

Abstract
MicroRNA-200 (miR-200) has emerged as a regulator of the PI3K/AKT pathway and cancer cell growth. It was reported that miR-200 can activate PI3K/AKT by targeting FOG2 (friend of GATA 2), which directly binds to the p85α regulatory subunit of PI3K. We found that miR-200 was elevated in early stage lung adenocarcinomas compared to normal lung tissues, and the expression of miR-200 promoted the tumor spheroid growth of lung adenocarcinoma cells. We show that AKT activation was essential for such oncogenic action of miR-200. However, depletion of FOG2 had little effect on AKT activation. By performing a reverse phase protein array, we found that miR-200 not only activated AKT, but also concomitantly inactivated S6K and increased IRS-1, an S6K substrate that is increased upon S6K inactivation. Depletion of IRS-1 partially inhibited the miR-200-dependent AKT activation. Taken together, our results suggest that miR-200 may activate AKT in lung adenocarcinoma cells through a FOG2-independent mechanism involving IRS-1. Our findings also provide evidence that increased miR-200 expression may contribute to early lung tumorigenesis, and AKT inhibitors may be useful for the treatment of miR-200-dependent tumor cell growth.
Keywords: microRNA-200, AKT, signaling, lung cancer
INTRODUCTION
MicroRNA-200 (miR-200) is a family of five clustered microRNAs (miR-141, 200a, 200b, 200c, and 429) that are highly expressed in epithelial tumor cells and play critical roles in multiple biological processes, including epithelial-mesenchymal transition (EMT) (1–4), cell growth (5–8), and invasion and metastasis (9–12). In the human genome, the five miR-200 family members are encoded by two miR-200 gene clusters, namely the miR-200b/a/429 and miR-200a/141 clusters. Like many other microRNAs, miR-200 can bind to specific sequences on its target mRNAs’ 3′UTR and silence their expression. One of the key miR-200 targets in tumor cells is the ZEB family of zinc-finger E-box binding transcription repressors that promote EMT (1–4). By targeting the ZEB factors, miR-200 maintains epithelial and polarity-related gene expression, decreases mesenchymal gene expression, and prevents EMT and tumor cell dissemination. On the other hand, ZEB factors can also bind to the promoter of miR-200 and repress its expression. Therefore, a reciprocal regulation between miR-200 and ZEB factors forms a double-negative feedback loop in tumor cells which enables the cells to effectively respond to both intracellular and environmental cues and to transit between epithelial and mesenchymal states (13–15).
PI3K/AKT pathway is a fundamental signaling pathway in tumor cells that promotes proliferation and survival, suppresses cell death, and regulates intracellular cytoskeletal remodeling that is required for tumor invasion (16, 17). Activation of PI3K leads to the generation of the second messenger PIP3 that activates phosphotidylinositol-dependent kinase PDK1, which in turn activates AKT and downstream mediators, including the mTOR and p70S6K signaling cascades. Notably, activated p70S6K can phosphorylate insulin receptor substrate IRS-1, an activator of PI3K, and induce IRS-1 degradation, followed by the suppression of PI3K activity. Thus, the inhibition of p70S6K can increase IRS-1 to feedback activate PI3K/AKT, which in turn drives tumor cells to acquire resistance to rapamycin (18, 19). Recently, others and we have shown that the PI3K/AKT pathway may be a novel target for the miR-200/ZEB1 axis and may be implicated in miR-200-dependent transformation, proliferation, and tumor dissemination (20–24). However, how miR-200 regulates the PI3K/AKT pathway remains incompletely understood. A previous report has shown that miR-200 activates PI3K by repressing its target gene FOG2, which acts as a suppressor of the assembly of PI3K by binding to the p85α regulatory subunit of PI3K (21).
Herein, we report that miR-200 promoted lung adenocarcinoma cell growth by activating AKT in a FOG2-independent manner. Our results show that miR-200 had a dichotomous effect on the expression and activation of several key components of the PI3K pathway, including AKT and S6K, in lung adenocarcinoma cell. We show evidence that miR-200 may activate AKT through a novel mechanism involving the inactivation of S6K and an increase of the S6K substrate IRS-1. In vivo, the expression of miR-200 in early stage lung adenocarcinomas is higher than that in adjacent normal lung tissues. These results suggest that AKT may be a useful target for treatment of miR-200-dependent lung tumor cell growth.
