

Abstract
Nitrogen mustard (NM) is a bifunctional alkylating agent that causes acute injury to the lung that progresses to fibrosis. This is accompanied by a prominent infiltration of macrophages into the lung and upregulation of proinflammatory/profibrotic cytokines including tumor necrosis factor (TNF)α. In these studies, we analyzed the ability of anti-TNFα antibody to mitigate NM-induced lung injury, inflammation, and fibrosis. Treatment of rats with anti-TNFα antibody (15 mg/kg, iv, every 9 days) beginning 30 min after intratracheal administration of NM (0.125 mg/kg) reduced progressive histopathologic alterations in the lung including perivascular and peribronchial edema, macrophage/monocyte infiltration, interstitial thickening, bronchiolization of alveolar walls, fibrin deposition, emphysema, and fibrosis. NM-induced damage to the alveolar-epithelial barrier, measured by bronchoalveolar lavage (BAL) protein and cell content, was also reduced by anti-TNFα antibody, along with expression of the oxidative stress marker, heme oxygenase-1. Whereas the accumulation of proinflammatory/cytotoxic M1 macrophages in the lung in response to NM was suppressed by anti-TNFα antibody, anti-inflammatory/profibrotic M2 macrophages were increased or unchanged. Treatment of rats with anti-TNFα antibody also reduced NM-induced increases in expression of the profibrotic mediator, transforming growth factor-β. This was associated with a reduction in NM-induced collagen deposition in the lung. These data suggest that inhibiting TNFα may represent an efficacious approach to mitigating lung injury induced by mustards.
Keywords: alveolar macrophages, lung injury, vesicant, fibrosis
Sulfur mustard (SM) and nitrogen mustard (NM), are cytotoxic vesicants developed as chemical warfare agents, which target the respiratory tract (Ekstrand-Hammarstrom et al., 2011; Malaviya et al., 2012; Razavi et al., 2013; Sunil et al., 2011a; Weinberger et al., 2011). In general, pulmonary complications following exposure to SM and NM are similar and include both acute (eg, chest tightness, hacking cough, rhinorrhea) and chronic (eg, bronchiolitis, emphysema, fibrosis) pathologies, which are major determinants of mortality and long-term morbidity (Balali-Mood and Hefazi, 2005; Razavi et al., 2013; Wang and Xia, 2007; Weinberger et al., 2011). In experimental models, SM and NM produce analogous histopathological alterations in the lung, most notably inflammation, perivascular and peribronchial edema, bronchiolization of alveolar walls, emphysema, bronchiectasis, squamous cell metaplasia, and fibrosis (Balali-Mood and Hefazi, 2005; Keyser et al., 2014; Malaviya et al., 2010, 2012; Sunil et al., 2011a). Toxicity is largely due to the lipophilic nature of these vesicants that allows them to rapidly penetrate tissues and cells and alkylate and cross-link cellular macromolecules including nucleic acids and proteins. This causes oxidative and nitrosative stress, impairment of cellular functioning, DNA damage, apoptosis, and autophagy (Malaviya et al., 2010, 2012; Shakarjian et al., 2010).
Evidence suggests that cytotoxic/proinflammatory mediators, released in large part by macrophages infiltrating into the lung in response to vesicants, play a role in the pathogenesis of pulmonary injury (Malaviya et al., 2010, 2012; Sunil et al., 2011a, 2014; Wigenstam et al., 2012). Of particular interest is the pleiotropic cytokine, tumor necrosis factor (TNF)α, which has been reported to increase in the lung following vesicant exposure (Emad and Emad, 2007; Ghabili et al., 2011; Malaviya et al., 2010; Mishra et al., 2012; Weinberger et al., 2011). TNFα is known to stimulate the release of reactive oxygen species (ROS) and reactive nitrogen species (RNS), deplete cellular glutathione, induce inflammatory cell and epithelial cell proliferation, cytotoxicity and apoptosis, and to promote pulmonary fibrosis (Aggarwal, 2003; Bradley, 2008; Mukhopadhyay et al., 2006a; Thrall et al., 1997). TNFα also induces focal accumulation of fibroblasts and collagen deposition (Piguet et al., 1990). The actions of TNFα are mediated by signaling through two functionally distinct cell surface receptors, TNFR1 (p55) and TNFR2 (p75) (Aggarwal, 2003; Bradley, 2008). Although TNFR1 mediates proinflammatory and cell death pathways associated with tissue injury, TNFR2 is involved in tissue repair and angiogenesis (Bradley, 2008). In previous studies, we reported that mice lacking TNFR1 are protected from lung toxicity induced by the half mustard, 2-chloroethyl ethyl sulfide (Sunil et al., 2011b). These findings prompted us to assess the effects of anti-TNFα antibody on lung injury induced by vesicants, using NM as a model. Our findings that inhibition of TNFα reduced lung injury, inflammation, and collagen deposition induced by NM suggest that blocking TNFα may represent an efficacious strategy to mitigate pulmonary toxicity induced by mustards.
MATERIALS AND METHODS
Animals and treatments
Male Wistar rats (175–250 g) were purchased from Harlan Laboratories (Indianapolis, Indiana). Animals were housed in filter top microisolation cages and provided food and water ad libitum; they received humane care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Rats were treated with control (PBS) or freshly prepared NM (0.125 mg/kg mechlorethamine hydrochloride, Sigma-Aldrich, St Louis, Missouri) by intratracheal instillation, as previously described (Sunil et al., 2011a). In earlier studies, we found that this dose of NM produces progressive pathologic changes in the lung, which are similar to those observed in humans after mustard exposure (Balali-Mood and Hefazi, 2005; Malaviya et al., 2012; Sunil et al., 2011a). Preparation and instillation of NM, which included the use of double gloves, safety glasses, and masks, were performed in a designated room under a chemical hood following Rutgers University Environmental Health and Safety guidelines. Rats were treated iv with vehicle (PBS) or recombinant mouse IgG2ακ monoclonal anti-rat TNFα antibody (Janssen Research & Development, Spring House, Pennsylvania), once every 9 days beginning 30 min after NM or control. Dose-response studies performed with anti-TNFα antibody demonstrated that optimal decreases in bronchoalveolar lavage (BAL) protein and cell accumulation were observed using 15 mg/kg administered once every 9 days (Supplementary Fig. S1 and not shown), and this was used in all subsequent experiments. Time-matched controls were run in all experiments.
Sample collection
Animals were euthanized 3, 7, or 28 days after administration of NM or control by ip injection of Sleepaway (2 ml/kg; Fort Dodge Animal Health, Fort Dodge, Iowa). These post exposure times were selected for analysis as they allowed us to assess acute lung injury, oxidative stress, initiation of tissue repair/remodeling, and fulminant fibrosis (Malaviya et al., 2012). For collection of BAL, the trachea was exposed and cannulated with a 16 gauge blunted needle. The left lobe was ligated and right lobes were infused with 8 ml of PBS with a constant infusion pressure maintained by a syringe. BAL was collected and cell and protein content assayed as previously described (Malaviya et al., 2012). For preparation of histological sections, the left lobe of the lung was instilled with 3% paraformaldehyde in PBS under equivalent inflation pressures using a syringe, removed and fixed in 3% paraformaldehyde overnight at 4°C, and then transferred to 50% ethanol. The remaining lung lobes were removed and stored at −80°C until analysis.
Histology
Sections (5 μm) were stained with hematoxylin and eosin or Gomori Trichrome stain and examined by light microscopy. Histopathologic changes in the lung were assessed blindly by a veterinary pathologist (LeRoy Hall, PhD, DVM). Semiquantitative grades (0–4) were assigned to specimens, with grade 0 indicating no changes; grade 1, minimal or small changes; grade 2, medium changes; grade 3, moderate changes; and grade 4, extensive changes, relative to PBS controls. Images were acquired using an Olympus VS-120 scanner or an Olympus light microscope using DP controller software (Ver. 3.3.1.292) (Olympus Corporation, Center Valley, Pennsylvania).
