

Abstract
Ischemic preconditioning (IPC) is a robust neuroprotective phenomenon whereby brief ischemic exposure confers tolerance to a subsequent ischemic challenge. IPC has not been studied selectively in CNS white matter (WM), although stroke frequently involves WM. We determined whether IPC is present in WM and, if so, its mechanism. We delivered a brief in vivo preconditioning ischemic insult (unilateral common carotid artery ligation) to 12- to 14-week-old mice and determined WM ischemic vulnerability [oxygen–glucose deprivation (OGD)] 72 h later, using acutely isolated optic nerves (CNS WM tracts) from the preconditioned (ipsilateral) and control (contralateral) hemispheres. Functional and structural recovery was assessed by quantitative measurement of compound action potentials (CAPs) and immunofluorescent microscopy. Preconditioned mouse optic nerves (MONs) showed better functional recovery after OGD than the non-preconditioned MONs (31 ± 3 vs 17 ± 3% normalized CAP area, p < 0.01). Preconditioned MONs also showed improved axon integrity and reduced oligodendrocyte injury compared with non-preconditioned MONs. Toll-like receptor-4 (TLR4) and type 1 interferon receptor (IFNAR1), key receptors in innate immune response, are implicated in gray matter preconditioning. Strikingly, IPC-mediated WM protection was abolished in both TLR4−/− and IFNAR1−/− mice. In addition, IPC-mediated protection in WM was also abolished in IFNAR1fl/fl LysMcre, but not in IFNAR1fl/fl control, mice. These findings demonstrated for the first time that IPC was robust in WM, the phenomenon being intrinsic to WM itself. Furthermore, WM IPC was dependent on innate immune cell signaling pathways. Finally, these data demonstrated that microglial-specific expression of IFNAR1 plays an indispensable role in WM IPC.
SIGNIFICANCE STATEMENT Ischemic preconditioning (IPC) has been studied predominantly in gray matter, but stroke in humans frequently involves white matter (WM) as well. Here we describe a novel, combined in vivo/ex vivo mouse model to determine whether IPC occurs in WM. It does. Using genetically altered mice, we identified two innate immune cell receptors, Toll-like receptor 4 and type 1 interferon receptor (IFNAR1), that are required for IPC-mediated protection in WM. Furthermore, using microglia-targeted IFNAR1 knockdown, we demonstrate that interferon signaling specifically in microglia is essential for this protection. The discovery of IPC as an intrinsic capability of WM is novel and important. This is also the first in vivo demonstration that cell-type-specific expression of an individual gene plays an indispensable role in IPC-mediated protection.
Keywords: interferon, ischemic preconditioning, microglia, toll-like receptor-4, white matter
Introduction
Ischemic preconditioning (IPC) is a brief period of cerebral ischemia that confers transient tolerance to subsequent prolonged ischemic challenge (Kirino et al., 1996). The protective effect of IPC is robust, requires 24–72 h to develop, lasts weeks to months, and has been confirmed in numerous animal models (Kirino et al., 1996). Elucidating the mechanisms of IPC is a critical challenge in stroke research (Gidday, 2006). Clinical studies suggest that stroke patients who suffered a recent transient ischemic attack in the same vascular distribution as their stroke have reduced infarct volumes and improved clinical outcomes (Moncayo et al., 2000; Wegener et al., 2004). These findings imply that a human clinical correlate to experimental IPC exists (Moncayo et al., 2000; Wegener et al., 2004). IPC requires gene activation and de novo protein synthesis and is achieved by the attenuation of injury-inducing mechanisms (Gidday, 2006). IPC genetically reprograms the response of the brain to ischemia, forming the basis of ischemic tolerance (Stenzel-Poore et al., 2003, 2004).
