

ABSTRACT
Linear ubiquitination is an atypical posttranslational modification catalyzed by the linear-ubiquitin-chain assembly complex (LUBAC), containing HOIP, HOIL-1L, and Sharpin. LUBAC facilitates NF-κB activation and inflammation upon receptor stimulation by ligating linear ubiquitin chains to critical signaling molecules. Indeed, linear-ubiquitination-dependent signaling is essential to prevent pyogenic bacterial infections that can lead to death. While linear ubiquitination is essential for intracellular receptor signaling upon microbial infection, this response must be measured and stopped to avoid tissue damage and autoimmunity. While LUBAC is activated upon bacterial stimulation, the mechanisms regulating LUBAC activity in response to bacterial stimuli have remained elusive. We demonstrate that LUBAC activity itself is downregulated through ubiquitination, specifically, ubiquitination of the catalytic subunit HOIP at the carboxyl-terminal lysine 1056. Ubiquitination of Lys1056 dynamically altered HOIP conformation, resulting in the suppression of its catalytic activity. Consequently, HOIP Lys1056-to-Arg mutation led not only to persistent LUBAC activity but also to prolonged NF-κB activation induced by bacterial lipopolysaccharide-mediated Toll-like receptor 4 (TLR4) stimulation, whereas it showed no effect on NF-κB activation induced by CD40 stimulation. This study describes a novel posttranslational regulation of LUBAC-mediated linear ubiquitination that is critical for specifically directing TLR4-mediated NF-κB activation.
IMPORTANCE
Posttranslational modification of proteins enables cells to respond quickly to infections and immune stimuli in a tightly controlled manner. Specifically, covalent modification of proteins with the small protein ubiquitin is essential for cells to initiate and terminate immune signaling in response to bacterial and viral infection. This process is controlled by ubiquitin ligase enzymes, which themselves must be regulated to prevent persistent and deleterious immune signaling. However, how this regulation is achieved is poorly understood. This paper reports a novel ubiquitination event of the atypical ubiquitin ligase HOIP that is required to terminate bacterial lipopolysaccharide (LPS)-induced TLR4 immune signaling. Ubiquitination causes the HOIP ligase to undergo a conformational change, which blocks its enzymatic activity and ultimately terminates LPS-induced TLR4 signaling. These findings provide a new mechanism for controlling HOIP ligase activity that is vital to properly regulate a proinflammatory immune response.
INTRODUCTION
Mutations in genes required for regulating antimicrobial or inflammatory signaling frequently lead to auto-inflammatory diseases and impair the immune system’s ability to fight infection (1, 2). In particular, mounting evidence indicates that proper functioning of the immune signaling regulatory complex called the linear-ubiquitin-chain assembly complex (LUBAC), consisting of heme-oxidized IRP2 ubiquitin ligase-1 (HOIL-1L), HOIL-1L interacting protein (HOIP), and SHANK-associated RH domain interactor (Sharpin), is required for maintenance of a functional immune system (3, 4). In humans and mice, mutations in LUBAC result in deregulation of immune signaling. For example, human patients with null mutations in the immunoregulatory RBCK1 gene, which codes for the protein HOIL-1L, exhibit a combined autoinflammatory/immunodeficient phenotype characterized by elevated serum levels of the proinflammatory cytokine interleukin 6 (IL-6) and chronic invasive bacterial infections that ultimately result in death (1). In accordance with immunodeficiency seen in humans, HOIL-1L-deficient mice are resistant to lipopolysaccharide (LPS)-induced lethal inflammation, suggesting that HOIL-1L is required to mount a systemic innate immune inflammatory response to bacterial infection (5). In mice, null mutations in SHARPIN cause chronic proliferative dermatitis as well as eosinophilic inflammation and impaired lymphoidogenesis (6). Genetic deletion of HOIP is embryonic lethal; however, the B-cell specific ablation of HOIP results in defective antibody responses to thymus-dependent and independent antigens in mice (7). Additionally, mutations in RNF31, the gene encoding HOIP, alter LUBAC activity and are enriched in certain B-cell lymphomas in humans (8). Collectively, genetic studies have demonstrated that LUBAC is required for numerous antimicrobial and inflammatory pathways, including NLRP3, IL-1 receptor (IL-1R), Toll-like receptor 4 (TLR4), TLR2, tumor necrosis factor (TNF), NOD2, and CD40 (5, 7, 9–12). Thus, the tight regulation of LUBAC activity is necessary to maintain a healthy immune response. Aberrant LUBAC activity contributes to the development of diseases resulting from autoinflammation and immunodeficiency.
LUBAC function is critical for NF-κB activation, during which it attaches linear ubiquitin to multiple molecules. The first-characterized function of LUBAC was linear ubiquitination of the NF-κB essential modulator (NEMO), which is required for NF-κB activation (11). During TNF-α-mediated NF-κB activation, RIP1 is modified with linear ubiquitin in the TNF receptor-signalizing complex (6). In contrast to direct substrate modification, during IL-1R- and TLR1/2-mediated NF-κB activation, LUBAC conjugates linear ubiquitin to existing lysine 63-linked ubiquitin chains on IRAK1, IRAK4, and MyD88 (13). In addition to NF-κB activation, LUBAC mediates induction of other host immune signaling pathways, including inflammasome activation, by attaching linear ubiquitin chains to key signaling molecules. During NLRP3 activation, LUBAC modifies the adaptor protein ASC, promoting ASC/NLRP3 binding and oligomerization (5). This directly leads to secretion of inflammatory cytokines. Thus, LUBAC facilitates signaling by modifying substrates, both directly and indirectly, to acutely modulate innate immunity.
LUBAC is a trimeric complex composed of Sharpin, HOIL-1L, and HOIP, where HOIP is the primary E3 ubiquitin ligase and the only enzyme known to catalyze linear-ubiquitin-chain conjugation. HOIP is a member of the RING-between-RING (RBR) family of E3 ubiquitin ligase. Interestingly, RBR ligases often exist in autoinhibited conformations, requiring protein-protein interactions or posttranslational modifications for conformational change and activation (14). For instance, posttranslational modifications of the well-studied RBR ligase Parkin can both positively and negatively regulate its activity by altering protein conformation (15, 16). In fact, mutations that enhance HOIP and HOIL-1L binding, and thus increase LUBAC enzymatic activity, are associated with human disease (8), suggesting that the strict negative regulation of linear-ubiquitin-chain formation is required for the maintenance of immune homeostasis.
Upon stimulation of immune receptors, including TNF-α receptors, TLR4, and CD40 receptors, LUBAC enzymatic activity is rapidly activated and subsequently inactivated through previously uncharacterized mechanisms. We report that the enzymatically critical LUBAC subunit HOIP is modified by ubiquitin at multiple lysine residues. Targeted mutations to block HOIP ubiquitination revealed that a lysine 1056-to-arginine (K1056R) mutation within the carboxyl-terminal linear-ubiquitin-chain-determining domain (LDD) resulted in elevated HOIP activity. TLR4 stimulation induced HOIP ubiquitination, which altered HOIP conformation and decreased LUBAC activity. Furthermore, blocking HOIP ubiquitination with the K1056R mutation resulted in elevated and persistent expression of NF-κB gene expression. Our results suggest that the carboxyl-terminal ubiquitination of HOIP inactivates LUBAC activity upon TLR4 stimulation to prevent persistent NF-κB activation, indicating conformational rearrangement as a primary regulatory mechanism for RBR ligases.
RESULTS
Identification of HOIP ubiquitination sites.
To investigate a role for posttranslational modifications in controlling HOIP function, full-length HOIP was purified from 293T cells under denaturing conditions, and mass spectrometry was used to identify ubiquitinated residues. Two residues, lysine 640 and lysine 1056, were assigned with high confidence as being modified covalently by ubiquitin (Fig. 1A). Mass spectrometry also identified various ubiquitin peptides corresponding to linear, lysine 11-linked, lysine 48-linked, and lysine 63-linked ubiquitin chains comigrating with HOIP. Specific ubiquitin chain linkages can have different functions when attached to substrate proteins. Lysine 11 and lysine 48 ubiquitin chains can lead to protein degradation, while methionine 1 and lysine 63 can regulate signal transduction (17). All four of these ubiquitin chain types have been implicated in LUBAC-mediated signaling pathways (6). To verify that lysine 640 and lysine 1056 are sites for HOIP ubiquitination, mutant HOIP constructs were generated in which the identified lysine residues were mutated to arginine, and these mutants were then transfected into 293T cells to examine their respective HOIP ubiquitination levels (Fig. 1B). A previously identified HOIP ubiquitination site was mutated as a positive control (K330R) (18, 19), and the enzymatically inactive CS (C885S) mutant, which is incapable of linear ubiquitination or autoubiquitination, was included as a negative control. The HOIP K330R and K1056R mutants both showed reduced ubiquitination levels relative to the wild type (WT). The K640R and CS mutants did not show altered HOIP ubiquitination levels, indicating that these mutations did not affect HOIP ubiquitination and that autoubiquitination is not the primary source of ubiquitin attached to HOIP under these conditions. Collectively, this demonstrates that HOIP is ubiquitinated at multiple residues and that ubiquitination of HOIP does not require the catalytic C885 residue.
