, CD14+ CD16+, and CD14lo CD16+. The middle two panels are representative histograms of CD64 and CD32 expression in P. vivax-infected patients and healthy donors (HD). Bottom panels show the mean fluorescence intensity (MFI) ratios of CD16/CD32 (left) and CD64/CD32 (right) of the three monocyte subsets from P. vivax-infected patients (n = 6) and in healthy donors (HD, n = 4). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Inflammasome specks and caspase-1 activation in monocytes stimulated with ICs from malaria patients.
The results presented in Fig. 5B and 6C indicated that monocytes are the main source of IL-1β in PBMCs. Hence, we investigated whether monocyte stimulation with ICs leads to activation of caspase-1. Highly purified monocytes were obtained from PBMCs of healthy donors and stimulated with ICs. Approximately 4% and 8% of stimulated monocytes expressed either the NOD-like receptor protein 3 (NLRP3) or absent in melanoma 2 (AIM2) inflammasome specks, respectively, while NLRC4 (NLR family, CARD domain-containing 4) specks were not detected (Fig. 7A). In addition, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) colocalized with either NLRP3 or AIM2 inflammasome specks. Our results also show that pro-caspase-1 was autocleaved into mature caspase-1 (p10 subunit) after monocyte stimulation with ICs (Fig. 7B). Importantly, we show that monocytes purified from PBMCs from patients undergoing acute episodes of P. vivax malaria express NLRP3+ ASC+ or AIM2+ ASC+ specks (Fig. 7C) as well as active caspase-1 (34). These findings suggest that ICs stimulate inflammasome assembly, caspase-1 activation, and IL-1β secretion in vivo during acute malaria infection.
FIG 7 .

Inflammasome specks containing NLRP3 and AIM2 after stimulation of monocytes with ICs. (A) Confocal analysis detected ASC (green), NLRP3 (red), and AIM2 (red) specks in CD14+ monocytes stimulated with 60 µg of ICs for 24 h. We also used antibodies to detect NLRC4 inflammasomes, but the results were negative. Reaction with secondary antibodies in the absence of primary antibody yielded negative results on confocal analysis. The bar graphs show the frequencies of specks in monocytes derived from healthy donors 24 h after stimulation with 60 µg of ICs. (B) Western blot assay showing pro-caspase-1 and active caspase-1 (p10) levels in enriched monocytes from healthy donors stimulated with 60 µg/ml of ICs for 24 h. (C) Double-positive NLRP3/ASC and AIM2/ASC inflammasome specks are found in circulating monocytes derived from P. vivax malaria patients but not from healthy donors.
DISCUSSION
Malaria is a devastating disease, infecting ~200 million people and killing close to 1 million children every year (36, 37). Paroxysm, a main pathophysiological response to Plasmodium infection, is characterized by cycles of sharp peaks of high fever accompanied by chills and rigors, which coincide with synchronized release of parasites from red blood cells (RBCs). While fever may aid in host defense, delaying the growth of pathogens, it is also associated with various pathological processes, such as respiratory distress, anemia, and neurological manifestations that cause morbidity and mortality in malaria. These clinical manifestations are associated with a systemic production of pyrogenic cytokines, such as TNF-α and IL-1β. However, many of the basic details of malaria-induced cytokinemia are not fully understood (38, 39). Here, we provide evidence that ICs carrying Plasmodium DNA are potent stimulators of cytokine synthesis by human monocytes through activation of innate immune receptors and may contribute to the pathogenesis of malaria.
Plasmodium-derived DNA and RNA stimulate nucleic-acid-sensing TLRs (NAS-TLRs) and AIM2 inflammasomes, as well as cytosolic sensors that activate the TBK1/STING/IRF3 pathway (1–3, 40, 41). As a consequence, they elicit both the NF-κΒ and type I IFN pathways. However, how parasite nucleic acids become accessible to these intracellular innate immune receptors is not completely understood. Recent studies from our group show that hemozoin, a detoxification polymer of heme released from hemoglobin digested by Plasmodium merozoites, may have an important role in this process. We have shown that phagocytosis of hemozoin, which is normally bound to Plasmodium DNA, makes parasite nucleic acids readily available to endosomal TLRs (1, 3). Furthermore, hemozoin crystals destabilize the phagolysosome membranes, releasing their contents, including parasite nucleic acids, into the host cell cytosol, culminating in activation of inflammasomes and other cytosolic sensors for DNA and RNA (3). Internalization and digestion of infected red blood cells (RBCs) also release hemozoin and parasite nucleic acids into phagolysosomes and subsequently into the cytosol, resulting in activation of innate immune receptors. Therefore, opsonization by IgGs efficiently mediates phagocytosis of infected RBCs as well as parasite components and may contribute to innate immune activation during malaria. The data presented here indicate that, as previously described for SLE (4, 6, 18, 19), DNA-containing ICs derived from malaria patients trigger monocytes to express an NF-κB transcriptional signature and produce high levels of proinflammatory cytokines. We hypothesize that ICs derived from malaria patients bind to Fc receptors and are internalized, releasing parasitic DNAs into phagolysosomes and subsequently into the monocyte cytosol, where they activate NAS-TLRs and AIM2, respectively.