Recent work has pointed to an important role for immune cell signaling in IPC. Specifically, IPC-mediated neuroprotection is attenuated markedly in mice deficient in key immune cell signaling molecules/pathways, including Toll-like receptor-4 (TLR4) (Pradillo et al., 2009) and interferon regulatory factors (IRFs) (Stevens et al., 2011). Furthermore, pretreatment of mice with either pharmacologic agonists targeting TLRs (Bahjat et al., 2011; Gesuete et al., 2012; Leung et al., 2012; Packard et al., 2012) or preexposure to interferon-β itself (Stevens et al., 2011) induces robust neuroprotection against subsequent cerebral ischemic injury. The role of microglia in IPC-mediated neuroprotection is unknown, although these CNS-resident innate immune cells are increasingly recognized as contributing to repair and regeneration from neural injury (Michell-Robinson et al., 2015). The function of innate immune signaling generally, and microglia specifically, in white matter (WM) ischemic injury and recovery is also unknown.
Stroke typically affects both gray matter (GM) and WM, although some strokes affect WM exclusively (Mohr, 2004). WM contains axons and glia, including astrocytes, oligodendrocytes, and microglia, without any neuronal cell bodies, dendrites, or conventional synapses (Matute et al., 2013). WM comprises approximately half of the brain volume in humans, a threefold to fourfold increase over rodents (Matute et al., 2013). Consequently, experimental brain ischemia in rodents is overwhelmingly an insult of GM (∼85% of stroke volume) (STAIR, 1999; Sozmen et al., 2012; Matute et al., 2013). Moreover, any WM that is rendered ischemic is in close proximity to GM, making it difficult to study in isolation. To circumvent this obstacle, “pure” CNS WM preparations are used, such as the acutely isolated corpus callosum (Tekkök and Ransom, 2004) or mouse optic nerve (MON; Stys et al., 1991; Bakiri et al., 2008; Sozmen et al., 2012; Matute et al., 2013). As is the case for experimental brain ischemia, most rodent models of IPC use infarct volume (predominantly GM) as their primary outcome measure. In some cases, the effects of IPC on specific CNS structures, such as the hippocampus (Tian et al., 2008; DeFazio et al., 2009) and retina (Fernandez et al., 2009; Dvoriantchikova et al., 2010), have been reported, but these structures are also predominantly GM. One important exception to models with limited WM expansion is the recent demonstration of pharmacologic preconditioning with a TLR agonist in Rhesus macaques (Bahjat et al., 2011). The study by Bahjat et al. (2011) provided proof of concept that TLR-mediated preconditioning can protect nonhuman primate brains, with volumetric ratios of GM/WM similar to humans, from cerebrovascular injury.
An intriguing open question is whether or not IPC protects WM. To address this, we developed a novel model of IPC in WM. After in vivo induction of IPC, we determined the extent of recovery from ischemic insult in acutely isolated (ex vivo) MONs. By allowing both the initial IPC pulse and the subsequent cellular, immunological, and gene expression changes to occur in situ, our aim was to more closely mimic the in vivo physiology that occurs in WM after IPC.
Materials and Methods
Animals.
All experiments were done in accordance with the University of Washington Institutional Animal Care and Use Committee. All mice were adult male, 12–14 week old, C57BL/6 wild-type (WT), TLR4 systemic knock-out (TLR4−/−; (Hoshino et al., 1999), type 1 interferon receptor systemic knock-out (IFNAR1−/−; Müller et al., 1994), cell-type-specific IFNAR1 knockdown (IFNAR1fl/fl LysMcre; Kamphuis et al., 2006; Diamond et al., 2011), or IFNAR1fl/fl control (Kamphuis et al., 2006; Diamond et al., 2011). The IFNAR1fl/fl LysMcre mouse strain has been characterized previously (Prinz et al., 2008) and shown to have a high degree of IFNAR1 knockdown in myeloid cells compared with the IFNAR1fl/fl control strain. To confirm cell-type specificity of IFNAR1 knockdown in the IFNAR1fl/fl LysMcre mice, we used flow cytometry to quantify differences in specific binding of an anti-IFNAR1 antibody in primary microglia derived from either IFNAR1fl/fl or IFNAR1fl/fl LysMcre mice. In unstimulated cells, we found a 93% reduction in expression of cell-surface IFNAR1 in the IFNAR1fl/fl LysMcre microglia relative to IFNAR1fl/fl (Fig. 1A). We also confirmed specificity of our anti-IFNAR1 antibody by demonstrating that treatment with IFNβ (1000 U/ml for 24 h) eliminated 89% of specific binding in IFNAR1fl/fl-derived microglia (Fig. 1A). We also quantified cell-type-specific knockdown of ifnar1 expression in ex vivo sorted cortical microglia from naive 12- to 16-week-old IFNAR1fl/fl and IFNAR1fl/fl LysMcre mice. We found ∼70% reduction in steady-state levels of ifnar1 mRNA in microglia isolated from the IFNAR1fl/fl LysMcre mice (Fig. 1B).