Immunohistochemistry
Paraffin sections (5 μm) were deparaffinized and endogenous peroxidase quenched using 3% hydrogen peroxide diluted in methanol. Antigen retrieval was performed by boiling the specimens for 10 min in 10 mM sodium citrate buffer (pH 6.0) containing 0.05% Tween-20. To block nonspecific binding, sections were incubated for 1–2 h at room temperature in buffer (Tris-buffered saline/Tween-20) containing horse, goat, or rabbit serum. Sections were then incubated overnight at 4°C in a humidified chamber with mouse monoclonal anti-CD68 (Bio-Rad Labs, Hercules, California; 1:400) or anti-CD163 antibodies (AbD Serotec, Raleigh, North Carolina; 1:400), or with rabbit polyclonal anti-heme oxygenase (HO)-1 (Assay Designs, Ann Arbor, Michigan; 1:1000), anti-transforming growth factor (TGF)-β (1:1500), anti-inducible nitric oxide synthase (iNOS) (1:1000) or anti-cyclooxygenase (COX)-2 (1:1500) antibodies (Abcam Inc, Cambridge, Massachusetts), rabbit monoclonal anti-CD11b antibody (Abcam Inc; 1:500) or anti-Ym1 antibody (Stemcell Technologies, Vancouver, Canada; 1:300), or goat polyclonal anti-galectin (Gal)-3 antibody (R&D Systems, Minneapolis, Minnesota; 1:4000), anti-TNFα antibody, raised against a peptide mapping at the N-terminus of mouse TNFα (Santa Cruz Biotechnology Inc., Dallas, Texas; 1:100), or appropriate IgG controls diluted in blocking buffer. Sections were then rinsed and incubated at room temperature for 30 min with biotinylated secondary antibody (Vectastain Elite ABC kit, Vector Labs, Burlingame, California). Binding was visualized using an avidin-biotinylated enzyme complex (Vectastain Elite ABC kit) with 3, 3′-diaminobenzidine as the substrate (Vector Labs). Random sections from at least 3 animals per treatment group were evaluated blindly by 2 individuals. Semiquantitative grades (0–4) were assigned, depending on the intensity of staining, with grade 0 indicating no staining; grade 1, minimal staining; grade 2, medium staining; grade 3, moderate staining; grade 4, extensive staining. The average grades were calculated and used to select representative sections for presentation (Supplementary Table S1).
Statistical analysis
Each experimental group consisted of 3–7 animals. Data were analyzed using GraphPad Prism V6.01 (GraphPad Software Inc, La Jolla, California). A one-way ANOVA with no matching between groups and Tukey’s multiple comparison, or unpaired t test (unequal variance) was used to analyze differences between groups. A p value of ≤.05 was considered statistically significant.
RESULTS
Effects of Anti-TNFα Antibody on NM-Induced Alterations in Lung Histology and Oxidative Stress
Consistent with previous studies (Malaviya et al., 2012), we found that NM caused progressive histopathological changes in the lung. These included multifocal inflammatory lesions, macrophage accumulation, perivascular and peribronchial edema, bronchial epithelial and goblet cell hyperplasia, interstitial thickening, bronchiolization of alveolar walls, and fibrin deposition (Fig. 1A and 1B, Supplementary Fig. S2, and Table 1). These changes were evident within 3 days of NM exposure and persisted for at least 7 days. Bronchiectasis, characterized by dilation and inflammation of the bronchi, squamous cell metaplasia, mesothelial cell proliferation, and emphysema-like changes were also observed in the lung at 3, 7, and 28 days post NM exposure (Table 1). Fibrotic changes in the lungs including multiple foci of trichrome staining in the alveolar septal wall were evident at 3 and 7 days post NM exposure; by 28 days prominent collagen deposits were noted in subpleural regions of the lung (Fig. 2, Table 1). Treatment of rats with anti-TNFα antibody reduced NM-induced structural alterations in the lung at all post exposure times (Fig. 1A and 1B Supplementary Fig. S2, and Table 1). Thus, NM-induced lesions were decreased in size and intensity, and fewer deposits of plasma proteins were noted in the lung parenchyma. Acute inflammation, edema, bronchoalveolar hyperplasia and hypertrophy, bronchiectasis, and goblet cells were also decreased, as well as NM-induced interstitial thickening, macrophage accumulation and increases in squamous cell metaplasia, mesothelial cell proliferation, and emphysema (Table 1). NM-induced peribronchial and parenchymal fibrotic alterations in the lung were also attenuated by anti-TNFα antibody. This was correlated with reduced trichrome staining (Fig. 2). Anti-TNFα antibody, by itself, had no significant effect on lung histology or collagen deposition in control (PBS treated) rats at any post exposure time point (Table 1 and not shown). Histopathological alterations in the lung induced by NM were associated with alveolar epithelial damage, measured by increases in BAL protein and cell content at all post NM exposure times (Fig. 3). This was suppressed by anti-TNFα antibody (Fig. 3).
FIG. 1.

Effects of anti-tumor necrosis factor (TNF)α antibody on nitrogen mustard (NM)-induced structural changes in the lung. Sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with H&E. Panel A, original magnification, ×4; Panel B, original magnification, ×200. Arrows, bronchial epithelium hypertrophy and hyperplasia; b, bronchiolization of alveolar septal wall; f, fibrin deposits; e, perivascular edema; em, emphysema; fib, fibrosis. Representative sections from 1 of 3–7 rats/treatment group are shown. CTL, 3 days post PBS exposure.
TABLE 1.
Summary of Effects of Anti-TNFα Treatment on NM-Induced Lung Histopathology
| CTL |
Days Post NM |
|||||||
|---|---|---|---|---|---|---|---|---|
| 3 |
7 |
28 |
||||||
| PBS | Anti-TNFα | PBS | Anti-TNFα | PBS | Anti-TNFα | PBS | Anti-TNFα | |
| No. of rats | 11 | 7 | 7 | 5 | 7 | 5 | 4 | 5 |
| Acute inflammation | 0.3 ± 0.1 | 0.1 ± 0.1 | 2.7 ± 0.2a | 1.4 ± 0.6 | 1.9 ± 0.5a | 1.2 ± 0.4 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Macrophage infiltration | 1.3 ± 0.1 | 1.0 ± 0.3 | 2.9 ± 0.1a | 1.8 ± 0.4 | 2.9 ± 0.1a | 2.2 ± 0.2 | 2.5 ± 0.5a | 2.0 ± 0.0 |
| Interstitial thickening | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.7 ± 0.2a | 1.8 ± 0.6 | 3.6 ± 0.2a | 2.2 ± 0.2b | 2.5 ± 0.3a | 1.4 ± 0.5 |
| Edema | 0.5 ± 0.2 | 0.4 ± 0.2 | 2.4 ± 0.2a | 1.8 ± 0.4 | 2.9 ± 0.1a | 2.6 ± 0.2 | 1.8 ± 0.3 | 0.7 ± 0.5 |
| Goblet cell hyperplasia | 0.0 ± 0.0 | 0.1 ± 0.1 | 2.3 ± 0.3a | 1.6 ± 0.6 | 2.6 ± 0.2a | 0.8 ± 0.4b | 1.5 ± 0.6a | 0.4 ± 0.4 |
| Bronchial epithelium hypertrophy and hyperplasia | 0.0 ± 0.0 | 0.1 ± 0.1 | 2.4 ± 0.2a | 1.4 ± 0.7 | 3.0 ± 0.2a | 1.0 ± 0.5b | 2.8 ± 0.3a | 1.2 ± 0.6 |
| Fibrin | 0.2 ± 0.1 | 0.0 ± 0.0 | 2.9 ± 0.1a | 1.6 ± 0.7 | 1.7 ± 0.6 | 1.0 ± 0.6 | 1.8 ± 0.8 | 1.0 ± 0.6 |
| Squamous cell metaplasia | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.4 ± 0.2a | 1.8 ± 0.7 | 3.0 ± 0.6a | 1.0 ± 0.6b | 1.5 ± 0.5 | 0.8 ± 0.4 |
| Mesothelial proliferation | 0.1 ± 0.1 | 0.0 ± 0.0 | 1.6 ± 0.2a | 0.6 ± 0.2 | 2.4 ± 0.4a | 0.8 ± 0.4b | 1.0 ± 0.4 | 0.2 ± 0.2 |
| Emphysema | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.1 ± 0.4a | 1.4 ± 0.6 | 3.4 ± 0.2a | 1.4 ± 0.5b | 2.8 ± 0.3a | 2.0 ± 0.4 |
| Bronchiectasis | 0.0 ± 0.0 | 0.1 ± 0.1 | 2.4 ± 0.2a | 1.6 ± 0.7 | 3.1 ± 0.5a | 0.8 ± 0.6b | 2.8 ± 0.3a | 2.2 ± 0.6 |
| Fibrosis | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.3 ± 0.3a | 0.6 ± 0.4b | 2.0 ± 0.4a | 1.4 ± 0.5 |
| Collagen deposits | 0.0 ± 0.0 | 0.0 ± 0.0 | 2.1 ± 0.2a | 1.4 ± 0.2 | 1.6 ± 0.2a | 1.0 ± 0.0 | 2.5 ± 0.3a | 1.2 ± 0.3b |
Histopathologic changes were quantified in H&E stained sections as described in the Materials and Methods. Collagen deposits were assessed in trichrome stained sections. CTL, combined means ± SE of data collected 3, 7, and 28 days post PBS control (CTL) treatment of rats administered PBS or PBS + Anti-TNFα. Days post NM, mean ± SE of NM or NM + Anti-TNFα treated rats 3, 7, or 28 days post NM exposure. One-way ANOVA comparing mean of each group to other groups using Tukey’s test was used to calculate p values.