FIG 1 .

Identification and characterization of HOIP ubiquitination. (A) LUBAC was purified and separated on an SDS-PAGE gel. Bands corresponding to elevated-molecular-weight HOIP species were cut, and protein was digested with trypsin for mass spectrometry analysis. A search for posttranslationally modified LUBAC peptides revealed that HOIP was modified by ubiquitin at lysine 640 and lysine 1056. While lysine 640 is not conserved between mouse and human, lysine 1056 is highly conserved in vertebrates and is located in the linear-ubiquitin chain-determining domain (LDD) of HOIP. (B) Flag-tagged HOIP constructs with various lysine-to-arginine mutations were cotransfected in human 293T cells with HA-ubiquitin constructs. Cells were lysed under denaturing conditions (1% SDS), followed by 10-fold dilution with lysis buffer for renaturation. Flag-HOIP was immunoprecipitated with anti-Flag and immunoblotted with anti-HA to detect the ubiquitinated forms of HOIP. Lysates were also used for immunoblotting with anti-Flag (bottom). (C) V5-tagged C terminus HOIP constructs were cotransfected in human 293T cells with HA-ubiquitin lysine point mutant (ubiquitin-K48R or ubiquitin-K63R) constructs. Lysates were immunoprecipitated with anti-V5, followed by immunoblotting with anti-HA. Lysates were also used for immunoblotting with anti-V5 or anti-HA. (D) V5-tagged C-terminal HOIP constructs were cotransfected in human 293T cells with HA-ubiquitin lysine mutant constructs where all lysines, except for the indicated residue, were mutated to arginine: ubiquitin-K48 or ubiquitin-K63 can form only K48- or K63-linked ubiquitin chains, respectively. Experiment was performed as described for panel B. (E) V5-tagged C-terminal HOIP constructs were cotransfected in human 293T cells with HA-ubiquitin constructs. Immunoprecipitation and immunoblotting were conducted as described for panel B. Samples were probed with ubiquitin linkage-specific antibodies to determine ubiquitin chain linkages modifying HOIP. Data are representative of three independent experiments.
Interestingly, the K1056 residue is highly evolutionarily conserved among vertebrates and is located in the carboxyl LDD of HOIP, which is essential for the protein’s enzymatic activity (Fig. 1A, 20). Specifically, the crystal structure of the HOIP enzymatic region revealed that the K1056 residue is located on an exposed surface (see Fig. S1A in the supplemental material) (21). As HOIP has already been shown to be ubiquitinated at multiple sites in its amino terminus (19), we generated the carboxyl-terminal enzymatic domain constructs of HOIP and found that the K1056 is the primary site for ubiquitination of the HOIP C terminus (see Fig. S1B and C in the supplemental material). We next tested whether the HOIP C terminus was capable of autoubiquitination by performing an in vitro ubiquitination assay using recombinant HOIP C terminus. No high-molecular-weight HOIP bands were detected in this assay, suggesting that the HOIP C terminus is not efficiently autoubiquitinated in vitro (see Fig. S1D in the supplemental material). To investigate the ubiquitin chain linkage at the K1056 of HOIP, we used WT ubiquitin or the K48R or K63R mutant. While both ubiquitin mutants could still ubiquitinate HOIP, neither was as efficient as WT ubiquitin (Fig. 1C). Next, we used the K48-only or K63-only ubiquitin mutant, where all the lysines except for K48 or K63 have been mutated to arginine. Neither K48-only nor K63-only ubiquitin was capable of efficiently ubiquitinating HOIP (Fig. 1D). Finally, we used ubiquitin-specific antibodies to compare the ubiquitin linkages on the C termini of HOIP-WT and HOIP-K1056R (22). HOIP-WT was modified primarily by M1-, K48-, and K63-linked ubiquitin chains, which were among the linkages identified by mass spectrometry (Fig. 1E). In contrast, the HOIP-K1056R mutant had dramatically reduced levels of all types of ubiquitin linkages observed on HOIP-WT (Fig. 1E; also, see Fig. S1E in the supplemental material), suggesting that K1056 is the primary site for ubiquitination in the carboxyl terminus of HOIP. Finally, to rule out the possibility that the decreased ubiquitination of the HOIP K1056R mutant occurs because mutating K1056 disrupts HOIP structure and renders HOIP nonfunctional, we examined its enzymatic activity in vitro. We purified full-length HOIP-WT and HOIP-K1056R from bacteria using dual-expression constructs carrying both HOIL-1L and HOIP. HOIP-K1056R had in vitro activity equivalent to that of HOIP-WT, indicating that this mutation does not alter the intrinsic enzymatic activity of HOIP (see Fig. S1F in the supplemental material). These experiments collectively demonstrate that HOIP is ubiquitinated at the K1056 by multiple types of ubiquitin chains and that a K1056R mutation disrupts HOIP ubiquitination without disrupting its intrinsic enzymatic activity.
LUBAC kinetics and function in B cells.
We next established a system to study the function of HOIP-K1056 ubiquitination in the context of immune receptor stimulation. To measure the kinetics of LUBAC activation and inactivation, we used the A20.2J mouse B-cell lymphoma cell line, which has previously been used to characterize LUBAC-dependent immune signaling (10). A20 cells were treated with LPS to activate TLR4, which requires HOIP enzymatic activity for efficient downstream signaling (7). To measure LUBAC activation, we examined linear ubiquitination before and after TLR4 activation by purifying linear ubiquitin chains from cells using two approaches. In the first approach, linear ubiquitin chains were purified from cells lysed under denaturing conditions using a linear-ubiquitin-specific antibody. In the second approach, linear ubiquitin chains were purified from cells lysed under nondenaturing conditions using the linear-ubiquitin-chain-binding coiled-zinc finger (CoZi) domain of NEMO (23). Upon treatment with LPS, linear-ubiquitin levels dramatically increased and peaked by 2 h poststimulation (Fig. 2A). By 8 h after LPS treatment, linear ubiquitination declined to levels comparable to unstimulated levels (Fig. 2A; also, see Fig. S2 in the supplemental material). Interestingly, the linear-ubiquitin-specific antibody, but not the CoZi domain, pulled down linear ubiquitin chains after 4 h of stimulation (compare Fig. 2A and B to Fig. S2 in the supplemental material). Unlike the CoZi pulldown, the immunoprecipitation was performed under denaturing conditions. It is possible that the linear ubiquitin chains present in cells at an earlier time point are not accessible for CoZi binding, which could explain this discrepancy and suggests that the linear-ubiquitin antibody is a more accurate tool to measure linear ubiquitination. We next examined the kinetics of LUBAC substrate modification. The linear ubiquitination of IRAK1, a LUBAC substrate, which is modified by mixed M1/K63 ubiquitin chains in MyD88-dependent signaling complexes (13), also peaked at 2 h poststimulation in A20.2J cells (Fig. 2B), as shown by the IRAK1 smear in the immunoprecipitation (IP) sample. These results demonstrate similar temporal regulation of both LUBAC activity and LUBAC substrate modification upon TLR4 activation in A20.2J B-cells.
FIG 2 .