Figure 1.
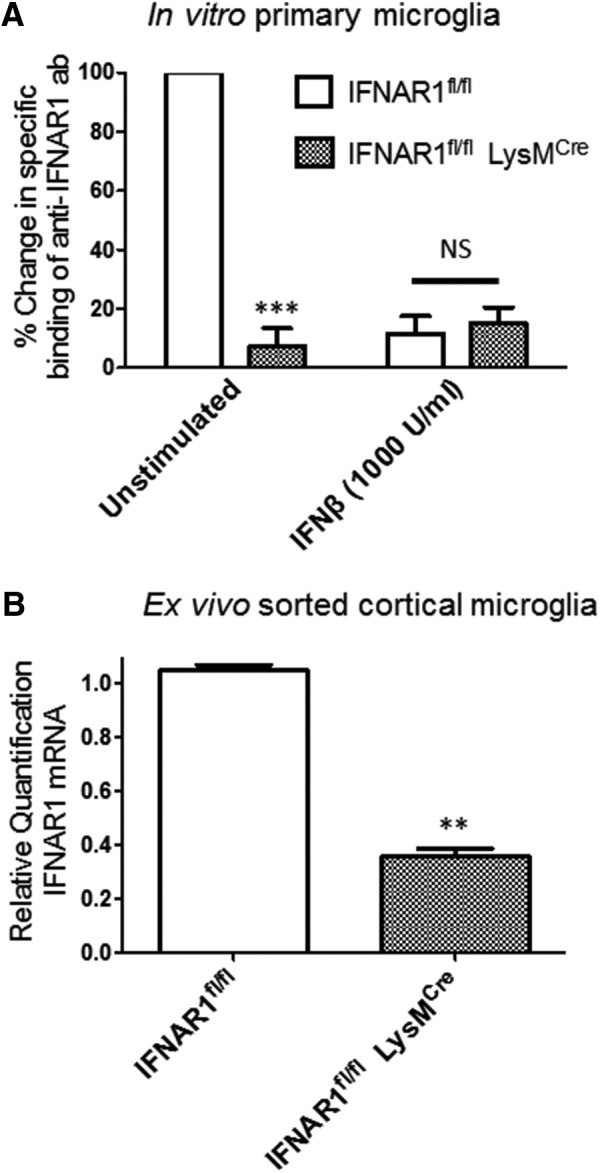
IFNAR1 knockdown in both in vitro cultured and ex vivo sorted microglia from IFNAR1fl/fl and IFNAR1fl/fl LysMcre mice. A, Primary microglia were derived from either IFNAR1fl/fl or IFNAR1fl/fl LysMcre mice as described in Materials and Methods, serum starved, and then either unstimulated or treated with IFNβ (1000 U/ml) for 24 h. Normalized specific binding (mean ± SEM) of anti-IFNAR1 antibody was quantified by flow cytometry as described in Materials and Methods. ***p < 0.001 compared with unstimulated IFNAR1fl/fl microglia. n = 3 experiments. B, Results of qRT-PCR quantifying steady-state levels of IFNAR1 mRNA in ex vivo sorted cortical microglia from IFNAR1fl/fl or IFNAR1fl/fl LysMcre mice. **p < 0.01 compared with IFNAR1fl/fl. n = 9 mice, 3 separate ex vivo flow cytometry preparations.