An important finding of this study is that malaria ICs induced the formation of NLRP3+ ASC+- as well as AIM2+ ASC+-containing inflammasomes in primary human monocytes. Induction of inflammasome assembly and caspase-1 activation requires activation of the NF-κB pathway, for instance, by TLR agonists or cytokines, and consequent expression of inflammasome components, e.g., different members of the NLR family, pro-caspase-1, and pro-IL-1β (42). We believe that ICs derived from the plasma of malaria patients carry Plasmodium DNA and activate intracellular NAS-TLRs, most likely TLR8 and TLR9, which may serve as the first signal for inflammasome activation. IC-induced instability of phagolysosomes and release of their contents into the cytosol may trigger the formation of NLRP3 inflammasomes. In addition, AIM2, a DNA sensor, is activated by Plasmodium DNA in mouse cells and may also provide the second signal for assembly of inflammasome platforms during malaria (3). Hence, the results presented here indicate that circulating ICs from malaria patients trigger the assembly of NLRP3+/ASC+ and AIM2+/ASC+ in human monocytes.
Monocytes are a heterogeneous population, which reflects different developmental stages or activation status with distinct physiological roles, such as migration to lesions or entry to normal tissues, production of proinflammatory or anti-inflammatory cytokines, and antimicrobial effector functions (43). These monocyte subsets can be distinguished based on expression of CD14 and different members of the FcγR family that may up- or downregulate cell responsiveness to ICs. For example, activation of FcγRIIIA (CD16a) or FcγRI (CD64) promotes a proinflammatory response, whereas FcγRIIB (CD32) triggers an anti-inflammatory response (23, 44). Interestingly, the frequency of a single nucleotide polymorphism in the FcγRIIB (CD32) gene that abrogates the receptor function is augmented in populations from areas of endemicity, suggesting a survival advantage against malaria. This same polymorphism is associated with susceptibility to SLE, further suggesting that CD32 is a negative regulator of the proinflammatory response and protects against this autoimmune disease. Thus, the simultaneous signaling of activating versus inhibitory Fcγ receptors sets the threshold for cellular activation and prevents an excessive inflammatory response. Another important finding of our study is that upon IC stimulation, CD14+ CD16 (FcγRIIIA)+ CD32 (FcγRIIB)low monocytes are the main source of proinflammatory cytokines in PBMCs from malaria patients. Hence, the enhanced expression of FcγRIIIA and/or FcγRI associated with decreased expression of FcγRIIB+ in monocytes from malaria patients seems to skew the balance of cytokine production toward a proinflammatory response. Further research is needed to determine whether activation of FcγRIIIA or FcγRI by ICs directly induces cytokine production by monocytes or simply licenses these cells to produce large amounts of proinflammatory cytokines upon activation of intracellular DNA/RNA sensors.
Furthermore, the cytokine milieu is also an important determinant of the capacity of ICs to stimulate either a pro- or anti-inflammatory response (23, 44). This dichotomy is regulated by cytokines, such as IFN-γ and TNF-α, which favor cellular activation, or IL-10, which modulates activation of professional phagocytes. For instance, depending on the clinical form of the disease, Leishmania infection induces a pronounced proinflammatory response (45). In contrast, induction of IL-10 by ICs promotes an anti-inflammatory response leading to establishment of Leishmania parasitism and disease (46). We found that in clinically ill patients undergoing acute malaria episodes, ICs trigger monocytes to produce high levels of TNF-α and IL-1β and low levels of IL-10. This pattern of cytokine production changes to an anti-inflammatory one after treatment and parasitological cure of malaria patients. Likewise, monocytes from healthy donors stimulated with ICs produce high IL-10 levels and low TNF-α and IL-1β levels, whereas after IFN-γ priming the production of the proinflammatory cytokines becomes dominant. These findings are likely to be relevant to the cytokine storm during malaria; a hallmark of this inflammatory response is IFN-γ-induced priming of innate immune cells (30, 34).
It is noteworthy that our studies were conducted in Brazil, where malaria is an occupational disease and affects mainly adults. In addition, both P. vivax and P. falciparum forms of malaria in Brazil are not as severe as those in Africa, where mostly children <5 years of age develop the disease. Hence, significant differences are likely to be found among populations from different geographic regions and different age groups as well as in pregnant women. It is notable that different studies described TLR9 polymorphisms associated with severity of disease in both P. vivax and P. falciparum malaria patients from Brazil and Africa, respectively (47–49). Furthermore, higher levels of circulating ICs were found in African children with severe malaria than in adults and children with moderate clinical symptoms (50). Moreover, studies have shown that symptomatic adults in Brazil as well as African children infected with P. falciparum are hyperresponsive to TLR agonists, including oligonucleotides containing unmethylated CpG motifs (30, 51). Hence, as reported here for adult malaria patients, children infected with P. falciparum are likely to be more responsive to IC stimulation. In contrast, immune individuals in Africa, who are continuously exposed to P. falciparum, are more tolerant to disease and less responsive to a TLR9 agonist and may be less responsive to malaria-induced ICs (52, 53).
In conclusion, many of the signs and symptoms of malaria are a result of the excessive activation of innate immune cells. This study provides evidence that ICs are important players in this process. ICs from malaria patients appear to carry parasite DNAs to the inner compartment of host cells, making them accessible to the intracellular innate immune sensors, including NAS-TLRs and AIM2 inflammasomes. Furthermore, our results suggest that the ratio of FcγRI and FcγRIIIA (stimulatory) expression to FcγRIIB (inhibitory) expression influences the magnitude of cytokine response by IC-activated monocytes. Hence, the IgG response during the early stage of malaria has an important role in the activation of the innate immune response and the pathogenesis of malaria.