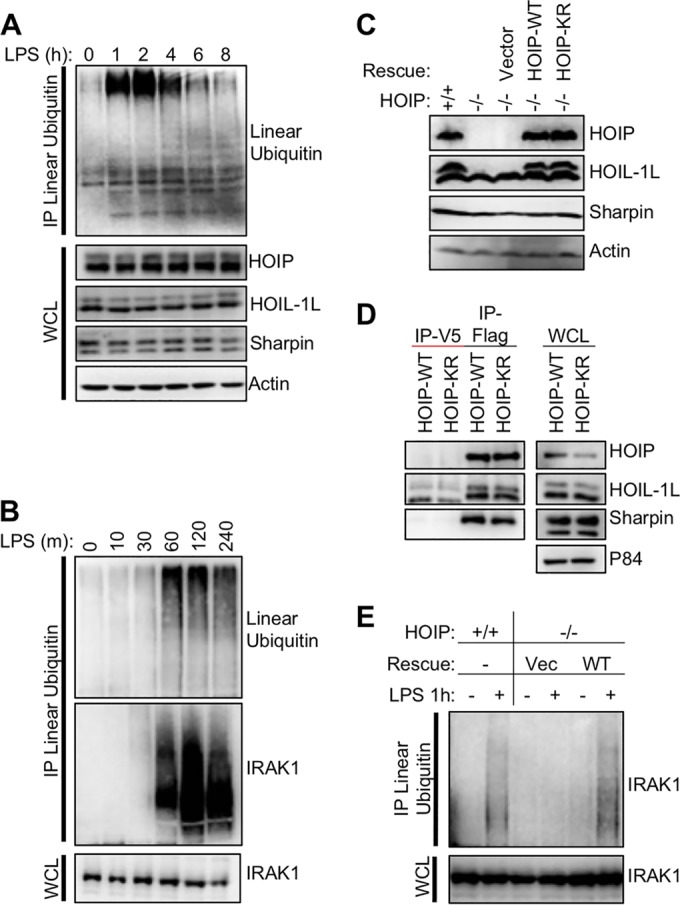
Kinetics of LUBAC activity and complementation of HOIP-deficient cells. (A) Mouse A20.2J B cells were stimulated with 10 µg/ml LPS to activate TLR4 signaling and induce LUBAC enzymatic activity. Cellular linear-ubiquitin levels were determined by immunoprecipitation with a linear-ubiquitin-specific antibody. Resting cells have low levels of linear ubiquitin relative to cells stimulated with LPS for 1 h. (B) As for panel A but with earlier time points as well as immunoblotting for IRAK1. (C) Mouse A20.2J HOIP−/− cells were complemented with vector, HOIP-WT, or HOIP-KR by lentiviral infection. LUBAC expression in complemented cells was compared to that in A20.2J HOIP+/+ cells by immunoblot analysis. (D) HOIP-Flag was immunoprecipitated from lysates of cell lines generated for panel A using Flag or IgG control antibody to determine LUBAC formation. (E) To verify function of complement cell lines, cells were stimulated with 10 µg/ml LPS for 1 h and then lysed in LUIP buffer. A linear-ubiquitin-specific antibody was used to precipitate linear ubiquitin chains, followed by immunoblotting with anti-IRAK1. Data are representative of three independent experiments.
To further test the role of HOIP ubiquitination upon TLR4 stimulation, we complemented A20.2J cells carrying a homozygous HOIP deletion (HOIP−/−) (10) with lentiviral constructs expressing either mouse Flag-tagged HOIP-WT or Flag-tagged HOIP-K1052R (homologous to human K1056 and referred to as HOIP-KR). HOIP-WT and HOIP-KR expression was equivalent to that of endogenous HOIP and both stabilized HOIL-1L expression (Fig. 2C). Furthermore, both HOIP-WT and HOIP-KR had similar abilities to associate with endogenous LUBAC components HOIL-1L and Sharpin (Fig. 2D). Next, HOIP+/+ and HOIP−/− cells complemented with HOIP-WT or vector alone were compared in their ability to induce the linear ubiquitination of IRAK1 upon LPS treatment. Complemented cells expressing HOIP-WT, but not cells expressing vector alone, were able to trigger IRAK1 linear ubiquitination at equivalent levels to HOIP+/+ cells (Fig. 2E). This demonstrates that HOIP−/− cells complemented with exogenous Flag-tagged HOIP behave similarly to HOIP+/+ cells and exhibit stimulation-dependent LUBAC activity, providing a system to study the function of HOIP ubiquitination.
HOIP is ubiquitinated upon TLR4 stimulation.
We next used the A20.2J complement cell system to examine the ubiquitination of HOIP in the context of immune receptor signaling. TLR4 stimulation led to the appearance of high-molecular-weight HOIP species, suggesting that HOIP underwent posttranslational modification (Fig. 3A). Interestingly, these high-molecular-weight bands appeared after total linear-ubiquitination levels peaked (Fig. 2A). To investigate whether this modification was ubiquitination at lysine 1056, we compared the ubiquitination status of HOIP in HOIP-WT and HOIP-KR complemented cells upon TLR4 stimulation. Indeed, HOIP-WT but not HOIP-KR underwent ubiquitination, as shown by the appearance of high-molecular-weight HOIP bands as well as ubiquitinated HOIP species (Fig. 3B). Collectively these data indicate that HOIP is ubiquitinated at lysine 1056 upon TLR4 stimulation and that this modification correlates with a decrease in total linear-ubiquitin levels.
FIG 3 .

HOIP is ubiquitinated upon TLR4 stimulation. (A) A20.2J mouse B cells were stimulated with LPS and lysed under denaturing conditions. Endogenous HOIP was immunoprecipitated, separated by SDS-PAGE, and analyzed by immunoblotting. High-molecular-weight HOIP bands (>120,000) appeared after LPS stimulation. (B) Complemented mouse A20.2J HOIP−/− cells used for Fig. 2C were stimulated with LPS for the indicated times and lysed under denaturing conditions. Anti-IgG (control)- or anti-Flag (HOIP)-immunoprecipitated samples were prepared as described for panel A. A long exposure of the Flag immunoblot revealed the presence of high-molecular-weight HOIP bands. Data are representative of three independent experiments.
Mutation of HOIP lysine 1056 increases LUBAC enzymatic activity.
While HOIP-WT and HOIP-KR had similar in vitro activities (see Fig. S1F in the supplemental material), we wanted to determine if the ubiquitination of lysine 1056 altered HOIP enzymatic activity in cells. We used conditions where LUBAC activity is stimulation-dependent (Fig. 2A) as well as a system where HOIP is constitutively active. Transfection of HOIP and HOIL-1L in 293T cells leads to constitutively active LUBAC enzymatic activity independent of immune receptor simulation (5). Transfection of HOIP-K1056R, but not K640R, in 293T cells resulted in the elevation of linear ubiquitination relative to that of HOIP-WT (Fig. 4A), suggesting that ubiquitination at lysine 1056 could negatively regulate HOIP enzymatic activity. While HOIP could be modified with K48-ubiquitin chains for protein degradation, treatment with the proteasome inhibitor MG132 did not alter linear-ubiquitin levels, and the HOIP-KR mutant showed HOIL-1L binding affinity similar to that of HOIP-WT (Fig. 2D; also, see Fig. S3A and B in the supplemental material). This showed that enhanced linear-ubiquitin levels generated by HOIP-KR were independent of protein degradation and HOIL-1L binding activity. Together, these data suggest that ubiquitination at lysine 1056 could alter the intrinsic enzymatic activity of HOIP.
FIG 4 .

Mutation of HOIP lysine 1056 increases LUBAC enzymatic activity. (A) HOIP constructs were transfected into human 293T cells in the presence or absence of HOIL-1L. At 24 h after transfection, cells were lysed, and LUBAC protein expression and linear-ubiquitin-chain levels were determined by immunoblot analysis of cell lysates. (B) A20.2J HOIP−/− complemented cells from Fig. 2C were stimulated with 10 µg/ml LPS to activate TLR4 signaling and induce LUBAC enzymatic activity. Endogenous linear-ubiquitin levels were determined by immunoprecipitation with a linear-ubiquitin-specific antibody. (C) As described for panel B, but with LPS stimulation for various time points to compare linear-ubiquitin levels between WT and KR cells. (D) Densitometry analysis of the linear-ubiquitin immunoblot from panel C. All values are relative to WT cells prior to stimulation. (E) A20.2J HOIP−/− complemented cells stimulated as described for panel C were stained for linear ubiquitin using M1Ub-APC, and linear-ubiquitin-positive cells were quantified by flow cytometry. (F) A20.2J HOIP−/− complemented cells were stimulated as described for panel C. The immunoblot for IRAK1 shows the presence of linear ubiquitin chains on IRAK1 upon TLR4 activation. Data are representative of three independent experiments.
To confirm these results in the context of immune receptor stimulation, the complemented cell lines used for Fig. 2 were stimulated with LPS for various lengths of time. Similar to transfection in 293T cells, stable expression of the HOIP-KR mutant in A20.2J cells also showed elevated linear ubiquitination upon LPS stimulation relative to that of the HOIP-WT (Fig. 4B to D). Interestingly, A20.2J HOIP-WT cells showed an increase of intracellular linear ubiquitination upon LPS stimulation that peaked at 2 h, rapidly decreased at 4 h, increased slightly at 6 h, and then decreased at 8 h poststimulation (Fig. 4C). In striking contrast, A20.2J HOIP KR cells showed a continuous increase in linear ubiquitination upon LPS stimulation (Fig. 4C). Neither LUBAC protein levels nor LUBAC subunit mRNA levels were altered upon LPS stimulation in A20.2J cells (Fig. 4C and F; also, see Fig. S3C in the supplemental material), suggesting that changes in linear ubiquitination are not due to fluctuations in LUBAC expression levels. Thus, both transient and stable cell systems for measuring HOIP activity demonstrated the enhanced enzymatic activity of HOIP-KR compared to HOIP-WT.