Combined in vivo/ex vivo model of IPC in MON.
A schematic diagram outlines our experimental plan (Fig. 2). The ophthalmic artery (a branch of the internal carotid artery) supplies the MON with collateral blood flow through the orbit from the middle meningeal artery (a branch of the external carotid artery; Hayreh, 2006). Unilateral MON ischemia was produced by transient (15 min) occlusion of the ipsilateral common carotid artery (CCA). The contralateral MON received continuous blood flow and, along with MONs from sham-operated mice, served as the control nerves not receiving IPC. The MONs, ipsilateral and contralateral, were removed 72 h after IPC, an optimal time delay for inducing IPC-mediated neuroprotection (Stenzel-Poore et al., 2003; Zhang et al., 2008).
Figure 2.

Schematic diagram showing combined in vivo/ex vivo IPC experimental paradigm. WT, TLR4−/−, IFNAR1−/−, IFNAR1fl/fl, or IFNAR1fl/fl LysMcre 12- to 14-week-old male mice (all on C57BL/6 background) were subjected to either 15 min CCAO (our IPC pulse) or sham surgery. Seventy-two hours later, mice were killed by CO2 exposure and MONs were dissected out and placed in an interface perfusion chamber. Electrodes were put in place, and MONs were allowed to equilibrate in control conditions for 60 min, followed by 45 min exposure to OGD. Oxygen and glucose were then restored to baseline levels. CAPs were monitored before, during, and after OGD (for details, see Materials and Methods). After 5 h in normoxic/normoglycemic control (“recovery”) conditions, selected MONs were collected and processed for immunohistochemistry (A). The experimental groups are named in bold font according to the following: (1) position of the MON in question relative to CCAO surgery (i.e., ipsilateral or contralateral); (2) their initial in vivo exposure (i.e., sham surgery or CCAO); and (3) their subsequent ex vivo exposure [i.e., control (Con) or OGD] (B). IHC, Immunohistochemistry.
For the IPC procedure, anesthesia was induced by inhalation of 2–4% isoflurane (Animal Health International) in an NO2/O2 70/30% mixture and maintained by inhalation of 1.5–2.0% isoflurane. Body temperature was monitored throughout surgery (via a rectal probe) and maintained at 36–38°C using a heating blanket. A 2 cm midline incision was made on the ventral surface of the neck, and the right CCA was isolated and ligated with a 6.0 silk suture. After 15 min of CCA occlusion (CCAO), the CCA ligation was released to allow reperfusion. Sham-operated mice underwent the same procedure, except that the CCA was only exposed but not ligated/occluded. Laser Doppler flowmetry was used to monitor cerebral blood flow (CBF) in the middle cerebral artery (MCA) territory throughout the procedure and during the immediate recovery period. CCAO typically induced ∼50% reductions in ipsilateral MCA CBF, whereas release of the ligating suture resulted in reperfusion CBF values typically near 120% of the initial baseline. We excluded animals (<5% of total) with outlying CBF profiles (i.e., CBF measurements during either occlusion or reperfusion that were more than 2 SDs away from the respective mean values). Seventy-two hours after surgery, mice were killed using CO2, and their optic nerves were immediately dissected free for additional study ex vivo. The number of animals per experimental group was chosen based on a modified power analysis with improved stopping rules for design of efficient small sample experiments (Fitts, 2010) with >80% power and estimated effect size of 50% for IPC.
Electrophysiology.