MATERIALS AND METHODS
Ethics statement.
The protocols and consent forms were approved by the Institutional Review Board of the University of Massachusetts Medical School (IRB-UMMS H-10268) and the Ethical Committees on Human Experimentation from Centro de Pesquisa em Medicina Tropical (CEP-CEPEM 095/2009) and Centro de Pesquisas René Rachou-Fundação Oswaldo Cruz (CEP-CPqRR 2004), as well as by the National Ethical Committee (CONEP 15652) from the Ministry of Health, Brazil.
Patients.
Individuals were between 18 and 60 years old. Malaria patients infected with either P. vivax (see Table S2 in the supplemental material) or P. falciparum (see Table S3) were diagnosed by a thick blood smear and confirmed by PCR. The clinical manifestations of malaria were fever, myalgia, chills, nausea, vomiting, and/or diarrhea, but no patient had complicated malaria requiring hospitalization. Patients infected with P. falciparum received a fixed dose of the artemether (20-mg) and lumefantrine (120-mg) combination two times a day (four tablets each dose) for 3 days followed by a single dose of primaquine (45 mg) on the last day of treatment. Patients infected with P. vivax were treated with chloroquine (150 mg) every 8 h for 3 days and a single dose per day of primaquine (15 mg) for 2 weeks. The treatment schedule, dose, and drugs described above have been tested and are recommended by the Brazilian Ministry of Health. Healthy donors as well as patients before and after chemotherapy had 50 to 100 ml of total blood collected using EDTA anticoagulant tubes. Parasitemic cure was documented by PCR. Exclusionary criteria included any patient who had a malaria episode in the last 6 months, severe anemia (defined as a hematocrit of <35), severe disease (requiring admission to the hospital), any comorbidity, recent or concurrent treatment with anti-inflammatory or immunosuppressive drugs, or pregnancy.
IC quantitation.
The MicroVue CIC-C1q enzyme immunoassay (Quidel, San Diego, CA) was used to quantify circulating ICs in human serum according to the manufacturer’s instructions. Serum samples were diluted 1:50 and tested in duplicate. Results were obtained in reference to the standard curve and were expressed as microgram-equivalents per milliliter.
Anti-dsDNA and anti-ssDNA antibody ELISA.
Anti-dsDNA antibody levels in sera of patients and healthy controls were determined as previously described (54). Briefly, Costar 96-well half-area plates (Corning Inc., New York, NY) were dry coated overnight at 37°C with 25 µg/ml of sonicated calf thymus DNA (Sigma-Aldrich, St. Louis, MO) that had been filtered using a 0.45-µm Millex HA filter (Millipore, Germany) to remove ssDNA. Plates were blocked and then incubated with serum samples (1:50 and 1:100 dilutions, 25 µl/well). IgG+ or IgM+ anti-dsDNA antibodies were detected using alkaline phosphatase (AP)-labeled anti-human IgG or anti-human IgM antibody (Southern Biotechnology, Birmingham, AL) and developed with 50 µl/well AP substrate (Sigma-Aldrich). The optical density at 405 nm (OD405) was measured by using a Victor microplate reader (PerkinElmer, Waltham, MA). The enzyme-linked immunosorbent assay (ELISA) to determine the levels of anti-ssDNA antibodies was performed exactly as described above, except that the plates were coated with 25 µg/ml ssDNA (Sigma-Aldrich).
IC purification.
ICs from plasma were purified using the Affi-Gel protein A MAPS II kit (Bio-Rad, Hercules, CA). Plasma (1 ml) was thawed, homogenized, and diluted 1:1 using a filtered binding buffer at pH 9.0 (Bio-Rad). An Econo-Column chromatography column (1 by 10 cm) was packed with 1 ml of Affi-Gel protein A agarose and equilibrated with 5 bed volumes of binding buffer. The diluted sample was applied, and the column was washed with 15 bed volumes of binding buffer. IgG was eluted with 5 bed volumes of an elution buffer (pH 3.0), neutralized immediately after elution by the addition of 1 M Tris-HCl (pH 9.0), and quantified in a NanoDrop spectrophotometer.
Quantitative PCR.
DNA samples were extracted from plasma or ICs using the QIAamp circulating nucleic acid kit (Qiagen, Germantown, MD) according to the manufacturer’s instructions. Reverse transcription-quantitative PCRs (qRT-PCRs) were performed in a final volume of 20 µl, containing ~1 µl DNA, 0.15 µl primers (10 µM initial concentration; 0.075 µM final concentration), 10 µl SYBR green PCR core reagents (Applied Biosystems), and 8.7 µl sterile water. Primer sequences for the human CYCLIN D1 (CCND) gene were GCTCCTGGTGAACAAGCTCAA (F) and TTGGAGAGGAAGTGTTCAATGAAA (R), and those for the P. vivax 18S from rRNA (SSUrRNA) gene were ACGATCAGATACCGTCGTCGTAAT (F) and CAATCTAAGAATAAACTCCGAAGAGAAA (R). A standard curve was constructed by diluting each gene (from 101 to 107). Real-time monitoring of PCR amplification was performed using a 7500 Real-Time PCR system, and results were analyzed with 7500 software, v2.0.5. The temperature profile was 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min.