aSignificantly (p ≤ .05) different from CTL.
bSignificantly different (p ≤ .05) from NM + PBS-treated rats at the same post NM exposure time.
FIG. 2.
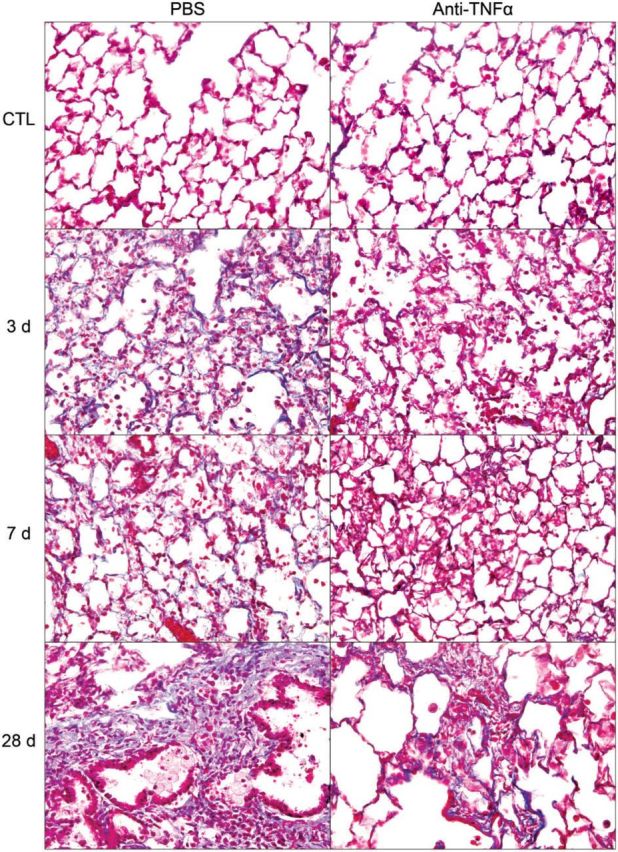
Effects of anti-TNFα antibody on NM-induced fibrosis. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with Gomori trichrome stain. Original magnification, ×400. Representative sections from 1 of 3–7 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 3.

Effects of anti-TNFα antibody on bronchoalveolar lavage (BAL) cell and protein content. BAL collected 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, was analyzed for BAL protein (upper panel) and cell content (lower panel). CTL, combined means ± SE of data collected 3, 7, and 28 days post PBS treatment of rats administered PBS or PBS + Anti-TNFα. Days post NM, mean ± SE of NM or NM + Anti-TNFα treated rats 3, 7, or 28 days post NM exposure. One-way ANOVA comparing mean of each group to other groups using Tukey’s test was used to calculate p values. aSignificantly (p ≤ .05) different from CTL; bSignificantly (p ≤ .05) different from PBS-treated rats at the same time post NM. CTL, 3 d post PBS exposure.
We also examined the effects of anti-TNFα antibody on NM-induced oxidative stress assessed by expression of the antioxidant, HO-1. In PBS-treated control rats, low-level expression of HO-1 was evident in alveolar macrophages (Fig. 4). Following NM exposure, an increase in HO-1+ macrophages was observed in the lung; this was most prominent 3 days post exposure. Anti-TNFα antibody reduced the effects of NM on macrophage expression of HO-1 at all post exposure times, with no effect on HO-1 expression in control rats (Fig. 4 and not shown).
FIG. 4.
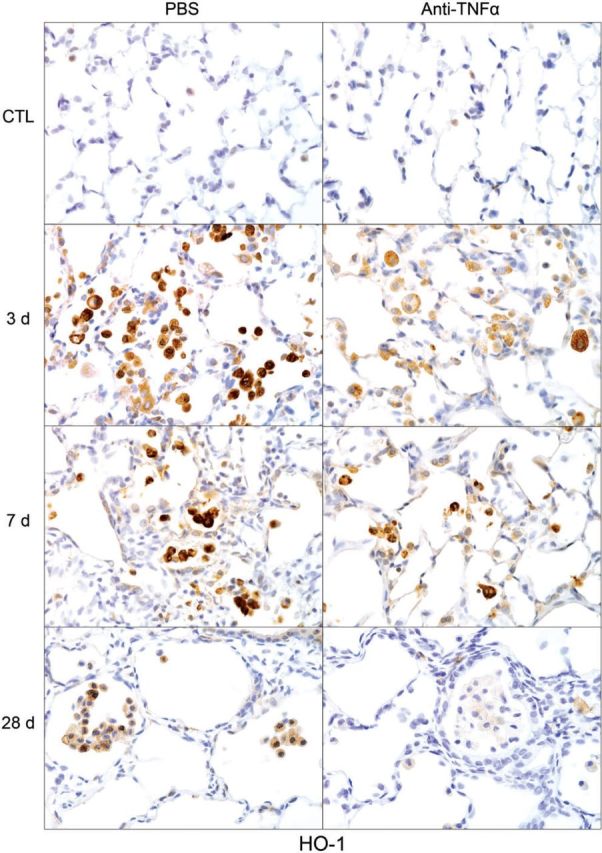
Effects of anti-TNFα antibody on NM-induced heme oxygenase (HO)-1 expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to HO-1. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–7 rats/treatment group are shown. CTL, 3 days post PBS exposure.
Effects of Anti-TNFα Antibody on NM-Induced Macrophage Accumulation and Inflammatory Mediator Expression in the Lung
CD11b is a β2 integrin expressed on infiltrating monocytes/macrophages (Mazzone and Ricevuti, 1995). Treatment of rats with NM resulted in increased numbers of CD11b+ macrophages in the lung, which were evident within 3 days (Fig. 5). With time following NM, CD11b+ macrophages became enlarged, and by 28 days, appeared mainly in clumps in the airways (Fig. 5). At this time, CD11b staining was localized predominantly in the cell membrane. Anti-TNFα antibody treatment had no significant effect on the accumulation of CD11b+ macrophages in the lung. To characterize these inflammatory cells, we assessed their expression of markers of proinflammatory/cytotoxic M1 (iNOS, COX-2, and TNFα) and antiinflammatory/profibrotic M2 (Ym1, Gal-3, CD68, and CD163) macrophages (Laskin et al., 2011; Martinez et al., 2008). In PBS-treated control animals, low-level expression of iNOS, COX-2, and TNFα was noted in macrophages, as well as in type II cells (Figs. 6–8). Following NM exposure, marked increases in iNOS+, COX-2+, and TNFα+ macrophages were observed in the lung within 3 days, remaining prominent for at least 7 days. At these times, low-level staining was also evident in alveolar epithelial cells. iNOS+ and COX-2+ macrophages were also noted in the lung at 28 days post NM exposure (Figs. 6 and 7). Treatment of rats with anti-TNFα antibody caused a marked reduction in numbers of iNOS+, COX-2+, and TNFα+ macrophages in the lung at all post NM times (Figs. 6–8). Anti-TNFα antibody treatment also resulted in reduced NM-induced expression of iNOS, COX-2, and TNFα in alveolar epithelial cells. In contrast, anti-TNFα antibody had no effect on expression of iNOS or COX-2 in lungs of PBS-treated control rats (Figs. 6–8 and not shown).