RESULTS
To explore the role of miR-200 in early lung tumorigenesis, we quantitated its expression in 33 pairs of early stage (stage I and II) lung adenocarcinomas and adjacent normal lung tissues (Table 1). Our results showed that the expression levels of both miR-200a and miR-200c (which represent the miR-200b/a/429 and miR-200a/141 clusters, respectively) were higher in tumors than those in normal lung tissues (Figure 1A and 1B), suggesting an oncogenic role for miR-200 in early stage lung adenocarcinoma. In support of such a role, the expression of miR-200 in the 344SQ lung adenocarcinoma cells, which do not express miR-200 (11), significantly promoted their tumor spheroid growth in matrigel culture (Figure 1C).
Table 1.
Demographic and clinical information for the 33 paired frozen lung adenocarcinoma and adjacent normal lung tissues used in this study
| Total number of tissues: 41 pairs | |
|---|---|
| Age at surgery | |
| Mean | 67 |
| Range | 47–89 |
| Sex | |
| Male | 11 (33%) |
| Female | 22 (67%) |
| Race | |
| Caucasian | 30 (91%) |
| Unknown | 3 (9%) |
| Smoking status | |
| Never | 16 (48%) |
| Former | 10 (30%) |
| Current | 7 (22%) |
| Stage | |
| I | 16 (48%) |
| II | 17 (52%) |
Figure 1.
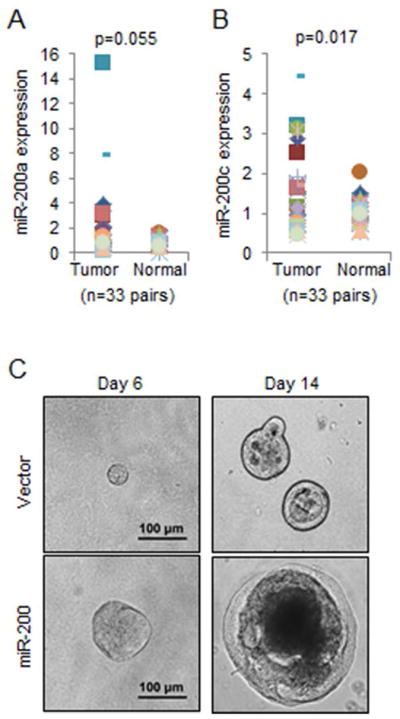
miR-200 promotes lung adenocarcinoma cell growth. (A and B) The expression levels of miR-200a (A) and miR-200c (B) were quantitated by qPCR and normalized to that of RNU24 (internal control for small non-coding RNAs). (C) Light microscopic images of tumor spheroids formed by 344SQ lung adenocarcinoma cells that stably express either an empty vector or miR-200b/a/429.
Previously, we have shown that PI3K signaling is essential for early lung tumorigenesis in mouse models of human lung adenocarcinoma (25, 26). To examine whether miR-200 regulates the PI3K signaling pathway, we performed western blotting for several key components of this pathway and found that miR-200 activated AKT phosphorylation but inactivated the phosphorylation of p70S6K and S6, a substrate of p70S6K (Figure 2A). On the contrary, the expression of the miR-200 repressor ZEB1 in 393P lung adenocarcinoma cells had a completely opposite effect on these molecules: it inactivated AKT phosphorylation but activated the phosphorylation of both p70S6K and S6K (Figure 2B). These results demonstrate that miR-200 distinctly regulates AKT and p70S6K in lung adenocarcinoma cells. To understand the physiological role of miR-200-mediated AKT activation, we treated the tumor spheroids formed by cells that express miR-200 with an AKT inhibitor (GSK2141795) or an mTOR inhibitor (rapamycin). The results showed that miR-200 significantly promoted tumor spheroid growth, which can be effectively inhibited by these inhibitors (Figure 2C). In a sharp contrast, these inhibitors had little effect on cells that do not express miR-200 (Figure 2C). These results are consistent with the observation that AKT was inactive in 344SQ cells (Figure 2A) and suggest that the activation of AKT by miR-200 promotes tumor cell growth.