Next, we used fluorescence-activated cell sorting (FACS) to quantify intracellular linear-ubiquitination levels using a linear-ubiquitin-specific antibody. A20.J2 HOIP−/−, HOIP-WT, and HOIP-KR cells were stimulated with LPS for various times and subjected to intracellular anti-linear-ubiquitin staining followed by FACS analysis. Concordant with Fig. 2A, this intracellular anti-linear-ubiquitin staining also showed that linear ubiquitination peaked at 2 h in HOIP WT cells upon LPS stimulation and declined thereafter, whereas no increase was detected in HOIP−/− cells (see Fig. S3D and E in the supplemental material). Interestingly, the signal in unstimulated HOIP−/− cells was lower than that in HOIP+/+ cells, suggesting the presence of low levels of linear ubiquitination in resting cells, as was seen by immunoprecipitation (Fig. 4F; also, see Fig. S3F in the supplemental material). Using these observations, we defined linear-ubiquitination-positive cells as cells with a fluorescence intensity higher than that of resting cells and then compared the linear-ubiquitination levels between HOIP-WT and HOIP-KR cells. This showed that the linear-ubiquitination levels were higher overall in HOIP-KR cells than in HOIP-WT cells after 2 h of LPS stimulation (Fig. 4E). We finally compared linear-ubiquitination levels of LUBAC substrate IRAK1, a key TLR4 signaling molecule (13). The linear-ubiquitination levels of IRAK1 not only were elevated after 2 h of LPS stimulation but also remained elevated after 2 h in HOIP-KR cells compared to those in HOIP-WT cells, (Fig. 4F). These data demonstrate that mutation of the HOIP lysine 1056 ubiquitination site results in sustained LUBAC enzymatic activity upon TLR4 stimulation.
HOIP ubiquitination triggers HOIP conformational change.
When we compared the ubiquitination status of HOIP-WT and HOIP-KR before and after TLR4 stimulation, the HOIP-KR mutant showed far less ubiquitination than HOIP-WT but showed protein stability similar to that of HOIP-WT (Fig. 3B), suggesting that ubiquitination may not trigger HOIP degradation. To address whether the ubiquitination at the HOIP K1056 residue affects LUBAC enzymatic activity by altering its conformation, we developed the conditions for a fluorescence resonance energy transfer (FRET) assay (24). FRET-HOIP constructs contained an amino-termina; enhanced yellow fluorescent protein (eYFP) and carboxyl-terminal enhanced cyan fluorescent protein (eCFP) fusion (Fig. 5A). The HOIP-WT-FRET fusion construct produced linear-ubiquitin chains in 293T cells (see Fig. S4A in the supplemental material) and induced IκBα phosphorylation and degradation in A20.2J cells (see Fig. S4B in the supplemental material), indicating that the FRET-HOIP-WT fusion protein is functionally similar to HOIP-WT in cells despite its reduced expression compared to HOIP-WT expression in A20.2J cells (see Fig. S4B and C in the supplemental material). Both FRET and linear-ubiquitination levels were higher in unstimulated 293T cells transfected with FRET-HOIP-KR than in those with FRET-HOIP-WT, indicating that FRET-HOIP-KR also is functionally similar in 293T cells to HOIP-KR (Fig. 5B). To compare FRET signals between the HOIP constructs, a direct YFP-CFP (FRET) fusion construct was transfected as a positive FRET control, and individual YFP and CFP-HOIP fusion constructs were used to correct for background FRET levels resulting from YFP emission in the CFP spectrum and CFP emission in the YFP spectrum, respectively. The ratio of fluorescent emission at 486 nm (CFP) to that at 535 nm (YFP) was measured in live cells. While FRET-HOIP-KR had a FRET signal equivalent to CFP-HOIP background levels, FRET-HOIP-WT exhibited a positive FRET signal (Fig. 5C). In order to determine whether there were conformational changes in HOIP upon LPS stimulation, FRET-HOIP-WT and FRET-HOIP-KR constructs were electroporated into HOIP−/− A20.2J cells, followed by LPS stimulation. CFP and YFP fluorescence intensity was measured hourly following LPS stimulation (Fig. 5D). The relative FRET intensity of the FRET-HOIP-WT construct rapidly increased upon LPS stimulation, reached a peak level at 2 h, and declined at 3 h after stimulation, suggesting that a conformational change occurs in HOIP-WT upon LPS stimulation. In striking contrast, the FRET intensity for FRET-HOIP-KR was not altered upon LPS stimulation. A control YFP-CFP (labeled FRET) fusion also showed no alteration of its FRET intensity upon LPS stimulation (Fig. 5D). Collectively, this suggests that HOIP dynamically undergoes conformational change upon LPS-induced TLR4 activation in a K1056 ubiquitination-dependent manner.
FIG 5 .

TLR4 stimulation induces HOIP conformational change. (A) Diagram of HOIP-FRET constructs with amino-terminal YFP fusions and carboxyl-terminal CFP fusions. (B) Function of HOIP-FRET constructs as determined by immunoblot for linear ubiquitins in 293T cell lysates containing either HOIP-WT or HOIP-FRET. (C) HOIP-FRET constructs were transfected into 293T cells. At 24 h after transfection, cells were lysed and FRET was measured. FRET values were calculated by subtracting background fluorescence and dividing fluorescence of YFP by fluorescence of CFP. Data are representative of three independent experiments. (D) HOIP-FRET constructs were transfected into A20.2J HOIP−/− cells by using the Neon system (Life Technologies). At 24 h after transfection, cells were resuspended in FluoroBrite medium, and fluorescence was measured using an Envision plate reader. After addition of LPS, fluorescence readings were taken at the indicated times. Relative FRET was calculated as FRET(x min)/FRET(0 min). HOIP-eCFP and HOIP-eYFP constructs were used as negative controls. Data are representative of three independent experiments.
Ubiquitination of HOIP-K1056 blocks NF-κB activation.
As HOIP enzymatic activity is required for efficient TLR4- and CD40-mediated NF-κB activation in B cells (6, 7), we compared the levels of NF-κB activation between A20.2J HOIP-WT and HOIP-KR complemented cells. Interestingly, no obvious differences in NF-κB activation were detected between WT and KR cells during the initial response (30 min) to LPS treatment, as measured by IκBα phosphorylation and degradation (see Fig. S5A in the supplemental material). In contrast, HOIP-KR cells had considerably elevated levels of IκBα phosphorylation compared to HOIP-WT cells during extended LPS treatment (60 to 480 min) (Fig. 6). Next, the mRNA levels for the NF-κB-inducible genes tnf (TNF-α), icam1 (ICAM1), nfkbia (IκBα), and tnfaip3 (A20) were determined by quantitative reverse transcription-PCR (qRT-PCR). The mRNA levels for NF-κB-inducible genes in HOIP-WT cells rapidly increased at 1 h after LPS stimulation and immediately decreased to levels comparable to those seen with the initial unstimulated conditions (Fig. 6B). Strikingly, HOIP-KR cells showing sustained and enhanced linear ubiquitination exhibited continuously elevated mRNA levels of NF-κB-induced genes upon LPS stimulation (Fig. 6B). However, this difference occurred specifically during TLR4 stimulation but not CD40 stimulation, since comparable levels of CD40 stimulation-induced NF-κB activation were observed in both HOIP-WT and HOIP-KR cells as measured by IκBα phosphorylation and NF-κB-dependent gene expression (Fig. 6C and D; also, see Fig. S5B in the supplemental material). We also attempted to use A20.2J cells to investigate HOIP ubiquitination in TNF-α signaling. However, due to the lack of TNF-α receptor surface expression of A20.2J cells (see Fig. S5C to E in the supplemental material), we were not able to examine NF-κB activation and HOIP ubiquitination upon TNF-α signaling. Collectively, these data strongly suggest that TLR4-induced NF-κB activation is negatively regulated by ubiquitination of the HOIP K1056 residue, while CD40-induced NF-kB activation is independent of HOIP K1056 ubiquitination.
FIG 6 .

TLR4 stimulation leads to higher NF-κB activation in HOIP-KR cells than in HOIP-WT cells. (A) A20.2J cells were stimulated with 10 µg/ml LPS and lysed at intervals up to 480 min after treatment with LPS. (B) Quantitative PCR using cDNA reverse transcribed from cells stimulated as described for panel A for the indicated times. Values were first calculated relative to 18S and then were normalized to vector cells prior to stimulation. (C) A20.2J cells were stimulated with 10 µg/ml CD40L, and lysates were analyzed up to 240 min poststimulation. (D) Quantitative PCR using cDNA reverse transcribed from A20.2J cells stimulated as described for panel C for the indicated times. Values were first calculated relative to 18S and then were normalized to vector cells prior to stimulation. Data are representative of three independent experiments.