PBMC and monocyte assays.
Blood from patients or healthy donors was diluted 1:1 in phosphate-buffered saline (PBS), layered onto Ficoll-Paque gradients with a density of 1.078 g/ml (GE Healthcare, United Kingdom), and centrifuged for 15 min at 800 × g. The mononuclear cells at the interface were aspirated, washed, and resuspended at 3 × 105 cells/well in complete RPMI medium containing 10% fetal calf serum (FCS). CD14+ cells were purified using the EasySep positive selection kit according to the manufacturer’s protocol (StemCell Technologies, Canada). Cells were stained with anti-CD14 (allophycocyanin [APC]) (eBioscience, San Diego, CA) and anti-CD16 (phycoerythrin [PE]) (Becton, Dickinson, Franklin Lakes, NJ), and purity was checked by fluorescence-activated cell sorting (FACS).
NanoString analysis.
After stimulation with 60 µg/ml of purified ICs, human monocytes were lysed in RLT buffer (Qiagen) supplemented with β-mercaptoethanol and used to determine mRNA abundance by NanoString technology as previously described (53). In brief, lysates were hybridized with capture and reporter probes overnight at 65°C and loaded onto the nCounter preparation station. Purified target/probe complexes were eluted and immobilized in the cartridge for data collection and quantification by the nCounter digital analyzer. For side-by-side comparisons of nCounter experiments, data were normalized for small variations using internal positive controls and seven housekeeping genes included in the CodeSet. The heat map was constructed using the Tiger Multi Experiment Viewer software, version 4.8.1.
Cytokine assays.
Levels of human TNF-α, IL-1β, IL-6, IL-8, and IL-10 were measured in PBMC culture supernatants or plasma by the cytometric bead array (CBA) human inflammatory cytokine kit (Becton, Dickinson) and ELISA for IL-1β (R&D Systems, Minneapolis, MN).
Flow cytometry.
For intracellular measurement of cytokines, PBMCs were isolated and cultured for 8 h in the presence of ICs (60 µg/ml) and brefeldin A (GolgiPlug; Becton, Dickinson). Surface markers were stained with anti-CD11b, -CD32, -CD35, -CD64, and -CD14 (eBioscience, San Diego, CA) or anti-CD16 (Becton, Dickinson), fixed and permeabilized with Cytofix (Becton, Dickinson), and incubated with phycoerythrin–anti-TNF-α or –anti-IL-1β (Becton, Dickinson). Subsequently, cells were washed and analyzed by flow cytometry on a FACScan cytometer (Becton, Dickinson).
Immunoblotting assays for caspase-1.
Radioimmunoprecipitation assay (RIPA) buffer (250 ml) plus protease inhibitor was added to a pellet containing 1 × 106 stimulated PBMCs. After 15 min on ice, lysates were centrifuged at 13,000 × g for 20 min at 4°C. Supernatants were separated on a 15% acrylamide SDS-PAGE gel and transferred onto nitrocellulose membranes. The membranes were incubated with pro-caspase-1- or caspase-1-specific antibodies and visualized with HRP-conjugated antibody and the ECL system (Amersham, Bucks, United Kingdom).
Confocal analysis.
Monocytes obtained from either healthy donors or malaria patients were analyzed ex vivo or after in vitro stimulation with ICs for 24 h. Monocytes were fixed with 4% paraformaldehyde, permeabilized using Triton X-100, and stained with anti-NLRP3 (fluorescein isothiocyanate [FITC] or Texas Red), anti-NLRC4 (Texas Red), or anti-AIM2 (Texas Red) (all from Abcam [United Kingdom]) and anti-ASC (FITC) (Santa Cruz, Dallas, TX). Images were acquired using an LSM510 microscope (Zeiss, Germany) and analyzed by ImageJ software (National Institutes of Health). Dual-color images were acquired by consecutive scanning with only one laser line active per scan to avoid cross-excitation.
Statistical analysis.
All data were analyzed using GraphPad InStat 6.0 software. Comparisons were performed using a one-way analysis of variance (ANOVA) and Student’s t test. The paired t test was used in experiments where we compared the same patients before and after treatment. Mann-Whitney U testing was used for nonparametric analysis when data did not fit a Gaussian distribution.