Figure 2.

miR-200 and ZEB1 reversely regulate key signaling components of the PI3K/AKT pathway. (A and B) Western blotting of pAKT (pS473), AKT, pp70S6K (pT389), p70S6K, pS6 (S235/S236), and S6 for 344SQ lung adenocarcinoma cells that express either an empty vector or a miR-200b/a/429 plasmid (A) and 393P lung adenocarcinoma cells that express either an empty vector or ZEB1 (B). (C) Tumor spheroids formed by 344SQ lung adenocarcinoma cells expressing either an empty vector or miR-200b/a/429 were treated with DMSO, GSK2141795 (2 μM), and rapamycin (500 nM). Tumor spheroids were counted and expressed as means ± S.D. * indicates that the t-test’s p value is less than 0.05 (statistically significant).
A recent study showed that FOG2 is a direct target of miR-200 and acts as a PI3K antagonist by binding to its regulatory subunit p85α (21). Thus, it is possible that miR-200 activates AKT phosphorylation by repressing FOG2. Unexpectedly, our results showed that expression of miR-200 only marginally decreased the level of FOG2 (Figure 3A), and knockdown of FOG2 failed to activate AKT (Figure 3B) and had little effect on cell growth (Figure 3C), indicating that FOG2 is not required for miR-200-dependent AKT activation in lung adenocarcinoma cells. To understand the mechanism by which miR-200 activates AKT, we performed a reverse phase protein array (RPPA) for cells that stably express miR-200. Consistent with our above-mentioned results in Figure 2, the RPPA data showed that miR-200 strongly activated AKT but inactivated p70S6K (Figure 4A; indicated by arrows). Interestingly, the data also revealed a significant increase of IRS-1 in response to miR-200 expression (Figure 4A; indicated by the symbol #). As IRS-1 is a well-characterized p70S6K substrate that is degraded upon p70S6K-mediated phosphorylation (18, 19), we posit that miR-200 increases IRS-1 by inactivating p70S6K. Importantly, IRS-1 can activate PI3K by recruiting p85α, suggesting that it may be required for AKT activation by miR-200. In support of this idea, knockdown of either IRS-1 (Figure 4B) or p85α (Figure 4C) suppressed the phosphorylation of AKT. Notably, knockdown of IRS-1 only partially inhibited AKT phosphorylation, suggesting that miR-200 may activate AKT through multiple mediators, including IRS-1.
Figure 3.
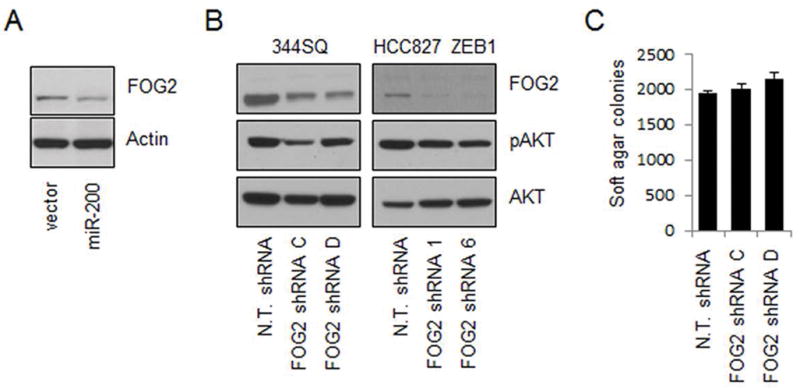
FOG2 is not required for AKT activation. (A) Western blotting of FOG2 and Actin (as a loading control) for 393Pzeb1 lung adenocarcinoma cells. (B) Western blotting of FOG2, pAKT (pS473), and AKT for 344SQ (left panels) and HCC827zeb1 (right panels) lung adenocarcinoma cells that stably express either non-targeting shRNA (N.T. shRNA) or FOG2 specific shRNAs. (C) Knockdown of FOG2 had no effect on 344SQ soft agar colony growth.
Figure 4.