DISCUSSION
While ubiquitination of HOIP has been detected at multiple residues (18, 19), detailed functions of these ubiquitinations have not been described. Here, we demonstrate that HOIP dynamically undergoes a conformational change upon LPS-induced TLR4 activation in a K1056 ubiquitination-dependent manner and that the enzymatic activity of HOIP is negatively regulated through the ubiquitination of K1056. Interestingly, HOIP was modified by multiple ubiquitin chain linkages, including linear (M1), K11-linked, K48-linked, and K63-linked ubiquitin chains, which were all drastically reduced in the HOIP K1056R mutant. A previous report demonstrated the presence of hybrid K63 and linear ubiquitin chains in IL-1R signaling, which activates NF-κB through a mechanism similar to TLR4 stimulation (13). This indicates that while mixed ubiquitin chains facilitate NF-κB activation by recruiting adaptor molecules, they may also inhibit NF-κB activation by suppressing HOIP function. Thus, mixed ubiquitin chains could have substrate-specific functions. Furthermore, previous mass spectrometry of HOIP found ubiquitination at the K99 residue in the region that binds the linear-ubiquitin deubiquitinase Otulin (25, 26), suggesting that the K99 ubiquitination may regulate the HOIP-Otulin interaction. Thus, HOIP itself undergoes various ubiquitinations, which ultimately regulates LUBAC activity for intracellular signal transduction.
Our study found that ubiquitination of HOIP at K1056 alters HOIP conformation and subsequently decreases its enzymatic activity. The K1056 residue of HOIP exhibits a high degree of evolutional conservation among vertebrates (from Homo sapiens to Danio rerio) and is located in the C-terminal LDD of HOIP, which is essential for its enzymatic activity. Our data suggest that when the K1056 residue undergoes ubiquitination, HOIP adopts a conformational change where its amino and carboxyl termini are in close proximity and that this conformational change inhibits the ability of HOIP to synthesize linear ubiquitin chains and/or modify substrates with linear ubiquitin chains. Specifically, the Npl4 zinc finger (NZF) domain is a compact zinc-binding module found in many proteins involved in ubiquitin-dependent processes, including HOIP, and has been shown to bind K63-ubiquitin chains (27). It is thus possible that the K1056 linked ubiquitin chain on HOIP may bind its central NZF domain, resulting in the closed conformation of the carboxyl RBR enzymatic domain, thereby reducing its enzymatic activity (Fig. 7). In fact, while the RBR family ligases have been shown to adopt autoinhibited conformations under resting conditions, little is known about the mechanistic similarities during RBR inactivation (14). While the full-length structure of HOIP is not known, structural studies suggest that RBR family member Parkin has a high degree of flexibility and that significant rearrangement occurs during enzymatic activation (28, 29). As conformational change is essential for enzyme activation, it is reasonable to expect that rearrangement could subsequently impair enzymatic activity. Thus, together with the known mechanism of Parkin RBR ligase inactivation (15), our study on HOIP suggests that posttranslational modification and subsequent conformational change may be a general mechanism to inactivate the enzymatic activity of RBR-family E3 ligases.
FIG 7 .
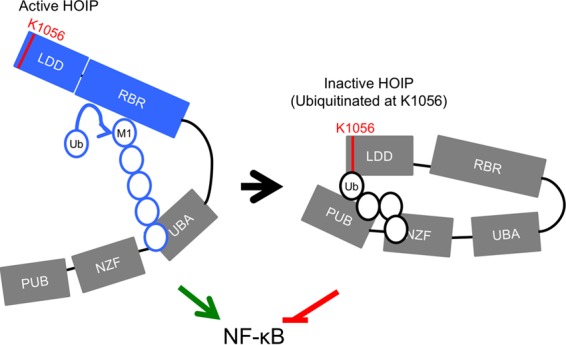
Model of HOIP structural alteration upon TLR4 stimulation. TLR4 stimulation activates HOIP enzymatic activity. The synthesis of linear ubiquitin chains by HOIP promotes NF-κB activation. This signal, however, must be terminated to prevent sustained signaling. Ubiquitination of HOIP at lysine 1056 is triggered by TLR4 stimulation and alters HOIP conformation. The change in conformation inactivates HOIP enzymatic activity, which in turn removes a signal required for NF-κB activation, thus facilitating termination of the immune response.
LUBAC has immune receptor and cell type-specific signaling functions (30). Intriguingly, we observed a specific role for the K1056 ubiquitination of HOIP in TLR4, but not CD40, signaling. CD40 is a member of the TNF receptor (TNFR) superfamily that shares signaling components with TNFR1, including cIAP1/2, which is required for LUBAC recruitment to receptor signaling complexes (31). In contrast, TLR4 is functionally similar to IL-1R, since both require the signaling molecules MyD88, IRAK1, and IRAK4, all of which are the known linear ubiquitination substrates of LUBAC (13). Further, IL-1R signaling, but not TNF signaling, requires K63-linked ubiquitin chains for NF-κB activation and LUBAC activation (13, 32). These functional differences required for LUBAC activation between these two pathways support the idea of receptor-specific negative regulation of LUBAC activity. Indeed, TLR and IL-1R have negative regulatory mechanisms distinct from TNF signaling (33). Thus, like seen in many receptor mediated signaling pathways, the regulation of LUBAC activity, through posttranslational modification at a specific site of HOIP, is receptor-specific. Future studies could investigate the receptor specific regulatory roles of posttranslational modification of HOIP.
It is intriguing that during TLR4 stimulation, the difference in signal transduction between HOIP-WT and HOIP-KR cells became evident 2 h after stimulation, which coincided with HOIP ubiquitination. This suggests that the KR mutation does not impact the initial response to TLR4 activation but rather prevents the inactivation of intracellular signaling by permitting continued linear ubiquitination of substrates. As HOIP activity appears to be terminated after the first round of NF-κB gene induction, it is plausible to hypothesize that one or more NF-κB-inducible E3 ligases mediate the K1056 ubiquitination of HOIP. It will be interesting to investigate which E3 ligase targets the HOIP K1056 specifically in TLR4 signaling. Further work on HOIP ubiquitination could investigate how HOIP activity is regulated in different immune signaling pathways, as our work suggests the presence of multiple regulatory mechanisms. This idea is supported by the fact that multiple HOIP isoforms have been detected in humans and, specifically, that one of these lacks the Otulin-binding PUB domain, implying a need for Otulin-independent regulation of LUBAC activity (34). However, even in this context, Otulin activity may still be required to disassemble linear ubiquitin chains after HOIP activity has been terminated. Knowledge of the precise regulatory mechanisms of LUBAC activity could present a strategy to therapeutically target inflammatory signaling with high specificity.
MATERIALS AND METHODS
Cell culture, stimulation, and complement cell generation.
The mouse A20.2J B-cell line was described previously (10). Cells were cultured in RPMI supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 µg/ml streptomycin, and 10 µm 2-mercaptoethanol (2ME). Cells were stimulated with 10 µg/ml LPS (E. coli 0127:B8; Sigma-Aldrich) for various times. To induce CD40 stimulation, cells were treated with 5 µg/ml of CD40L (Peprotech) for various times. TNF-α (BioLegend) was added to cells at 20 ng/ml. 293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. For complemented-cell generation, lentiviruses were produced in 293T cells by calcium phosphate transfection using the pCDH-CMV-MCS-EF1-hygro system (System Biosciences). Virus was collected 2 days after transfection, filtered, and concentrated by ultracentrifugation at 20,000 rpm for 2 h. A20.2J cells were spin-infected at 2,000 rpm for 30 min in RPMI containing 8 µg/ml Polybrene and 100 µl concentrated virus. Two days after infection, the medium was changed to include 200 µg/ml hygromycin B.
qRT-PCR.
RNA was isolated from A20.2J cells with 1 ml Tri Reagent (Sigma-Aldrich). DNA was removed by treatment with DNase I (Sigma-Aldrich). RNA was reverse-transcribed to generate cDNA using iScript (Bio-Rad Laboratories). cDNA was diluted 1:10 with water and quantified by qRT-PCR using the iQ SYBR green Supermix kit (Bio-Rad Laboratories). Reactions were cycled at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s and 60°C for 30 s, followed by melt curve analysis and analyzed on a CFX96 PCR machine (Bio-Rad Laboratories). Threshold cycle (CT) ratios were determined by normalizing to 18S RNA and a vector complementation control sample using the following equation: relative expression = 100 × 2−ΔΔCT, where ΔΔCT = (CT_gene − CT_18S) − (ΔCT_Vec unstimulated). All values were normalized to the unstimulated vector condition. Primer sequences were validated previously (5).
In vitro linear-ubiquitin-chain formation assay.