SUPPLEMENTAL MATERIAL
High levels of circulating cytokines in plasma from patients acutely infected with P. vivax or P. falciparum. IL-8, IL-10, and IL-6 levels in plasma of P. vivax-infected (n = 56) (A) and P. falciparum-infected (n = 14) (B) subjects were measured before (closed circles) and 30 to 45 days after (open circles) treatment. Limits of detection for IL-6, IL-8, and IL10, measured by cytometric bead array (CBA), were 2.5, 3.6, and 3.3 pg/ml, respectively. Significant differences are indicated with P values using the Mann-Whitney test. Download
E6446 blocks the immunostimulatory activity of ICs from malaria patients. (A) PBMCs isolated from healthy donors were stimulated with different concentrations of ICs, and cytokine levels were measured 24 h later in tissue culture supernatants. (B) PBMCs were treated with E6446 (2 µM or 20 µM) or E5564 (2 µM) for 3 h, before stimulation with 60 µg/ml of ICs for 24 h, and cytokine levels were measured. Bars represent percentages of cytokine inhibition in the supernatants. TNF-α, IL-1β, and IL-10 were measured by cytometric bead array (CBA). In similar experiments performed with PBMCs from three different patients, inhibition of IC-induced IL-1β, TNF-α, and IL-10 by 20 µM E6446 varied from 88 to 100%, 61 to 90%, and 88 to 100%, respectively. Download
IFN-γ induces expression of CD64 and inhibits expression of CD32 in monocytes from healthy donors. PBMCs were either unprimed or primed with 100 ng of IFN-γ overnight and stimulated with ICs for 24 h. FACS analysis was performed by gating on CD14+ cells and then evaluating the levels of expression (mean fluorescence intensity [MFI]) of CD16 (FcγRIIIA), CD32 (FcγRIIB), CD64 (FcγRIA), CD11b (CR3), and CD35 (CR1). The presented results are averages from three patients and representative of two experiments that yielded similar results. Asterisks indicate that differences are significant, as determined by one-way ANOVA: **, P < 0.01; ****, P < 0.001. Download
Expression of FcγR in monocyte subsets in whole blood from P. vivax malaria patients. (A) Dot plots showing the gating strategy for CD14+ CD16 (FcγRIIIA)+ and CD14dim CD16+ monocytes are shown. Mean fluorescence intensity (MFI) of CD16 was evaluated on CD14dim CD16+ monocytes in whole blood from P. vivax malaria patients (n = 5), before and 30 to 45 days after treatment, as well as in three healthy donors (n = 3). Asterisks indicate that differences are significant, as determined by Mann-Whitney U test: *, 0.01 > P < 0.05. (B) Increased expression of CD64 in monocytes from P. vivax malaria patients. Mean fluorescence intensity (MFI) of CD64 (FcγRI), CD32 (FcγRIIB), and CD16 (FcγRIIIA), of the three monocyte subsets in P. vivax-infected patients (n = 8 to 15) and in healthy donors (n = 4). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Download
Expression of complement receptors by monocytes from malaria patients infected with either P. vivax or P. falciparum. Mean fluorescence intensity (MFI) of CD11b and CD35 was evaluated on monocytes in whole blood from P. vivax (n = 11) and P. falciparum (n = 5) malaria patients, before and 30 to 45 days after treatment, as well as from five healthy donors. No significant differences were indicated using the paired t test. Download
Genes with augmented expression in monocytes stimulated with ICs. Asterisks indicate that differences are significant, as determined by Student’s t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Plasmodium vivax malaria patients. *, the number of malaria episodes for each patient was determined according to individual history and detailed anamnesis; **, parasitemia level was not available.
Plasmodium falciparum malaria patients. *, the number of malaria episodes for each patient was determined according to individual history and detailed anamnesis. **, parasitemia level was not available.
ACKNOWLEDGEMENTS
We are grateful to Melanie Trombly for critically reviewing the manuscript.
This work was supported by the U.S. National Institutes of Health (AI079293), the National Institute of Science and Technology for Vaccines (CNPq and FAPEMIG), and the Rede Mineira de Biomoléculas (Fapemig). R.T.G., D.T.G., M.A.A., and B.C.R. received fellowships from CNPq. I.C.H. received a fellowship from CAPES. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
I.C.H., M.A.A., B.D., S.R., D.T.G., and R.T.G. conceived and designed the experiments. J.V. provided samples. I.C.H., C.G.-M., M.A.A., H.G., W.A.A., B.C.R., R.B.D.O., and D.B.P. performed the experiments. I.C.H., B.D., S.R., J.V., D.T.G., and R.T.G. analyzed the data. I.C.H., D.T.G., and R.T.G. wrote the paper.
Footnotes
Citation Hirako IC, Gallego-Marin C, Ataide MA, Andrade WA, Gravina H, Rocha BC, de Oliveira RB, Pereira DB, Vinetz J, Diamond B, Ram S, Golenbock DT, Gazzinelli RT. 2015. DNA-containing immunocomplexes promote inflammasome assembly and release of pyrogenic cytokines by CD14+ CD16+ CD64high CD32low inflammatory monocytes from malaria patients. mBio 6(6):e01605-15. doi:10.1128/mBio.01605-15.