FIG. 5.
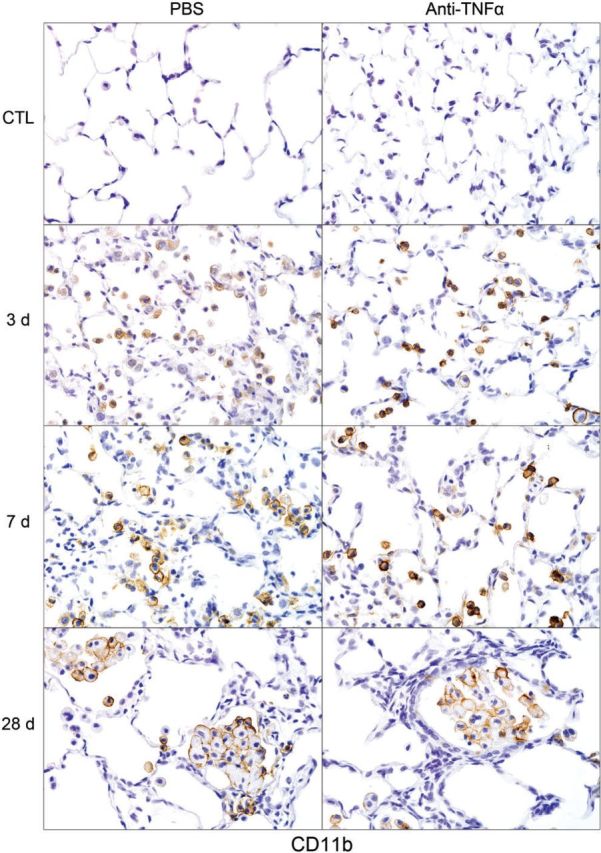
Effects of anti-TNFα antibody on NM-induced CD11b+ macrophage accumulation in the lung. Sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to CD11b. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 6.
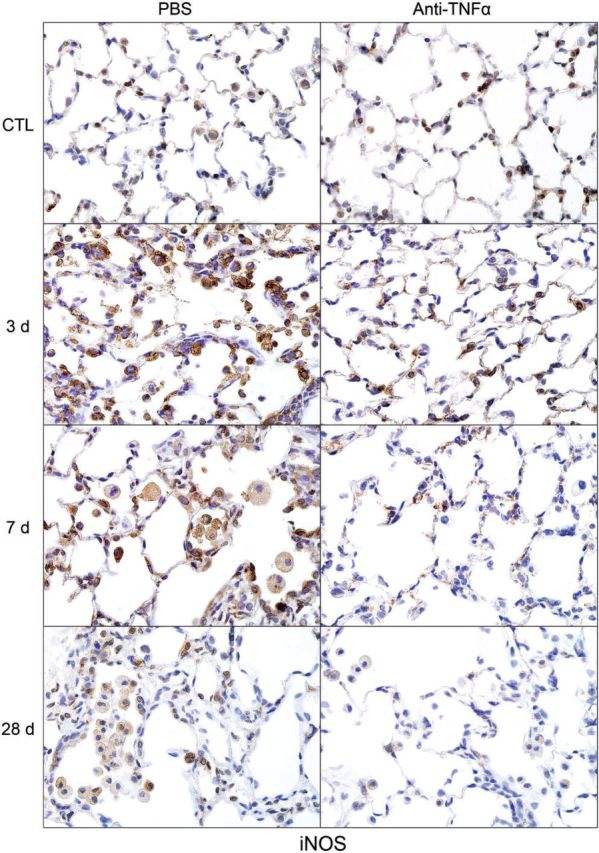
Effects of anti-TNFα antibody on NM-induced inducible nitric oxide synthase (iNOS) expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to iNOS. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–5 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 7.
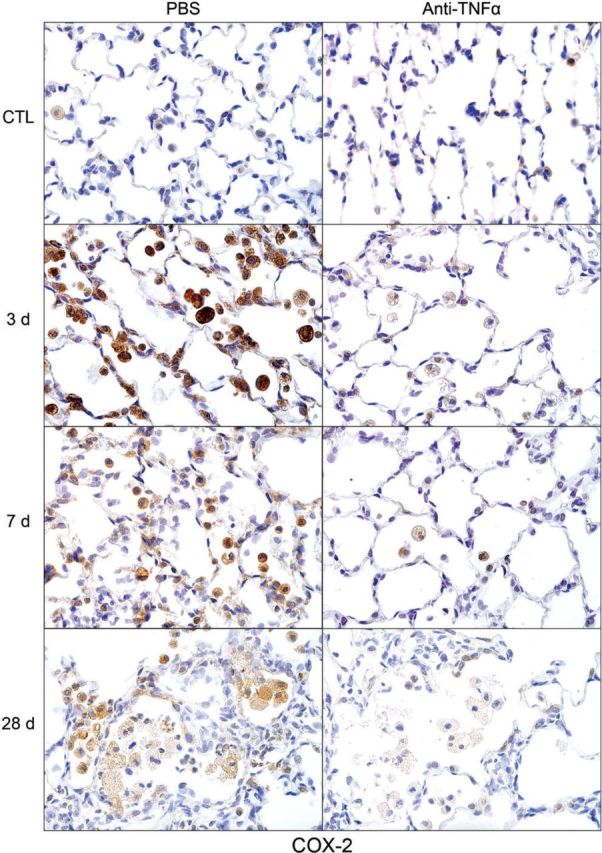
Effects of anti-TNFα antibody on NM-induced cyclooxygenase (COX)-2 expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to COX-2. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–5 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 8.
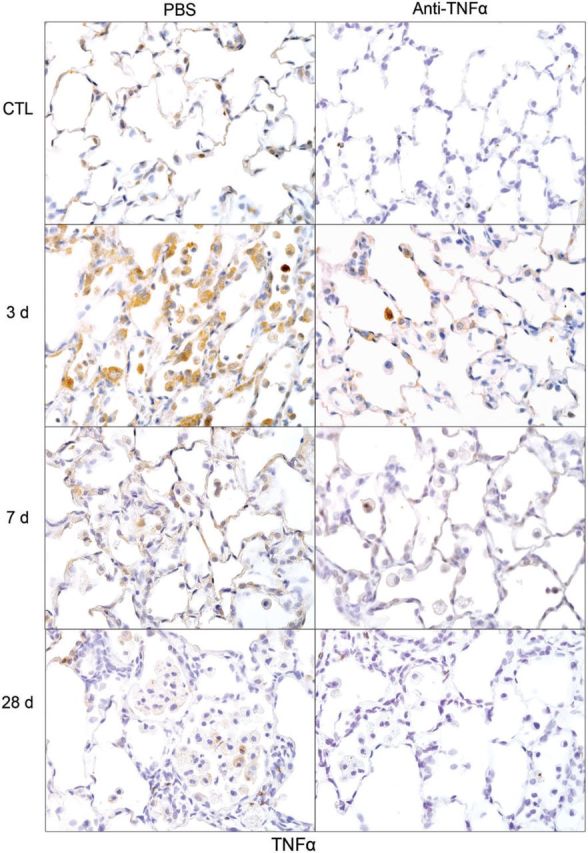
Effects of anti-TNFα antibody on NM-induced TNFα expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to TNFα. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–4 rats/treatment group are shown. CTL, 3 days post PBS exposure.