IRS-1 is partially involved in miR-200-dependent AKT activation. (A) RPPA array for 344SQ lung adenocarcinoma cells that stably express either an empty vector or miR-200b/a/429 plasmid. * and ** indicate that the t-test’s p values are less than 0.05 or 0.01, respectively (statistically significant or very significant). (B) Western blotting of IRS-1, pAKT (pS473), AKT and Actin for 344SQ lung adenocarcinoma cells expressing an empty vector or miR-200b/a/429 that transfected with control non-targeting siRNA (N.T. siRNA) or IRS-1 siRNA. (C) Western blotting of p85α, pAKT (pS473), and Actin showing knockdown of p85α suppresses AKT phosphorylation.
DISCUSSION
In this report, we studied the impact of miR-200 on the PI3K/AKT signaling and the mechanism of miR-200-mediated AKT activation. A previous report showed that FOG2, a transcription co-factor for the GATA family of transcription factors (27, 28), is a miR-200 target that antagonizes PI3K/AKT by binding to the p85α PI3K subunit in liver cancer cells (21). However, our results (Figure 3) showed that the depletion of FOG2 in lung cancer cells had little effect on AKT activation, suggesting that miR-200 may utilize cellular context-dependent mechanisms to regulate AKT in different types of cancer cells.
Interestingly, our data (Figures 2 and 4) showed that miR-200 concomitantly induced the activation of AKT and inactivation of p70S6K, demonstrating a dichotomous effect of miR-200 on the PI3K/AKT signaling. More importantly, we found that IRS-1 was increased by miR-200 and partially involved in AKT activation. These findings provide evidence for a novel mechanism by which miR-200 activates AKT in lung cancer cells. Given that p70S6K phosphorylates IRS-1 and induces its degradation (18, 19), we postulate that the inactivation of p70S6K by miR-200 in lung cancer cells may feedback activate AKT by increasing IRS-1 (Figure 5).
Figure 5.
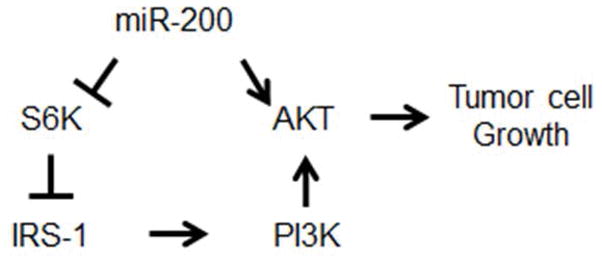
Schematic for the mechanism of miR-200-mediated AKT activation and tumor cell growth.
Our results also showed that miR-200 expression level was higher in lung adenocarcinomas than that in normal lung tissues. Functional studies showed that miR-200 promoted tumor spheroid growth and that the AKT/mTOR inhibitors inhibited the tumor spheroid growth of cells expressing miR-200, but not of those that do not express miR-200, suggesting that the presence or absence of miR-200 in lung cancer cells may induce a switch of AKT’s growth regulating function. Therefore, our data also suggest that AKT antagonists may be useful for treatment of miR-200-dependent lung tumors.
MATERIALS AND METHODS
Cell culture and transfection: all cells are cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and in a humidified 5% CO2 incubator. Synthesized siRNAs (Cell Signaling) were transiently transfected into the cells using RNAiMAX reagent from Life Technologies, and gene expression was examined by western blotting 48 hours post transfection. To stably knock down target gene expression, specific shRNA expression plasmids (Sigma) were transfected into the cells using lipofectamine 2000 transfection reagent (Invitrogen). After 48 hours, cells were re-suspended and put into culture medium containing 15 μg/ml puromycin (Sigma). Cells that stably express shRNA plasmids were generally acquired within 6–8 weeks post antibiotic selection.
Reagents: rabbit anti-pAKT (9271), AKT (9272), p70S6K (9202), pS6 (2211), S6 (2212), and IRS-1 (2382), and mouse-anti-pp70S6K (9206), and siRNA against mouse IRS-1 (6346) were from Cell Signaling. Goat anti-Actin (sc-1616), rabbit anti-FOG2 (sc-10755), and mouse anti-p85α (sc-1637) were from Santa Cruz.