REFERENCES
- 1.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. 2007. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A 104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma S, DeOliveira R, Kalantari P, Parroche P, Goutagny N, Jiang Z, Chan J, Bartholomeu D, Lauw F, Hall J, Barber G, Gazzinelli R, Fitzgerald K, Golenbock D. 2011. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35:194–207. doi: 10.1016/j.immuni.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalantari P, DeOliveira R, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli R, Golenbock D, Fitzgerald K. 2014. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep 6:196–210. doi: 10.1016/j.celrep.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barbalat R, Ewald SE, Mouchess ML, Barton GM. 2011. Nucleic acid recognition by the innate immune system. Annu Rev Immunol 29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 5.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. 2006. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward J, Flavell RA, Bolland S. 2007. Control of Toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. 2007. Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford) 46:1052–1056. doi: 10.1093/rheumatology/kem112. [DOI] [PubMed] [Google Scholar]
- 8.Barrat FJ, Coffman RL. 2008. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev 223:271–283. doi: 10.1111/j.1600-065X.2008.00630.x. [DOI] [PubMed] [Google Scholar]
- 9.Coban C, Ishii KJ, Uematsu S, Arisue N, Sato S, Yamamoto M, Kawai T, Takeuchi O, Hisaeda H, Horii T, Akira S. 2007. Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol 19:67–79. doi: 10.1093/intimm/dxl123. [DOI] [PubMed] [Google Scholar]
- 10.Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, Zheng W, Ataide MA, de Oliveira RB, Golenbock DT, Gazzinelli RT. 2011. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci U S A 108:3689–3694. doi: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baccarella A, Fontana MF, Chen EC, Kim CC. 2013. Toll-like receptor 7 mediates early innate immune responses to malaria. Infect Immun 81:4431–4442. doi: 10.1128/IAI.00923-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gowda NM, Wu X, Gowda DC. 2012. TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J Immunol 188:5073–5085. doi: 10.4049/jimmunol.1102143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rönnblom L, Elkon KB. 2010. Cytokines as therapeutic targets in SLE. Nat Rev Rheumatol 6:339–347. doi: 10.1038/nrrheum.2010.64. [DOI] [PubMed] [Google Scholar]
- 14.Grau G, Fajardo L, Piguet P, Allet B, Lambert P, Vassalli P. 1987. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237:1210–1212. doi: 10.1126/science.3306918. [DOI] [PubMed] [Google Scholar]
- 15.McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwjatkowski D. 1994. Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 371:508–510. doi: 10.1038/371508a0. [DOI] [PubMed] [Google Scholar]
- 16.Stevenson MM, Tam MF, Wolf SF, Sher A. 1995. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J Immunol 155:2545–2556. [PubMed] [Google Scholar]
- 17.Katz SJ, Russell AS. 2011. Re-evaluation of antimalarials in treating rheumatic diseases: re-appreciation and insights into new mechanisms of action. Curr Opin Rheumatol 23:278–281. doi: 10.1097/BOR.0b013e32834456bf. [DOI] [PubMed] [Google Scholar]
- 18.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cros J, Cagnard N, Woollard K, Patey N, Zhang S, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, Jais J, D’Cruz D, Casanova J, Trouillet C, Geissmann F. 2010. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mibei EK, Orago AS, Stoute JA. 2005. Immune complex levels in children with severe Plasmodium falciparum malaria. Am J Trop Med Hyg 72:593–599. [PubMed] [Google Scholar]
- 21.Fernandez-Arias C, Lopez JP, Hernandez-Perez JN, Bautista-Ojeda MD, Branch O, Rodriguez A. 2013. Malaria inhibits surface expression of complement receptor 1 in monocytes/macrophages, causing decreased immune complex internalization. J Immunol 190:3363–3372. doi: 10.4049/jimmunol.1103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Oliveira RB, Wang JP, Ram S, Gazzinelli RT, Finberg RW, Golenbock DT. 2014. Increased survival in B-cell-deficient mice during experimental cerebral malaria suggests a role for circulating immune complexes. mBio 5:e00949-14. doi: 10.1128/mBio.00949-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nimmerjahn F, Ravetch JV. 2008. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 24.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. 2002. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(−/−) mice. J Exp Med 195:1167–1174. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Ravetch JV, Diamond B. 2006. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med 203:2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clatworthy MR, Willcocks L, Urban B, Langhorne J, Williams TN, Peshu N, Watkins NA, Floto RA, Smith KGC. 2007. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc Natl Acad Sci U S A 104:7169–7174. doi: 10.1073/pnas.0608889104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niederer HA, Clatworthy MR, Willcocks LC, Smith KGC. 2010. FcgammaRIIB, FcgammaRIIIB, and systemic lupus erythematosus. Ann N Y Acad Sci 1183:69–88. doi: 10.1111/j.1749-6632.2009.05132.x. [DOI] [PubMed] [Google Scholar]