Following NM exposure, we also observed increased numbers of lung macrophages staining positively for M2 markers including Ym1, Gal-3, CD68, and CD163; however, the timing of their appearance in the tissue was distinct. Thus, although CD68+, Ym1+, and Gal-3+ macrophages were evident in the lung 3 days post NM, CD163+ macrophages were notable after 7–28 days. CD163+ macrophages were also smaller and less numerous than Ym1+, CD68+, or Gal-3+ macrophages (Figs. 9–12). At 28 days post NM, all positively staining M2 macrophages were enlarged, relative to macrophages in control lungs, and were foamy in appearance. Treatment of rats with anti-TNFα antibody resulted in a reduction in NM-induced increases in numbers of Ym1+ macrophages in the lung (Fig. 9). In contrast, numbers of CD163+ macrophages increased (Fig. 10), while numbers of Gal-3+ and CD68+ macrophages were unaltered (Figs. 11 and 12). Low numbers of Gal-3+ and CD68+ macrophages were identified in the lungs of control rats; these were unaffected by anti-TNFα antibody (Figs. 11 and 12 and not shown).
FIG. 9.
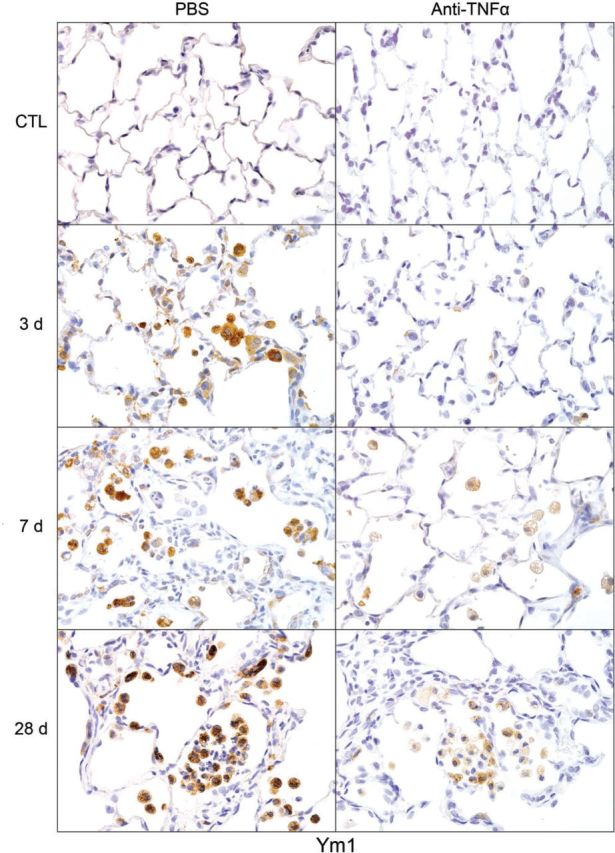
Effects of anti-TNFα antibody on NM-induced Ym1 expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to Ym1. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–5 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 10.
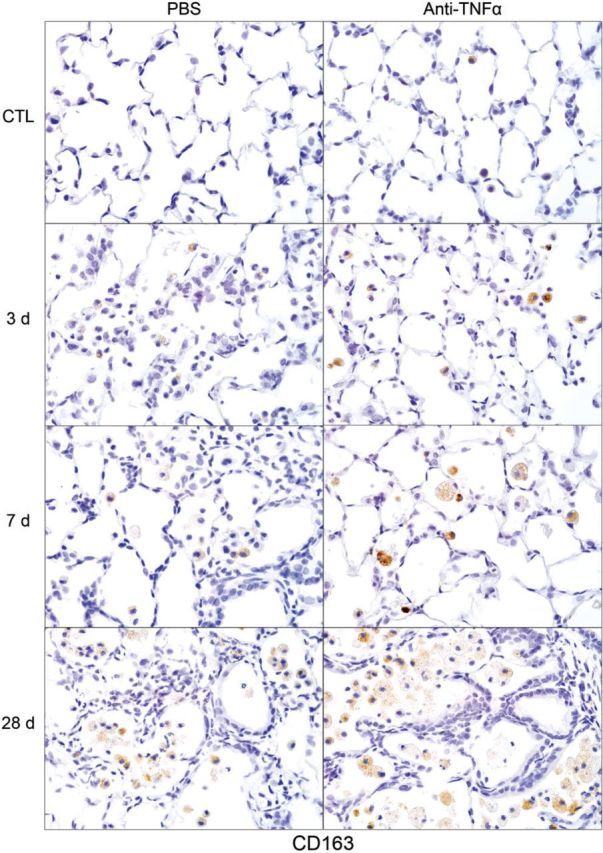
Effects of anti-TNFα antibody on NM-induced CD163 expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to CD163. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–4 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 11.
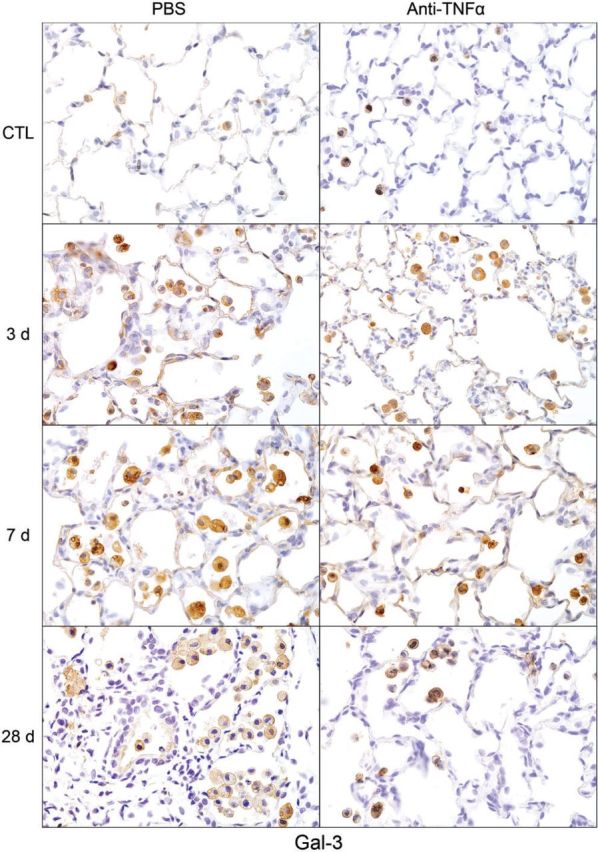
Effects of anti-TNFα antibody on NM-induced Gal-3 expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to Gal-3. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–4 rats/treatment group are shown. CTL, 3 days post PBS exposure.
FIG. 12.
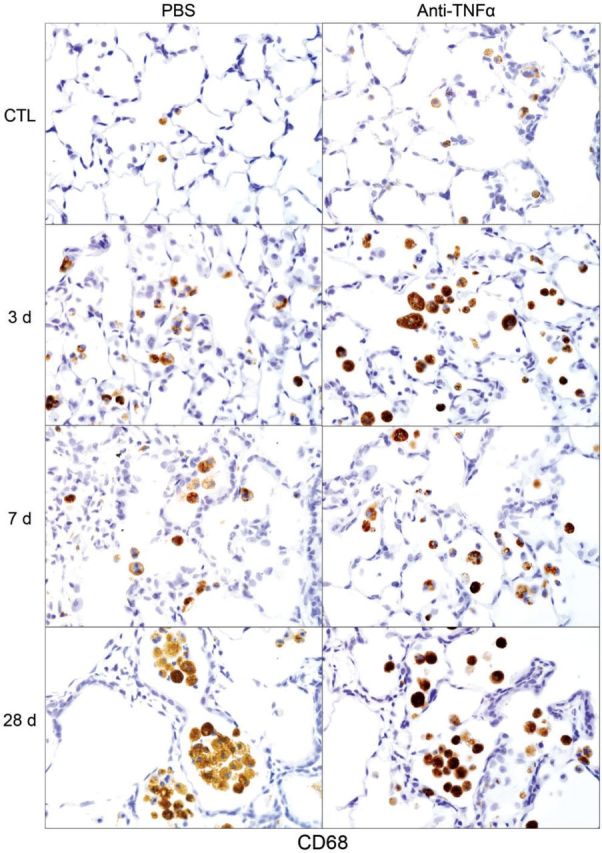
Effects of anti-TNFα antibody on NM-induced CD68 expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to CD68. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3–4 rats/treatment group are shown. CTL, 3 days post PBS exposure.