HOIL-1L-6xHIS and HOIP-K1056R-6xHIS were cloned into the pET-duet-1 vector, expressed in BL21(DE3) RIPL bacteria (Agilent Technologies), and purified from cultures as previously described (5). HOIP-Cterm (amino acids 699 to 1072), E1 (Ube1), E2 (UbcH5c), and ubiquitin were purchased from Boston Biochem. In vitro linear-ubiquitination assay mixtures contained 200 ng E1, 400 ng E2, 1 µg HOIL-1L/HOIP-6xHIS purification prep, 20 mM Tris (pH 7.5), 5 mM dithiothreitol (DTT), 5 mM MgCl2, 2 mM Mg-ATP (Boston Biochem), and 10 µg ubiquitin. Assay mixtures were incubated for various times at 37°C, and reactions were stopped by adding LDS (lithium dodecyl sulfate) sample buffer (Life Technologies) and heating at 70°C for 10 min.
Immunoprecipitation.
For linear-ubiquitin immunoprecipitation (IP), cells were lysed in linear-ubiquitination IP (LUIP) buffer (5 M urea, 135 mM NaCl, 1% Triton X-100, 1.5 mM MgCl2, 2 mM N-ethylmaleimide, and complete protease inhibitor cocktail [Roche]). A total of 5 × 106 cells were used per condition. Lysates were precleared with Sepharose beads for 1 h at room temperature. Linear ubiquitin antibody (0.25 µg) (Genentech) was added to each sample and incubated overnight at room temperature with end-over-end mixing. Protein A/G beads were then incubated with samples for 2 h at room temperature, and samples were washed with LUIP buffer twice and then with phosphate-buffered saline (PBS) twice. Samples were eluted for immunoblotting from beads by adding LDS sample buffer supplemented with 2.5 mM DTT and heating at 70°C for 10 min. For denaturing IP experiments to detect ubiquitinated HOIP species, cells were lysed in 100 µl of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 7.4], 1% NP-40, 0.5% sodium deoxycholate, 150 mM NaCl) supplemented with SDS at a final concentration of 1% to denature and disrupt protein-protein interactions. Samples were sonicated and then diluted to 1 ml in RIPA buffer. Lysates were precleared with Sepharose beads for 1 h at 4°C and then incubated with anti-Flag antibody (Sigma) overnight at 4°C. Protein A/G beads were incubated with lysate-antibody mixtures for 2 h before being washed three times in RIPA buffer and heated at 70°C in 2× Laemmli dye for 10 min to elute protein for immunoblotting. For precipitation of LUBAC, cells were lysed in IP buffer (1% NP-40, 50 mM Tris [pH 7.4], 150 mM NaCl, 0.5% sodium deoxycholate, and complete protease inhibitor cocktail [Roche]) and sonicated for 20 s at 10% amplitude. Lysates were precleared with Sepharose beads for 1 h at 4°C and then incubated with anti-Flag antibody (Sigma) overnight at 4°C. Protein A/G beads were incubated with lysate-antibody mixtures for 2 h before being washed three times in IP buffer and IP beads were heating at 70°C in 2× Laemmli dye for 10 min to elute protein for immunoblotting. Pulldown of linear ubiquitin with CoZi-GST was performed as previously described (5). Where 293T cells were used, cesium chloride-purified plasmid DNA preparations were transfected by PEI (polyethylenimine) as previously described (35). Cell lysates were collected 24 h after transfection.
Immunoblotting.
Cell lysates were collected in RIPA buffer and quantified by Bradford protein assay (Thermo, Fisher Scientific) to ensure equal amounts of protein loading. Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories) by semidry transfer at 25 V for 30 min. For linear-ubiquitin immunoblots, SDS-PAGE was transferred to nitrocellulose (Bio-Rad Laboratories) by wet transfer at 4°C for 2 h at 30V. Membranes were blocked in either 5% milk in phosphate-buffered saline–Tween (PBST) or 3% BSA in Tris-buffered saline–Tween (TBST) for pIκBα and probed overnight with the appropriate antibodies in 3% BSA, except for the linear-ubiquitin antibody, which was incubated at room temperature for 1 h. Primary antibodies included those against mouse IκBα (1:1,000; Cell Signaling Technology), mouse pIκBα (1:1,000; Cell Signaling Technology), actin (clone C4; Santa Cruz Biotechnology, Inc.), GAPDH (1:5000; GeneTex), p84 (1:2,000; GeneTex), V5 (1:2,000; Life Technologies), Flag (1:2,000; Sigma-Aldrich), and HA (1:2,000; Covance). Antibodies against HOIP, HOIL-1L, and Sharpin were described previously (11). The linear ubiquitin (M1), K11 ubiquitin, K48 ubiquitin, and K63 ubiquitin antibodies were a gift from Genentech and have been characterized in detail (22). Samples for specific ubiquitin chain immunoblots were separated by SDS-PAGE and transferred to nitrocellulose by wet transfer at 4°C and 30 V for 2 h. Membranes were blocked in 5% milk in PBST, probed with primary antibody (1:2,000) for 1 h at room temperature, washed, probed with TrueBlot-HRP (1:300; Thermo, Fisher Scientific), and then developed with enhanced chemiluminescence (ECL) reagent (Thermo, Fisher Scientific). For all other antibodies, appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies were incubated on membranes, and bands were developed with ECL reagent and imaged on an LAS-4000 imager (Fuji). For IP with Western blots, the Clean Blot HRP-conjugated secondary antibody (1:300; Thermo, Fisher Scientific) was substituted.
Flow cytometry.
A20.2J cells (1 × 106) in 1 ml of medium were treated with 10 µg/ml LPS for various times. Cells were collected by centrifugation and fixed with ice-cold methanol for 15 min at −20°C. After fixation, cells were washed with PBS and blocked with 3% BSA in PBST for 1 h at room temperature. Cells were stained with a linear-ubiquitin-specific antibody (Genentech) or human isotype control for 1 h at room temperature and then washed twice with PBS. Next, cells were incubated with allophycocyanin (APC)-conjugated anti-human IgG secondary antibody for 1 h. Data were acquired with a BD FACSCanto II flow cytometer and analyzed with FlowJo software.
Cloning.
Mouse HOIP was cloned from cDNA derived from B6 mouse bone marrow. It was PCR amplified to include an amino-terminal Flag tag along with 5′ NheI and 3′ NotI digestion sites and cloned into the lentiviral pCDH-CMV-MCS-EF1-hygro vector. To clone human and mouse HOIP-FRET constructs, we generated a pIRES-Flag-eYFP-eCFP (pIRES-FRET) construct. Enhanced yellow fluorescent protein (eYFP) was PCR amplified with 5′ NheI and 3′ EcoRI digestion sites and cloned into pIRES-Flag. Enhanced cyan fluorescent protein (eCFP) was PCR amplified with 5′ MluI and 3′ XbaI and cloned into both pIRES-Flag and pIRES-Flag-eYFP. Human and mouse HOIP were separately PCR amplified with 5′ EcoRI and 3′ MluI digestion sites and cloned into the following vectors: pIRES-Flag, pIRES-Flag-eYFP, pIRES-Flag-eCFP, and pIRES-Flag-eYFP-eCFP.
FRET assay.
For experiments in A20.2J HOIP−/− cells, Neon transfection (Life Technologies) was used to electroporate HOIP-FRET constructs. The 10-µl tip was used with cells at a concentration of 7.6 × 106 cells/ml in buffer R. A 1.5-µg portion of plasmid DNA was added per 10 µl of cells. The cells were pulsed with 1,300 V for 20 ms with 2 pulses. Transfection efficiency was verified by counting eYFP-positive cells 24 h posttransfection as well as by immunoblotting. For FRET analysis, four 10-µl transfections were used per condition. Cells were pelleted 24 h posttransfection and resuspended in 100 µl FluoroBrite DMEM (Life Technologies) supplemented with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin, and 10 µm 2ME. For 293T FRET experiments, cells were transfected with FRET-HOIP constructs. Twenty-four hours after transfection, cells were lysed with IP buffer, and the lysate was used for FRET analysis. Cells and lysates were imaged with the Envision multilabel reader (PerkinElmer), and emission readings were recorded at 486 nm for CFP and 535 nm for YFP. As a negative control, cells were transfected with either HOIP-eCFP alone or HOIP-eYFP alone. A direct eYFP-eCFP fusion FRET construct was used as a positive control. Sample emission was measured prior to stimulation and at various times after 10 µg/ml LPS was added to the medium. To correct for background in fluorescent emission values, the emission of HOIP-eCFP in the YFP channel was subtracted from the YFP emission value of all samples. The emission of HOIP-eYFP in the CFP channel was subtracted from the CFP emission value of all samples. The corrected values were used for the FRET ratio, which was the value of CFP emission divided by that of YFP emission. Values were then normalized to the eYFP-eCFP-positive FRET construct.
Mass spectrometry.