Human tissues
Human tissues were acquired from Mayo Clinic lung tissue registry with protocols approved by the Mayo Clinic internal review board (IRB) and human tissue subcommittee.
Reverse phase protein array (RPPA assay): the RPPA assay was performed by University of Texas M.D. Anderson Cancer Center RPPA core facility.
Quantitative RT-PCR: the expression levels of the five microRNA-200 members were examined by quantitative RT-PCR using TaqMan assays from Applied Biosystems. Briefly, total RNA was extracted from cells using the miRNeasy kit (217004) from Qiagen. 5 ng total RNA was subjected to reverse transcription, followed by quantitative PCR using a 7500 fast qPCR machine (Applied Biosystems). The levels of miR-200 members were then normalized to that of an internal control small non-coding RNA snoRNA-135.
Protein lysate preparation and Western blotting: cells were lysed in ice-cold RIPA buffer containing protein inhibitor cocktail (Sigma), 1 mM PMSF, and 1 mM orthovanadate, homogenized three times by 25 gauge needles, and centrifuged at 12,000 g for 15 minutes to remove cell debris and non-soluble components. The resulted supernatant (total protein lysate) was immediately subjected to protein concentration quantitation using a BCA protein assay kit from Pierce. SDS PAGE electrophoresis and immunoblotting were performed as we described before (Yang et al., 2014).
Soft agar colony formation and tumor spheroid growth assay: single cell suspension was prepared by Trypsin-EDTA digestion, followed by re-suspension of cells into RPMI-1640 medium containing 10% FBS and aspiration of cells through a 25 gauge needle. For tumor spheroid growth assay, cells (500 cells per well) were seeded in matrigel coated 8-well NuncLink chambers (Fisher Scientific) and incubated for two weeks. For soft agar formation assay, cells (10,000 cells per well of 6-well plates) were mixed well with 0.3% agar, which was then plated on 0.6% agar in RPMI 1640 medium containing 20% FBS. After two weeks, the colonies of tumor cells were visualized by MTT staining and counted and expressed as mean ± SD of the number of colonies.
Acknowledgments
GRANT SUPPORT
This work was partly supported by the Mayo Clinic foundation (CA197202RELIEF) and National Cancer Institute (R21CA184817, R01CA80127, and R01CA84354).
Footnotes
AUTHOR CONTRIBUTION
Y.Y. conceived the project. P.Y. and Q. L. contributed to the acquisition of human tissues and clinical information. L.G., J.W., T.Z., and Y.Y. performed experiments, collected and analyzed data, and wrote the manuscript. Y.Y. supervised the project.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The authors declare that there is no conflict of interest.
References
- 1.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008 Jun;9(6):582–9. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008 May;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 3.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008 May 30;283(22):14910–4. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008 Apr 1;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009 Dec;11(12):1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 6.Xia W, Li J, Chen L, Huang B, Li S, et al. Mol Cell Biochem. 2010 Nov;344(1–2):261–6. doi: 10.1007/s11010-010-0550-2. MicroRNA-200b regulates cyclin D1 expression and promotes S-phase entry by targeting RND3 in HeLa cells. [DOI] [PubMed] [Google Scholar]
- 7.Xiao Y, Wang J, Yan W, Zhou Y, Chen Y, et al. Dysregulated miR-124 and miR-200 expression contribute to cholangiocyte proliferation in the cholestatic liver by targeting IL-6/STAT3 signalling. J Hepatol. 2014 Oct 30; doi: 10.1016/j.jhep.2014.10.033. pii: S0168-8278(14)00799-5. [DOI] [PubMed] [Google Scholar]