We next analyzed the effects of anti-TNFα antibody on NM-induced expression of the profibrogenic mitogen, TGF-β. In PBS-treated control rats, low levels of TGF-β were noted in alveolar macrophages and epithelial cells (Fig. 13). NM administration resulted in increased numbers of TGF-β+ macrophages in the lung at 3 days. TGF-β staining was also noted in epithelial cells at this time. Treatment of rats with anti-TNFα antibody reduced NM-induced TGF-β expression in both macrophages and epithelial cells with no effect on control rats (Fig. 13 and not shown).
FIG. 13.
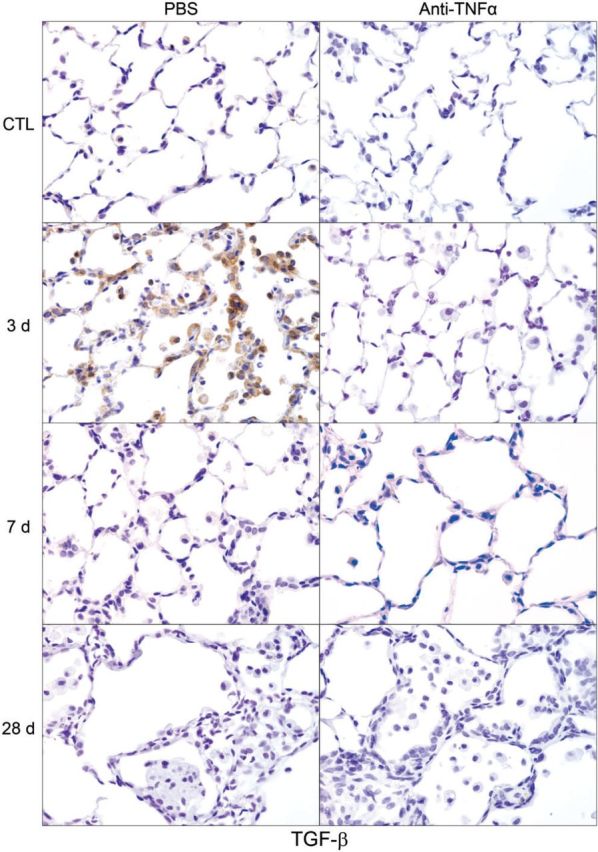
Effects of anti-TNFα antibody on NM-induced transforming growth factor (TGF)-β expression. Lung sections, prepared 3, 7, and 28 days after exposure of rats to NM or PBS control (CTL), followed by anti-TNFα antibody or PBS, were stained with antibody to TGF-β. Binding was visualized using a Vectastain kit. Original magnification, ×600. Representative sections from 1 of 3 rats/treatment group are shown. CTL, 3 days post PBS exposure.
DISCUSSION
TNFα is a proinflammatory cytokine generated mainly by macrophages in response to tissue injury (Gwyer Findlay and Hussell, 2012; Malaviya et al., 2010; Mukhopadhyay et al., 2006a; Sunil et al., 2011a). It has been implicated in acute and chronic inflammatory diseases including asthma, chronic bronchitis, chronic obstructive pulmonary disease, acute respiratory distress syndrome, and pulmonary fibrosis, pathologies associated with injury induced by mustards (Balali-Mood et al., 2011; Ding et al., 2002; Emad and Emad, 2007; Mukhopadhyay et al., 2006a; Shimabukuro et al., 2003). These studies demonstrate that anti-TNFα antibody, administered once every 9 days, was effective in reducing the development of lung injury, oxidative stress, and inflammation caused by exposure to NM; additionally NM-induced collagen deposition, a characteristic of fibrosis, was attenuated. This was correlated with a significant decrease in cytotoxic/proinflammatory macrophages in the lung, which are known to release mediators that contribute to pulmonary toxicity (Laskin et al., 2011). These findings suggest that targeting inflammatory macrophages and TNFα may prove beneficial in treating humans exposed to mustards.
Treatment of rats with anti-TNFα antibody beginning 30 min after NM significantly attenuated histopathological alterations in the lung including acute inflammation, fibrin deposition, bronchiectasis, alveolar and bronchiolar hyperplasia, and goblet cell alterations, along with interstitial thickening, squamous cell metaplasia, mesothelial proliferation, and emphysema. This was correlated with reduced alveolar-epithelial barrier dysfunction. Notably, anti-TNFα antibody significantly attenuated NM-induced collagen deposition, suggesting reduced fibrogenesis. Similar protective effects of anti-TNFα antibody have been described in models of lung injury induced by ozone, silica, or cigarette smoke, as well as in acute asthma and in bleomycin-induced pulmonary fibrosis, which are characterized by prominent inflammation, cytokine production, and collagen deposition (Abraham et al., 1995; Catal et al., 2015; Piguet and Vesin, 1994; Thrall et al., 1997). These findings are consistent with a key role of TNFα in acute and chronic inflammatory pathologies.
Oxidative stress is known to contribute to vesicant-induced lung injury (Keyser et al., 2014; Laskin et al., 2010; Mukhopadhyay et al., 2006b; Weinberger et al., 2011). In response to oxidative stress, cells upregulate HO-1, a microsomal antioxidant, that catalyzes heme degradation and protects cells from ROS-induced injury. Following NM exposure, we observed a persistent increase in HO-1+ macrophages in the lung, consistent with mustard-induced oxidative stress. Both numbers of HO-1+ macrophages and the intensity of HO-1 staining were reduced by anti-TNFα antibody indicating reduced oxidative stress. Evidence suggests a reciprocal relationship between the generation of ROS and TNFα. Thus, ROS promote the release of TNFα which in turn can stimulate the generation of ROS (Dapino et al., 1993; Kao et al., 2006; Tanner et al., 1992). Our findings that anti-TNFα antibody treatment reduced HO-1 in the lung may be due to decreased oxidative stress mediated in part by diminished levels of TNFα.
Macrophages are known to play a role in both the initiation and resolution of inflammatory responses to tissue injury (Laskin et al., 2011). These diverse biological activities are mediated by distinct macrophage subpopulations, broadly referred to as M1 and M2 macrophages. Thus, while early in the pathogenic response, M1 macrophages accumulate in the tissue and release mediators like ROS, RNS, and TNFα which promote tissue injury, later in the process, M2 macrophages appear, releasing mediators like interleukin-10 and TGF-β which suppress inflammation and initiate wound repair. Excessive release of mediators by M1 and M2 macrophages has been shown to contribute to disease pathogenesis and fibrosis (Laskin et al., 2011). Following NM exposure, we observed a persistent increase in CD11b+ inflammatory macrophages in the lung, consistent with a prolonged inflammatory response. Our findings that anti-TNFα antibody reduced lung inflammation but had no significant effect on CD11b+ macrophages in the lung suggest that TNFα exerted distinct effects on different macrophage subpopulations. To assess this possibility, we analyzed the effects of anti-TNFα antibody administration on M1 and M2 macrophages in the lung after NM. TNFα, iNOS, and COX-2 are proinflammatory proteins expressed by M1 macrophages (Laskin et al., 2011; Liu and Yang, 2013). In accord with previous reports (Malaviya et al., 2010, 2012; Sunil et al., 2011a), we observed an increase in TNFα+, iNOS+, and COX-2+ macrophages in the lung within 3 days of NM exposure, a response that persisted for 28 days, although at reduced levels. The accumulation of each of these M1 macrophage populations in the lung was suppressed by anti-TNFα antibody, in accord with the proinflammatory M1 inducing actions of TNFα (Aggarwal et al., 2014; Novak and Koh, 2013). Anti-TNFα antibody treatment has previously been reported to reduce numbers of M1 macrophages in lesional skin of patients with pustular psoriasis (Tang et al., 2012). Taken together, these findings suggest that the therapeutic response following anti-TNF antibody treatment may be due, in part, to downregulation of M1 macrophages.