Bands corresponding to high-molecular-weight HOIP species were cut from an 8% SDS-PAGE gel and sent to the Taplin Mass Spectrometry Core Facility (Harvard) for processing and analysis. Protein was digested with trypsin, and peptides were analyzed for modification by ubiquitination and phosphorylation.
Statistical analysis.
All experiments were performed a minimum of three times, and representative data are presented here. For experiments with statistical analysis, a two-tailed Student’s t test was done, and P values of <0.05 were considered significant.
SUPPLEMENTAL MATERIAL
K1056 plays an important role in HOIP ubiquitination but not in HOIP enzymatic activity. (A) Previously published HOIP crystal structure of the enzymatically active carboxyl terminus of HOIP in complex with ubiquitin. The structure reveals that lysine 1056 is on an exposed region of the active enzyme (arrow), suggesting that it is accessible for modification by another E3 ligase. (B) Regions of human HOIP included in carboxyl terminus constructs. (C) HOIP constructs tagged with HA (which contains no lysine residues) and containing the RBR and LDD domains were expressed in 293T cells along with Flag-ubiquitin. WT lanes contain HOIP-RBR-LDD-WT, and KR lanes contain HOIP-RBR-LDD-K1056R. Immunoprecipitation was conducted under denaturing conditions. Mutation of lysine 1056 to arginine dramatically reduced the presence of ubiquitinated HOIP species. Data are representative of three independent experiments. (D) Human recombinant HOIP (amino acids 699 to 1072) was incubated with ubiquitin, ATP, E1, and E2 enzymes for 2 h. The reaction was stopped with the addition of sample buffer, and samples were analyzed by immunoblot analysis. (E) Additional blots from experiment in Fig. 1D showing HA-ubiquitin and K11-ubiquitin. (F) HOIP was copurified with HOIL-1L from BL21(DE3) RIPL bacterial cells. Purified LUBAC was incubated with E1, E2, and ATP for the indicated times. Equal protein amounts of LUBAC containing either HOIP-WT or HOIP-KR were used for the reactions. Reaction mixtures were incubated for the indicated times. Download
Purification of linear ubiquitination by the CoZi domain of NEMO. Mouse A20.2J B-cells were stimulated with 10 µg/ml LPS to activate TLR4 signaling and induce LUBAC enzymatic activity. The ubiquitin-binding coiled-zinc finger (CoZi) domain of NEMO fused to GST was purified from bacteria and used to pull down linear ubiquitin from cell lysate. Data are representative of three independent experiments. Download
Increase of intracellular linear ubiquitination by HOIP KR. (A) Flag-HOIP was expressed with or without HOIL-1L-HA in 293T cells. The proteasome inhibitor MG-132 was added to the indicated samples for 4 h at 20 µM prior to cell lysis and immunoblot analysis. (B) HOIP and HOIL-1L constructs were transfected into 293T cells and cells were lysed with NP-40 buffer 24 h posttransfection. Immunoprecipitation was performed separately for HOIL-1L using HA antibody and HOIP using Flag antibody. Data are representative of three independent experiments. (C) Quantitative PCR using cDNA reverse transcribed from A20.2J B-cell mRNA isolated from cells stimulated with LPS for the indicated times. Samples were normalized to ribosomal 18S RNA. Each data point represents three biological and three analytical replicates. (D) A total of 1 × 106 A20.2J cells were stimulated with 10 µg/ml LPS for the indicated times, fixed, and then stained for linear ubiquitin (M1Ub-APC). Cells were analyzed by flow cytometry. (E) As for panel D. HOIP+/+ and HOIP−/− cells were stimulated with LPS and stained for linear ubiquitin (M1Ub-APC). Linear-ubiquitin staining was compared between cells at the indicated times. (F) Cells were stimulated as described for panel E for direct comparison between isotype staining and linear-ubiquitin staining in HOIP+/+ and HOIP−/− cells. Data are representative of three independent experiments. Download
HOIP ubiquitination and HOIP-FRET constructs. (A) Various HOIP fusion constructs were expressed in 293T cells. Linear-ubiquitin levels were determined using immunoblot analysis. (B) HOIP-FRET constructs were transfected using the Neon system (Life Technologies) into A20.2J HOIP−/− cells. Twenty-four hours after transfection, cells were stimulated with LPS. Cells were lysed and analyzed by immunoblotting. Arrows indicate FRET-HOIP and HOIP-WT proteins. (C) Quantitative PCR using cDNA reverse transcribed from cells prepared as described for panel B. Download
Immunoblot analysis upon LPS or CD40L stimulation. (A) Various A20.2J cell lines were stimulated with 10 µg/ml of LPS, and cell lysates were subjected to immunoblotting at intervals up to 30 min after stimulation. (B) Indicated cells were stimulated with 10 µg/ml CD40L for up to 30 min, and cell lysates were subjected to immunoblotting. (C) HOIP+/+ and HOIP−/− cells were stimulated with TNF-α (20 ng/ml), and cell lysates were subjected to immunoblotting at intervals up to 8 h after stimulation. (D) Quantitative PCR using cDNA reverse transcribed from cells stimulated as described for panel C. (E) A total of 1 × 106 A20.2J cells were stained for cell surface receptors using phycoerythrin-conjugated CD40 (CD40-PE), TNFAR-PE, or isotype control-PE. Cells were analyzed by flow cytometry. CD40 staining served as a positive control. Download
ACKNOWLEDGMENTS
This work was partly supported by CA82057, CA31363, CA115284, CA180779, DE023926, HL110609, AI073099, AI116585, DE021982, the Hastings Foundation, the Fletcher Jones Foundation, and the GRL Program.
We thank Vishva Dixit, Junying Yuan, Si-Yi Chen, and Blossom Damania for reagents. Also, we thank all the members of the Jung laboratory for discussion.
Footnotes
Citation Bowman J, Rodgers MA, Shi M, Amatya R, Hostager B, Iwai K, Gao SJ, Jung JU. 2015. Posttranslational modification of HOIP blocks Toll-like receptor 4-mediated linear-ubiquitin-chain formation. mBio 6(6):e01777-15. doi:10.1128/mBio.01777-15.
REFERENCES
- 1.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israël L, Trevejo-Nunez G, Bogunovic D, Cepika A, MacDuff D, Chrabieh M, Hubeau M, Bajolle F, Debré M, Mazzolari E, Vairo D, Agou F, Virgin HW, Bossuyt X, Rambaud C, Facchetti F, Bonnet D, Quartier P, Fournet JC, Pascual V, Chaussabel D, Notarangelo LD, Puel A, Israel A, Casanova JL, Picard C. 2012. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol 13:1178–1186. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Courtois G, Gilmore TD. 2006. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene 25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 3.Tokunaga F. 2013. Linear ubiquitination-mediated NF-kappaB regulation and its related disorders. J Biochem 154:313–323. doi: 10.1093/jb/mvt079. [DOI] [PubMed] [Google Scholar]
- 4.Popovic D, Vucic D, Dikic I. 2014. Ubiquitination in disease pathogenesis and treatment. Nat Med 20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 5.Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, Amatya R, Kelly TJ, Iwai K, Ting J, Jung JU. 2014. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211:1333–1347. doi: 10.1084/jem.20132486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong W-W, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, Walczak H. 2011. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki Y, Sano S, Nakahara M, Murata S, Kometani K, Aiba Y, Sakamoto S, Watanabe Y, Tanaka K, Kurosaki T, Iwai K. 2013. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. EMBO J 32:2463–2476. doi: 10.1038/emboj.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, Wright GW, Zhao H, Yang Y, Xu W, Rosenwald A, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Wiestner A, Kruhlak MJ, Iwai K, Bernal F, Staudt LM. 2014. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov 4:480–493. doi: 10.1158/2159-8290.CD-13-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damgaard R, Nachbur U, Yabal M, Wong W, Fiil B, Kastirr M, Rieser E, Rickard J, Bankovacki A, Peschel C, Ruland J, Bekker-Jensen S, Mailand N, Kaufmann T, Strasser A, Walczak H, Silke J, Jost P, Gyrd-Hansen M. 2012. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 46:746–758. doi: 10.1016/j.molcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Hostager BS, Kashiwada M, Colgan JD, Rothman PB. 2011. HOIL-1L interacting protein (HOIP) is essential for CD40 signaling. PLoS One 6:e23061. doi: 10.1371/journal.pone.0023061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. 2009. Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol 11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 12.Dubois SM, Alexia C, Wu Y, Leclair HM, Leveau C, Schol E, Fest T, Tarte K, Chen ZJ, Gavard J, Bidere N. 2014. A catalytic-independent role for the LUBAC in NF-kappaB activation upon antigen receptor engagement and in lymphoma cells. Blood 123:2199–2203. doi: 10.1182/blood-2013-05-504019. [DOI] [PubMed] [Google Scholar]