- 28.Waisberg M, Tarasenko T, Vickers BK, Scott BL, Willcocks LC, Molina-Cruz A, Pierce MA, Huang CY, Torres-Velez FJ, Smith KGC, Barillas-Mury C, Miller LH, Pierce SK, Bolland S. 2011. Genetic susceptibility to systemic lupus erythematosus protects against cerebral malaria in mice. Proc Natl Acad Sci U S A 108:1122–1127. doi: 10.1073/pnas.1017996108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leoratti FM, Trevelin SC, Cunha FQ, Rocha BC, Costa PA, Gravina HD, Tada MS, Pereira DB, Golenbock DT, Antonelli LR, Gazzinelli RT. 2012. Neutrophil paralysis in Plasmodium vivax malaria. PLoS Negl Trop Dis 6:e1710. doi: 10.1371/journal.pntd.0001710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franklin BS, Parroche P, Ataide MA, Lauw F, Ropert C, de Oliveira RB, Pereira D, Tada MS, Nogueira P, da Silva LHP, Bjorkbacka H, Golenbock DT, Gazzinelli RT. 2009. Malaria primes the innate immune response due to interferon-gamma induced enhancement of Toll-like receptor expression and function. Proc Natl Acad Sci U S A 106:5789–5794. doi: 10.1073/pnas.0809742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, Genest M, Yang H, Shaffer C, Zhao Y, Shen Y, Liu C, Liu D, Mempel TR, Rowbottom C, Chow J, Twine NC, Yu M, Gusovsky F, Ishizaka ST. 2014. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol 85:429–440. doi: 10.1124/mol.113.089821. [DOI] [PubMed] [Google Scholar]
- 32.Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, Przetak M, Chow J, Gusovsky F, Christ WJ, Rossignol DP. 2003. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther 304:1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- 33.McCall MBB, Netea MG, Hermsen CC, Jansen T, Jacobs L, Golenbock D, van der Ven AJAM, Sauerwein RW. 2007. Plasmodium falciparum infection causes proinflammatory priming of human TLR responses. J Immunol 179:162–171. doi: 10.4049/jimmunol.179.1.162. [DOI] [PubMed] [Google Scholar]
- 34.Ataide MA, Andrade WA, Zamboni DS, Wang D, Souza MDC, Franklin BS, Elian S, Martins FS, Pereira D, Reed G, Fitzgerald KA, Golenbock DT, Gazzinelli RT. 2014. Malaria-induced NLRP12/NLRP3-dependent caspase-1 activation mediates inflammation and hypersensitivity to bacterial superinfection. PLoS Pathog 10:e1003885. doi: 10.1371/journal.ppat.1003885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonelli LRV, Leoratti FMS, Costa PAC, Rocha BC, Diniz SQ, Tada MS, Pereira DB, Teixeira-Carvalho A, Golenbock DT, Gonçalves R, Gazzinelli RT. 2014. The CD14+ CD16+ inflammatory monocyte subset displays increased mitochondrial activity and effector function during acute Plasmodium vivax malaria. PLoS Pathog 10:e1004393. doi: 10.1371/journal.ppat.1004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. 2012. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 37.Gething PW, Elyazar IR, Moyes CL, Smith DL, Battle KE, Guerra CA, Patil AP, Tatem AJ, Howes RE, Myers MF, George DB, Horby P, Wertheim HF, Price RN, Mueller I, Baird JK, Hay SI. 2012. A long neglected world malaria map: Plasmodium vivax endemicity in 2010. PLoS Negl Trop Dis 6:e1814. doi: 10.1371/journal.pntd.0001814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anstey NM, Douglas NM, Poespoprodjo JR, Price RN. 2012. Plasmodium vivax: clinical spectrum, risk factors and pathogenesis. Adv Parasitol 80:151–201. doi: 10.1016/B978-0-12-397900-1.00003-7. [DOI] [PubMed] [Google Scholar]
- 39.Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. 2014. Innate sensing of malaria parasites. Nat Rev Immunol 14:744–757. doi: 10.1038/nri3742. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Tian L, Yu X, Pattaradilokrat S, Li J, Wang M, Yu W, Qi Y, Zeituni AE, Nair SC, Crampton SP, Orandle MS, Bolland SM, Qi CF, Long CA, Myers TG, Coligan JE, Wang R, Su XZ. 2014. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc Natl Acad Sci U S A 111:E511–E520. doi: 10.1073/pnas.1316467111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liehl P, Zuzarte-Luís V, Chan J, Zillinger T, Baptista F, Carapau D, Konert M, Hanson KK, Carret C, Lassnig C, Müller M, Kalinke U, Saeed M, Chora AF, Golenbock DT, Strobl B, Prudêncio M, Coelho LP, Kappe SH, Superti-Furga G, Pichlmair A, Vigario AM, Rice CM, Fitzgerald KA, Barchet W, Mota MM. 2014. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med 20:47–53. doi: 10.1038/nm.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Gordon S, Taylor PR. 2005. Monocyte and macrophage heterogeneity. Nat Rev Immunol 5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 44.Smith KGC, Clatworthy MR. 2010. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol 10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elshafie AI, Ahlin E, Mathsson L, ElGhazali G, Ronnelid J. 2007. Circulating immune complexes (IC) and IC-induced levels of GM-CSF are increased in Sudanese patients with acute visceral Leishmania donovani infection undergoing sodium stibogluconate treatment: implications for disease pathogenesis. J Immunol 178:5383–5389. doi: 10.4049/jimmunol.178.8.5383. [DOI] [PubMed] [Google Scholar]
- 46.Halstead SB, Mahalingam S, Marovich MA, Ubol S, Mosser DM. 2010. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis 10:712–722. doi: 10.1016/S1473-3099(10)70166-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leoratti F, Farias L, Alves F, Suarez-Mútis M, Coura J, Kalil J, Camargo E, Moraes S, Ramasawmy R. 2008. Variants in the toll-like receptor signaling pathway and clinical outcomes of malaria. J Infect Dis 198:772–780. doi: 10.1086/590440. [DOI] [PubMed] [Google Scholar]
- 48.Sam-Agudu NA, Greene JA, Opoka RO, Kazura JW, Boivin MJ, Zimmerman PA, Riedesel MA, Bergemann TL, Schimmenti LA, John CC. 2010. TLR9 polymorphisms are associated with altered IFNg levels in children with cerebral malaria. Am J Trop Med Hyg 82:548–555. doi: 10.4269/ajtmh.2010.09-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mockenhaupt F, Hamann L, von Gaertner C, Bedu-Addo G, Kleinsorge C, Schumann R, Bienzle U. 2006. Common polymorphisms of Toll-like receptors 4 and 9 are associated with the clinical manifestation of malaria during pregnancy. J Infect Dis 194:184–188. doi: 10.1086/505152. [DOI] [PubMed] [Google Scholar]