Anti-TNFα antibody was also found to reduce numbers of Ym1+ M2 macrophages in the lung. In contrast, it had no major effect on Gal-3+ or CD68+ M2 macrophages, and numbers of CD163+ M2 macrophages increased. These data are consistent with the concept that there are multiple heterogeneous subpopulations of M2 macrophages that play distinct roles in controlling inflammation, wound repair, and fibrosis; moreover, they are regulated differently (Aggarwal et al., 2014; Novak and Koh, 2013). Findings that CD163+ macrophages are increased after anti-TNFα antibody treatment are in accord with reports that TNFα suppresses CD163 expression (Buechler et al., 2000; Fabriek et al., 2005). Increases in CD163+ M2 macrophages may contribute to the resolution of inflammation, oxidative stress and wound repair in anti-TNFα treated animals (Finn et al., 2012). Conversely, decreases in Ym1+ macrophages may be important in attenuating fibrosis. In this regard, depletion of lung macrophages during fibrogenesis has been shown to reduce pulmonary fibrosis, a response associated with reduced numbers of Ym1+ cells in the lung (Gibbons et al., 2011).
Excessive release of growth factors such as TGF-β by overactivated M2 macrophages can contribute to the development of fibrosis (Aggarwal et al., 2014; Novak and Koh, 2013). Following NM exposure, we observed increased expression of TGF-β at 3 days, which is consistent with early onset of fibrogenic changes in the lung. The fact that fibrosis continues to progress in NM-treated rats, despite reduced levels of TGF-β, suggests that other fibrogenic mediators such as connective tissue growth factor, platelet-derived growth factor, and/or fibroblast growth factor contribute to its development (Araya and Nishimura, 2010; Padilla-Carlin et al., 2011). Downregulation of TGF-β expression and decreased bleomycin-induced lung fibrosis have been reported following local delivery of TNFα antisense nucleotide (Luo et al., 2013). Similarly, we found that anti-TNFα antibody reduced TGF-β expression in the lung, as well as collagen deposition. These data suggest that TNFα plays a role in the development of vesicant-induced fibrosis and that TGF-β mediates early fibrogenic changes in the lung (Araya and Nishimura, 2010).
Exposure to vesicants causes both acute and long-term pathologic consequences in the lung, including cytotoxicity, disruption of tissue architecture, myofibroblast activation, fibroblast proliferation, and collagen deposition resulting in fibrosis (Weinberger et al., 2011). Previous studies have demonstrated that TNFα is involved in the development of each of these pathologies (Bradley, 2008; Miyazaki et al., 1995; Mukhopadhyay et al., 2006a; Thrall et al., 1997). These studies show that blocking TNFα mitigates the acute, as well as the long-term effects of NM on the respiratory system. In a rodent model of acute asthma, anti-TNFα antibody therapy was found to reduce the severity of tissue damage by suppressing inflammatory cell infiltration into the lung (Catal et al., 2015). Similarly, etanercept, a recombinant soluble TNF receptor that binds to and neutralizes soluble TNFα, has been reported to reduce the progression of idiopathic pulmonary fibrosis in humans (Raghu et al., 2008). In our studies, the efficacy of anti-TNFα antibody may be attributed to its unique ability to bind to both soluble and transmembrane TNFα, and to cross-link transmembrane TNF trimers (Tracey et al., 2008). Taken together, these studies support the notion that TNFα is an important mediator of vesicant-induced pulmonary injury. Targeting TNFα with antibodies may be an efficacious approach to mitigating the toxic effects of mustards in humans.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
This work was supported by National Institutes of Health Grants (U54AR055073, K01HL096426, R01ES004738, R01CA132624, and P30ES005022).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr David Reimer (DVM, Laboratory Animal Services, Rutgers University, New Jersey) for performing animal instillations and Dr David Shealy (Immunology Discovery Research, Janssen Research & Development, Spring House, Pennsylvania) for providing anti-TNFα antibody.
REFERENCES
- Abraham E., Jesmok G., Tuder R., Allbee J., Chang Y. H. (1995). Contribution of tumor necrosis factor-alpha to pulmonary cytokine expression and lung injury after hemorrhage and resuscitation. Crit. Care Med. 23, 1319–1326. [DOI] [PubMed] [Google Scholar]
- Aggarwal B. B. (2003). Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 3, 745–756. [DOI] [PubMed] [Google Scholar]
- Aggarwal N. R., King L. S., D'Alessio F. R. (2014). Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L709–L725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J., Nishimura S. L. (2010). Fibrogenic reactions in lung disease. Annu. Rev. Pathol. 5, 77–98. [DOI] [PubMed] [Google Scholar]
- Balali-Mood M., Afshari R., Zojaji R., Kahrom H., Kamrani M., Attaran D., Mousavi S. R., Zare G. A. (2011). Delayed toxic effects of sulfur mustard on respiratory tract of Iranian veterans. Hum. Exp. Toxicol. 30, 1141–1149. [DOI] [PubMed] [Google Scholar]
- Balali-Mood M., Hefazi M. (2005). The pharmacology, toxicology, and medical treatment of sulphur mustard poisoning. Fundam. Clin. Pharmacol. 19, 297–315. [DOI] [PubMed] [Google Scholar]
- Bradley J. R. (2008). TNF-mediated inflammatory disease. J. Pathol. 214, 149–160. [DOI] [PubMed] [Google Scholar]
- Buechler C., Ritter M., Orso E., Langmann T., Klucken J., Schmitz G. (2000). Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 67, 97–103. [PubMed] [Google Scholar]
- Catal F., Mete E., Tayman C., Topal E., Albayrak A., Sert H. (2015). A human monoclonal anti-TNF alpha antibody (adalimumab) reduces airway inflammation and ameliorates lung histology in a murine model of acute asthma. Allergol. Immunopathol. (Madr). 43, 14–18. [DOI] [PubMed] [Google Scholar]
- Dapino P., Dallegri F., Ottonello L., Sacchetti C. (1993). Induction of neutrophil respiratory burst by tumour necrosis factor-alpha; priming effect of solid-phase fibronectin and intervention of CD11b-CD18 integrins. Clin. Exp. Immunol. 94, 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M., Chen F., Shi X., Yucesoy B., Mossman B., Vallyathan V. (2002). Diseases caused by silica: Mechanisms of injury and disease development. Int. Immunopharmacol. 2, 173–182. [DOI] [PubMed] [Google Scholar]
- Ekstrand-Hammarstrom B., Wigenstam E., Bucht A. (2011). Inhalation of alkylating mustard causes long-term T cell-dependent inflammation in airways and growth of connective tissue. Toxicology 280, 88–97. [DOI] [PubMed] [Google Scholar]
- Emad A., Emad Y. (2007). Levels of cytokine in bronchoalveolar lavage (BAL) fluid in patients with pulmonary fibrosis due to sulfur mustard gas inhalation. J. Interferon Cytokine Res. 27, 38–43. [DOI] [PubMed] [Google Scholar]
- Fabriek B. O., Dijkstra C. D., van den Berg T. K. (2005). The macrophage scavenger receptor CD163. Immunobiology 210, 153–160. [DOI] [PubMed] [Google Scholar]
- Finn A. V., Nakano M., Polavarapu R., Karmali V., Saeed O., Zhao X., Yazdani S., Otsuka F., Davis T., Habib A., et al. (2012). Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 59, 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabili K., Agutter P. S., Ghanei M., Ansarin K., Panahi Y., Shoja M. M. (2011). Sulfur mustard toxicity: History, chemistry, pharmacokinetics, and pharmacodynamics. Crit. Rev. Toxicol. 41, 384–403. [DOI] [PubMed] [Google Scholar]
- Gibbons M. A., MacKinnon A. C., Ramachandran P., Dhaliwal K., Duffin R., Phythian-Adams A. T., van Rooijen N., Haslett C., Howie S. E., Simpson A. J., et al. (2011). Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 184, 569–581. [DOI] [PubMed] [Google Scholar]