- 8.Yu J, Ohuchida K, Mizumoto K, Sato N, Kayashima T, et al. MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol Cancer. 2010 Jun 28;9:169. doi: 10.1186/1476-4598-9-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humphries B, Wang Z, Oom AL, Fisher T, Tan D, et al. MicroRNA-200b targets protein kinase Cα and suppresses triple-negative breast cancer metastasis. Carcinogenesis. 2014 Oct;35(10):2254–63. doi: 10.1093/carcin/bgu133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng F, Jiang J, Yu Y, Tian R, Guo X, et al. Direct targeting of SUZ12/ROCK2 by miR-200b/c inhibits cholangiocarcinoma tumourigenesis and metastasis. Br J Cancer. 2013 Dec 10;109(12):3092–104. doi: 10.1038/bjc.2013.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roybal JD, Zang Y, Ahn YH, Yang Y, Gibbons DL, et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol Cancer Res. 2011 Jan;9(1):25–35. doi: 10.1158/1541-7786.MCR-10-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams LV, Veliceasa D, Vinokour E, Volpert OV. miR-200b inhibits prostate cancer EMT, growth and metastasis. PLoS One. 2013 Dec 31;8(12):e83991. doi: 10.1371/journal.pone.0083991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008 Oct 1;68(19):7846–54. doi: 10.1158/0008-5472.CAN-08-1942. [DOI] [PubMed] [Google Scholar]
- 14.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009 Sep 15;23(18):2140–51. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. 2013 Feb 15;132(4):745–54. doi: 10.1002/ijc.27708. [DOI] [PubMed] [Google Scholar]
- 16.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006 Aug;7(8):606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 17.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015 Jan;15(1):7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santoni M, Pantano F, Amantini C, Nabissi M, Conti A, et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim Biophys Acta. 2014 Apr;1845(2):221–31. doi: 10.1016/j.bbcan.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Sheppard K, Kinross KM, Solomon B, Pearson RB, Phillips WA. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit Rev Oncog. 2012;17(1):69–95. doi: 10.1615/critrevoncog.v17.i1.60. [DOI] [PubMed] [Google Scholar]
- 20.Becker LE, Takwi AA, Lu Z, Li Y. The role of miR-200a in mammalian epithelial cell transformation. Carcinogenesis. 2015 Jan;36(1):2–12. doi: 10.1093/carcin/bgu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyun S, Lee JH, Jin H, Nam J, Namkoong B, et al. Conserved MicroRNA miR-8/miR-200 and its target USH/FOG2 control growth by regulating PI3K. Cell. 2009 Dec 11;139(6):1096–108. doi: 10.1016/j.cell.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 22.Park JT, Kato M, Yuan H, Castro N, Lanting L, et al. FOG2 protein down-regulation by transforming growth factor-β1-induced microRNA-200b/c leads to Akt kinase activation and glomerular mesangial hypertrophy related to diabetic nephropathy. J Biol Chem. 2013 Aug 2;288(31):22469–80. doi: 10.1074/jbc.M113.453043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Virtakoivu R, Pellinen T, Rantala JK, Perälä M, Ivaska J. Distinct roles of AKT isoforms in regulating β1-integrin activity, migration, and invasion in prostate cancer. Mol Biol Cell. 2012 Sep;23(17):3357–69. doi: 10.1091/mbc.E12-03-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Ahn YH, Chen Y, Tan X, Guo L, et al. ZEB1 sensitizes lung adenocarcinoma to metastasis suppression by PI3K antagonism. J Clin Invest. 2014 Jun;124(6):2696–708. doi: 10.1172/JCI72171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Iwanaga K, Raso MG, Wislez M, Hanna AE, et al. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-ras-induced lung cancer. PLoS One. 2008 May 21;3(5):e2220. doi: 10.1371/journal.pone.0002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwanaga K, Yang Y, Raso MG, Ma L, Hanna AE, et al. Pten inactivation accelerates oncogenic K-ras-initiated tumorigenesis in a mouse model of lung cancer. Cancer Res. 2008 Feb 15;68(4):1119–27. doi: 10.1158/0008-5472.CAN-07-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cantor AB, Orkin SH. Coregulation of GATA factors by the Friend of GATA (FOG) family of multitype zinc finger proteins. Semin Cell Dev Biol. 2005 Feb;16(1):117–28.36. doi: 10.1016/j.semcdb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Chlon TM, Crispino JD. Combinatorial regulation of tissue specification by GATA and FOG factors. Development. 2012 Nov;139(21):3905–16. doi: 10.1242/dev.080440. [DOI] [PMC free article] [PubMed] [Google Scholar]