- 50.Thomas B, Diallo D, Noumsi G, Moulds JM. 2012. Circulating immune complex levels are associated with disease severity and seasonality in children with malaria from Mali. Biomark Insights 7:81–86. doi: 10.4137/BMI.S9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartgers FC, Obeng BB, Voskamp A, Larbi IA, Amoah AS, Luty AJF, Boakye D, Yazdanbakhsh M. 2008. Enhanced Toll-like receptor responsiveness associated with mitogen-activated protein kinase activation in Plasmodium falciparum-infected children. Infect Immun 76:5149–5157. doi: 10.1128/IAI.01579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crompton PD, Mircetic M, Weiss G, Baughman A, Huang CY, Topham DJ, Treanor JJ, Sanz I, Lee FE, Durbin AP, Miura K, Narum DL, Ellis RD, Malkin E, Mullen GED, Miller LH, Martin LB, Pierce SK. 2009. The TLR9 ligand CpG promotes the acquisition of Plasmodium falciparum-specific memory B cells in malaria-naive individuals. J Immunol 182:3318–3326. doi: 10.4049/jimmunol.0803596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Traore B, Koné Y, Doumbo S, Doumtabé D, Traoré A, Crompton PD, Mircetic M, Huang C, Kayentao K, Dicko A, Sagara I, Ellis RD, Miura K, Guindo A, Miller LH, Doumbo OK, Pierce SK. 2009. The TLR9 agonist CpG fails to enhance the acquisition of Plasmodium falciparum-specific memory B cells in semi-immune adults in Mali. Vaccine 27:7299–7303. doi: 10.1016/j.vaccine.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ray SK, Putterman C, Diamond B. 1996. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: a paradigm for autoimmune disease. Proc Natl Acad Sci U S A 93:2019–2024. doi: 10.1073/pnas.93.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. 2008. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
High levels of circulating cytokines in plasma from patients acutely infected with P. vivax or P. falciparum. IL-8, IL-10, and IL-6 levels in plasma of P. vivax-infected (n = 56) (A) and P. falciparum-infected (n = 14) (B) subjects were measured before (closed circles) and 30 to 45 days after (open circles) treatment. Limits of detection for IL-6, IL-8, and IL10, measured by cytometric bead array (CBA), were 2.5, 3.6, and 3.3 pg/ml, respectively. Significant differences are indicated with P values using the Mann-Whitney test. Download
E6446 blocks the immunostimulatory activity of ICs from malaria patients. (A) PBMCs isolated from healthy donors were stimulated with different concentrations of ICs, and cytokine levels were measured 24 h later in tissue culture supernatants. (B) PBMCs were treated with E6446 (2 µM or 20 µM) or E5564 (2 µM) for 3 h, before stimulation with 60 µg/ml of ICs for 24 h, and cytokine levels were measured. Bars represent percentages of cytokine inhibition in the supernatants. TNF-α, IL-1β, and IL-10 were measured by cytometric bead array (CBA). In similar experiments performed with PBMCs from three different patients, inhibition of IC-induced IL-1β, TNF-α, and IL-10 by 20 µM E6446 varied from 88 to 100%, 61 to 90%, and 88 to 100%, respectively. Download
IFN-γ induces expression of CD64 and inhibits expression of CD32 in monocytes from healthy donors. PBMCs were either unprimed or primed with 100 ng of IFN-γ overnight and stimulated with ICs for 24 h. FACS analysis was performed by gating on CD14+ cells and then evaluating the levels of expression (mean fluorescence intensity [MFI]) of CD16 (FcγRIIIA), CD32 (FcγRIIB), CD64 (FcγRIA), CD11b (CR3), and CD35 (CR1). The presented results are averages from three patients and representative of two experiments that yielded similar results. Asterisks indicate that differences are significant, as determined by one-way ANOVA: **, P < 0.01; ****, P < 0.001. Download
Expression of FcγR in monocyte subsets in whole blood from P. vivax malaria patients. (A) Dot plots showing the gating strategy for CD14+ CD16 (FcγRIIIA)+ and CD14dim CD16+ monocytes are shown. Mean fluorescence intensity (MFI) of CD16 was evaluated on CD14dim CD16+ monocytes in whole blood from P. vivax malaria patients (n = 5), before and 30 to 45 days after treatment, as well as in three healthy donors (n = 3). Asterisks indicate that differences are significant, as determined by Mann-Whitney U test: *, 0.01 > P < 0.05. (B) Increased expression of CD64 in monocytes from P. vivax malaria patients. Mean fluorescence intensity (MFI) of CD64 (FcγRI), CD32 (FcγRIIB), and CD16 (FcγRIIIA), of the three monocyte subsets in P. vivax-infected patients (n = 8 to 15) and in healthy donors (n = 4). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Download
Expression of complement receptors by monocytes from malaria patients infected with either P. vivax or P. falciparum. Mean fluorescence intensity (MFI) of CD11b and CD35 was evaluated on monocytes in whole blood from P. vivax (n = 11) and P. falciparum (n = 5) malaria patients, before and 30 to 45 days after treatment, as well as from five healthy donors. No significant differences were indicated using the paired t test. Download
Genes with augmented expression in monocytes stimulated with ICs. Asterisks indicate that differences are significant, as determined by Student’s t test: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Plasmodium vivax malaria patients. *, the number of malaria episodes for each patient was determined according to individual history and detailed anamnesis; **, parasitemia level was not available.
Plasmodium falciparum malaria patients. *, the number of malaria episodes for each patient was determined according to individual history and detailed anamnesis. **, parasitemia level was not available.