- 13.Emmerich CH, Ordureau A, Strickson S, Arthur JSC, Pedrioli PGA, Komander D, Cohen P. 2013. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A 110:15247–15252. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smit JJ, Sixma TK. 2014. RBR E3-ligases at work. EMBO Rep 15:142–154. doi: 10.1002/embr.201338166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caulfield TR, Fiesel FC, Moussaud-Lamodière EL, Dourado DFAR, Flores SC, Springer W. 2014. Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput Biol 10:e1003935. doi: 10.1371/journal.pcbi.1003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko HS, Lee Y, Shin J-H, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM. 2010. Phosphorylation by the c-Abl protein tyrosine kinase inhibits Parkin’s ubiquitination and protective function. Proc Natl Acad Sci U S A 107:16691–16696. doi: 10.1073/pnas.1006083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komander D, Rape M. 2012. The ubiquitin code. Annu Rev Biochem 81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 18.Kim W, Bennett E, Huttlin E, Guo A, Li J, Possemato A, Sowa M, Rad R, Rush J, Comb M, Harper J, Gygi S. 2011. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, Choudhary C. 2011. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics 10:M111.013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smit JJ, Monteferrario D, Noordermeer SM, van Dijk WJ, van der Reijden BA, Sixma TK. 2012. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J 31:3833–3844. doi: 10.1038/emboj.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stieglitz B, Rana RR, Koliopoulos MG, Morris-Davies AC, Schaeffer V, Christodoulou E, Howell S, Brown NR, Dikic I, Rittinger K. 2013. Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature 503:422–426. doi: 10.1038/nature12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumoto ML, Dong KC, Yu C, Phu L, Gao X, Hannoush RN, Hymowitz SG, Kirkpatrick DS, Dixit VM, Kelley RF. 2012. Engineering and structural characterization of a linear polyubiquitin-specific antibody. J Mol Biol 418:134–144. doi: 10.1016/j.jmb.2011.12.053. [DOI] [PubMed] [Google Scholar]
- 23.Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, Dikic I. 2009. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Erickson JR, Patel R, Ferguson A, Bossuyt J, Bers DM. 2011. Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ Res 109:729–738. doi: 10.1161/CIRCRESAHA.111.247148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliott P, Nielsen S, Marco-Casanova P, Fiil B, Keusekotten K, Mailand N, Freund SV, Gyrd-Hansen M, Komander D. 2014. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol Cell 54:335–348. doi: 10.1016/j.molcel.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schaeffer V, Akutsu M, Olma M, Gomes L, Kawasaki M, Dikic I. 2014. Binding of OTULIN to the PUB domain of HOIP controls NF-kappaB signaling. Mol Cell 54:349–361. doi: 10.1016/j.molcel.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, Walczak H. 2009. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 28.Trempe J-F, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K. 2013. Structure of Parkin reveals mechanisms for ubiquitin ligase activation. Science 340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- 29.Wauer T, Komander D. 2013. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32:2099–2112. doi: 10.1038/emboj.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiil BK, Gyrd-Hansen M. 2014. Met1-linked ubiquitination in immune signalling. FEBS J 281:4337–4350. doi: 10.1111/febs.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emmerich CH, Schmukle AC, Walczak H. 2011. The emerging role of linear ubiquitination in cell signaling. Sci Signal 4:re5. doi: 10.1126/scisignal.2002187. [DOI] [PubMed] [Google Scholar]
- 32.Xu M, Skaug B, Zeng W, Chen ZJ. 2009. A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol Cell 36:302–314. doi: 10.1016/j.molcel.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi K, Hernandez LD, Galán JE, Janeway CA Jr., Medzhitov R, Flavell RA. 2002. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202. doi: 10.1016/S0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 34.Thompson HGR, Harris JW, Lin L, Brody JP. 2004. Identification of the protein Zibra, its genomic organization, regulation, and expression in breast cancer cells. Exp Cell Res 295:448–459. doi: 10.1016/j.yexcr.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 35.Inn K, Gack MU, Tokunaga F, Shi M, Wong L, Iwai K, Jung JU. 2011. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell 41:354–365. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
K1056 plays an important role in HOIP ubiquitination but not in HOIP enzymatic activity. (A) Previously published HOIP crystal structure of the enzymatically active carboxyl terminus of HOIP in complex with ubiquitin. The structure reveals that lysine 1056 is on an exposed region of the active enzyme (arrow), suggesting that it is accessible for modification by another E3 ligase. (B) Regions of human HOIP included in carboxyl terminus constructs. (C) HOIP constructs tagged with HA (which contains no lysine residues) and containing the RBR and LDD domains were expressed in 293T cells along with Flag-ubiquitin. WT lanes contain HOIP-RBR-LDD-WT, and KR lanes contain HOIP-RBR-LDD-K1056R. Immunoprecipitation was conducted under denaturing conditions. Mutation of lysine 1056 to arginine dramatically reduced the presence of ubiquitinated HOIP species. Data are representative of three independent experiments. (D) Human recombinant HOIP (amino acids 699 to 1072) was incubated with ubiquitin, ATP, E1, and E2 enzymes for 2 h. The reaction was stopped with the addition of sample buffer, and samples were analyzed by immunoblot analysis. (E) Additional blots from experiment in Fig. 1D showing HA-ubiquitin and K11-ubiquitin. (F) HOIP was copurified with HOIL-1L from BL21(DE3) RIPL bacterial cells. Purified LUBAC was incubated with E1, E2, and ATP for the indicated times. Equal protein amounts of LUBAC containing either HOIP-WT or HOIP-KR were used for the reactions. Reaction mixtures were incubated for the indicated times. Download
Purification of linear ubiquitination by the CoZi domain of NEMO. Mouse A20.2J B-cells were stimulated with 10 µg/ml LPS to activate TLR4 signaling and induce LUBAC enzymatic activity. The ubiquitin-binding coiled-zinc finger (CoZi) domain of NEMO fused to GST was purified from bacteria and used to pull down linear ubiquitin from cell lysate. Data are representative of three independent experiments. Download
Increase of intracellular linear ubiquitination by HOIP KR. (A) Flag-HOIP was expressed with or without HOIL-1L-HA in 293T cells. The proteasome inhibitor MG-132 was added to the indicated samples for 4 h at 20 µM prior to cell lysis and immunoblot analysis. (B) HOIP and HOIL-1L constructs were transfected into 293T cells and cells were lysed with NP-40 buffer 24 h posttransfection. Immunoprecipitation was performed separately for HOIL-1L using HA antibody and HOIP using Flag antibody. Data are representative of three independent experiments. (C) Quantitative PCR using cDNA reverse transcribed from A20.2J B-cell mRNA isolated from cells stimulated with LPS for the indicated times. Samples were normalized to ribosomal 18S RNA. Each data point represents three biological and three analytical replicates. (D) A total of 1 × 106 A20.2J cells were stimulated with 10 µg/ml LPS for the indicated times, fixed, and then stained for linear ubiquitin (M1Ub-APC). Cells were analyzed by flow cytometry. (E) As for panel D. HOIP+/+ and HOIP−/− cells were stimulated with LPS and stained for linear ubiquitin (M1Ub-APC). Linear-ubiquitin staining was compared between cells at the indicated times. (F) Cells were stimulated as described for panel E for direct comparison between isotype staining and linear-ubiquitin staining in HOIP+/+ and HOIP−/− cells. Data are representative of three independent experiments. Download
HOIP ubiquitination and HOIP-FRET constructs. (A) Various HOIP fusion constructs were expressed in 293T cells. Linear-ubiquitin levels were determined using immunoblot analysis. (B) HOIP-FRET constructs were transfected using the Neon system (Life Technologies) into A20.2J HOIP−/− cells. Twenty-four hours after transfection, cells were stimulated with LPS. Cells were lysed and analyzed by immunoblotting. Arrows indicate FRET-HOIP and HOIP-WT proteins. (C) Quantitative PCR using cDNA reverse transcribed from cells prepared as described for panel B. Download
Immunoblot analysis upon LPS or CD40L stimulation. (A) Various A20.2J cell lines were stimulated with 10 µg/ml of LPS, and cell lysates were subjected to immunoblotting at intervals up to 30 min after stimulation. (B) Indicated cells were stimulated with 10 µg/ml CD40L for up to 30 min, and cell lysates were subjected to immunoblotting. (C) HOIP+/+ and HOIP−/− cells were stimulated with TNF-α (20 ng/ml), and cell lysates were subjected to immunoblotting at intervals up to 8 h after stimulation. (D) Quantitative PCR using cDNA reverse transcribed from cells stimulated as described for panel C. (E) A total of 1 × 106 A20.2J cells were stained for cell surface receptors using phycoerythrin-conjugated CD40 (CD40-PE), TNFAR-PE, or isotype control-PE. Cells were analyzed by flow cytometry. CD40 staining served as a positive control. Download