- Gwyer Findlay E., Hussell T. (2012). Macrophage-mediated inflammation and disease: A focus on the lung. Mediators Inflamm. doi:10.1155/2012/140937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S. J., Wang D., Lin H. I., Chen H. I. (2006). N-acetylcysteine abrogates acute lung injury induced by endotoxin. Clin. Exp. Pharmacol. Physiol. 33, 33–40. [DOI] [PubMed] [Google Scholar]
- Keyser B. M., Andres D. K., Holmes W. W., Paradiso D., Appell A., Letukas V. A., Benton B., Clark O. E., Gao X., Ray P., et al. (2014). Mustard Gas Inhalation Injury: Therapeutic Strategy. Int. J. Toxicol. 33, 271–281. [DOI] [PubMed] [Google Scholar]
- Laskin D. L., Sunil V. R., Gardner C. R., Laskin J. D. (2011). Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 51, 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin J. D., Black A. T., Jan Y. H., Sinko P. J., Heindel N. D., Sunil V., Heck D. E., Laskin D. L. (2010). Oxidants and antioxidants in sulfur mustard-induced injury. Ann. N Y Acad. Sci. 1203, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Yang H. (2013). Modulation of macrophage activation and programming in immunity. J. Cell. Physiol. 228, 502–512. [DOI] [PubMed] [Google Scholar]
- Luo Y., Wang M., Pang Z., Jiang F., Chen J., Zhang J. (2013). Locally instilled tumor necrosis factor α antisense oligonucleotide contributes to inhibition of TH2-driven pulmonary fibrosis via induced CD4+CD25+Foxp3+ regulatory T cells. J. Gene Med. 15, 441–452. [DOI] [PubMed] [Google Scholar]
- Malaviya R., Sunil V. R., Cervelli J., Anderson D. R., Holmes W. W., Conti M. L., Gordon R. E., Laskin J. D., Laskin D. L. (2010). Inflammatory effects of inhaled sulfur mustard in rat lung. Toxicol. Appl. Pharmacol. 248, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R., Venosa A., Hall L., Gow A. J., Sinko P. J., Laskin J. D., Laskin D. L. (2012). Attenuation of acute nitrogen mustard-induced lung injury, inflammation and fibrogenesis by a nitric oxide synthase inhibitor. Toxicol. Appl. Pharmacol. 265, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez F. O., Sica A., Mantovani A., Locati M. (2008). Macrophage activation and polarization. Front. Biosci. 13, 453–461. [DOI] [PubMed] [Google Scholar]
- Mazzone A., Ricevuti G. (1995). Leukocyte CD11/CD18 integrins: Biological and clinical relevance. Haematologica 80, 161–175. [PubMed] [Google Scholar]
- Mishra N. C., Rir-Sima-Ah J., Grotendorst G. R., Langley R. J., Singh S. P., Gundavarapu S., Weber W. M., Pena-Philippides J. C., Duncan M. R., Sopori M. L. (2012). Inhalation of sulfur mustard causes long-term T cell-dependent inflammation: Possible role of Th17 cells in chronic lung pathology. Int. Immunopharmacol. 13, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y., Araki K., Vesin C., Garcia I., Kapanci Y., Whitsett J. A., Piguet P. F., Vassalli P. (1995). Expression of a tumor necrosis factor-α transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J. Clin. Invest. 96, 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Hoidal J. R., Mukherjee T. K. (2006a). Role of TNFα in pulmonary pathophysiology. Respir. Res. 7, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Rajaratnam V., Mukherjee S., Smith M., Das S. K. (2006b). Modulation of the expression of superoxide dismutase gene in lung injury by 2-chloroethyl ethyl sulfide, a mustard analog. J. Biochem. Mol. Toxicol. 20, 142–149. [DOI] [PubMed] [Google Scholar]
- Novak M. L., Koh T. J. (2013). Macrophage phenotypes during tissue repair. J. Leukoc. Biol. 93, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Carlin D. J., Schladweiler M. C., Shannahan J. H., Kodavanti U. P., Nyska A., Burgoon L. D., Gavett S. H. (2011). Pulmonary inflammatory and fibrotic responses in Fischer 344 rats after intratracheal instillation exposure to Libby amphibole. J. Toxicol. Environ. Health A. 74, 1111–1132. [DOI] [PubMed] [Google Scholar]
- Piguet P. F., Grau G. E., Vassalli P. (1990). Subcutaneous perfusion of tumor necrosis factor induces local proliferation of fibroblasts, capillaries, and epidermal cells, or massive tissue necrosis. Am. J. Pathol. 136, 103–110. [PMC free article] [PubMed] [Google Scholar]
- Piguet P. F., Vesin C. (1994). Treatment by human recombinant soluble TNF receptor of pulmonary fibrosis induced by bleomycin or silica in mice. Eur. Respir. J. 7, 515–518. [DOI] [PubMed] [Google Scholar]
- Raghu G., Brown K. K., Costabel U., Cottin V., du Bois R. M., Lasky J. A., Thomeer M., Utz J. P., Khandker R. K., McDermott L., et al. (2008). Treatment of idiopathic pulmonary fibrosis with etanercept: An exploratory, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 178, 948–955. [DOI] [PubMed] [Google Scholar]
- Razavi S. M., Ghanei M., Salamati P., Safiabadi M. (2013). Long-term effects of mustard gas on respiratory system of Iranian veterans after Iraq-Iran war: A review. Chin. J. Traumatol. 16, 163–168. [PubMed] [Google Scholar]
- Shakarjian M. P., Heck D. E., Gray J. P., Sinko P. J., Gordon M. K., Casillas R. P., Heindel N. D., Gerecke D. R., Laskin D. L., Laskin J. D. (2010). Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol. Sci. 114, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimabukuro D. W., Sawa T., Gropper M. A. (2003). Injury and repair in lung and airways. Crit. Care Med. 31, S524–S531. [DOI] [PubMed] [Google Scholar]
- Sunil V. R., Patel K. J., Shen J., Reimer D., Gow A. J., Laskin J. D., Laskin D. L. (2011a). Functional and inflammatory alterations in the lung following exposure of rats to nitrogen mustard. Toxicol. Appl. Pharmacol. 250, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil V. R., Patel-Vayas K., Shen J., Gow A. J., Laskin J. D., Laskin D. L. (2011b). Role of TNFR1 in lung injury and altered lung function induced by the model sulfur mustard vesicant, 2-chloroethyl ethyl sulfide. Toxicol. Appl. Pharmacol. 250, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil V. R., Vayas K. N., Cervelli J. A., Malaviya R., Hall L., Massa C. B., Gow A. J., Laskin J. D., Laskin D. L. (2014). Pentoxifylline attenuates nitrogen mustard-induced acute lung injury, oxidative stress and inflammation. Exp. Mol. Pathol. 97, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M. M., Spanou Z., Tang H., Schibler F., Pelivani N., Yawalkar N. (2012). Rapid downregulation of innate immune cells, interleukin-12 and interleukin-23 in generalized pustular psoriasis with infliximab in combination with acitretin. Dermatology 225, 338–343. [DOI] [PubMed] [Google Scholar]
- Tanner W. G., Welborn M. B., Shepherd V. L. (1992). Tumor necrosis factor-α and interleukin-1α synergistically enhance phorbol myristate acetate-induced superoxide production by rat bone marrow-derived macrophages. Am. J. Respir. Cell Mol. Biol. 7, 379–384. [DOI] [PubMed] [Google Scholar]
- Thrall R. S., Vogel S. N., Evans R., Shultz L. D. (1997). Role of tumor necrosis factor-α in the spontaneous development of pulmonary fibrosis in viable motheaten mutant mice. Am. J. Pathol. 151, 1303–1310. [PMC free article] [PubMed] [Google Scholar]
- Tracey D., Klareskog L., Sasso E. H., Salfeld J. G., Tak P. P. (2008). Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 117, 244–279. [DOI] [PubMed] [Google Scholar]
- Wang G. Q., Xia Z. F. (2007). Tissue injury by hot fluid containing nitrogen mustard. Burns 33, 923–926. [DOI] [PubMed] [Google Scholar]
- Weinberger B., Laskin J. D., Sunil V. R., Sinko P. J., Heck D. E., Laskin D. L. (2011). Sulfur mustard-induced pulmonary injury: Therapeutic approaches to mitigating toxicity. Pulm. Pharmacol. Ther. 24, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigenstam E., Jonasson S., Koch B., Bucht A. (2012). Corticosteroid treatment inhibits airway hyperresponsiveness and lung injury in a murine model of chemical-induced airway inflammation. Toxicology 301, 66–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.