Ipsilateral and contralateral MONs were placed in an interface perfusion chamber (Harvard Apparatus) and maintained at 37°C (Fig. 2; Hamner et al., 2011). MONs were superfused with artificial CSF (ACSF) containing the following (in mmol/L): 125 NaCl, 3.0 KCl, 2.0 CaCl2, 2.0 MgSO4 · 7H2O, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.45. The ACSF was bubbled with an O2-free gas mixture (95%N2/5%CO2). A humidified gas mixture of 95%O2/5% CO2 continuously aerated the chamber under control conditions (i.e., at all times except during ischemic insult). Two sets of suction electrodes backfilled with ACSF were used for stimulating and recording from each of the two MONs. The stimulating electrode was attached to the retinal end of the nerve, and the recording electrode was attached to the chiasmatic end of the nerve. This electrode arrangement means that evoked compound action potentials (CAPs) travel in the orthodromic direction. The suction electrodes were positioned carefully to ensure maximum seal resistance judged by stable CAP amplitudes over the 60 min preinjury recording period. Stimulus pulse strength (30 μs duration; Isostim 520; WPI) was adjusted to evoke the maximum CAP and then increased another 25% to ensure supramaximal stimulation throughout the recording period; supramaximal CAPs were elicited every 30 s. The recording electrode was connected to an amplifier (model SR 560; Standford Research Systems), filtered at 30 kHz, and acquired at 20 kHz.
To ensure stable baseline readings, MONs were allowed to equilibrate for 30–60 min before injury was induced (Stys et al., 1991). Ischemia was simulated by oxygen–glucose deprivation (OGD) for 45 min. In brief, ACSF was switched to a solution lacking glucose and replaced by 10 mm sucrose to maintain osmolarity, and anoxia was achieved by switching to 95%N2/5%CO2. Control conditions (i.e., normal ACSF and O2) were restored quickly at the end of the injury period.
Data analysis.
MON function was determined quantitatively by integrating the area under the CAP. Data were acquired online (Digidata 1440A; Molecular Devices) using proprietary software (Clampex; Molecular Devices). CAP area was calculated using pClamp (Molecular Devices) and was normalized by averaging the baseline CAP area over a period of 15 min and setting this value to 1.0. As such, the normalized CAP area at any time is proportional to the relative number of functioning axons (Buchtal and Rosenfalck, 1966; Cummins et al., 1979; Stys et al., 1991). Data are presented as the mean ± SEM of several normalized traces. Statistical significance was determined by ANOVA at 60 min of recovery. p values <0.05 were considered significant.
Glutamate and aspartate measurements.
Release of the excitatory amino acids (EAAs) glutamate and aspartate from MON was measured by collecting the superfusate during 60 min OGD and quantified using HPLC (Ye et al., 2003; Yang et al., 2014). Experiments were designed to monitor glutamate and aspartate release simultaneously from one to two pairs of MONs. Glutamate and aspartate measurements, normalized to respective baseline release levels, were made from MONs treated identically (with the exception of a slightly longer period of OGD) to those studied electrophysiologically, and the results were plotted against time. The rate and release pattern for both EAAs was monitored for 30 min before OGD and was continued for at least 30 min after OGD. Glutamate and aspartate concentrations were measured from the bath perfusate passing over the MONs in aliquots taken every 2–4 min (Ye et al., 2003). Actual amounts of glutamate and aspartate measured in nanomoles were determined by comparing the measurements with a standard sample.
Immunofluorescent microscopy.
MONs were processed for immunofluorescent microscopy as described previously (Baltan et al., 2008). In brief, MONs were placed in 4% paraformaldehyde in PBS overnight, cryoprotected, frozen, and sectioned into 12-μm-thick slices. Sections were blocked and permeabilized. All primary antibodies were applied in the same solution. Phosphorylated neurofilaments were stained with anti-neurofilament H phosphorylated monoclonal (clone SMI-31) antibody (Covance), astrocytes with anti-glial fibrillary acidic protein (GFAP) antibody (ImmunoStar), microglia/macrophages with anti-ionized calcium-binding adapter molecule 1 (Iba1) antibody (Wako Chemicals), and oligodendrocytes with anti-adenomatous polyposis coli (APC) antibody CC-1 clone (Calbiochem/EMD Biosciences). With regard to the latter antibody (specifically the CC-1 clone), it should be noted that the staining pattern for this antibody does not reflect endogenous expression of APC, but it does stain oligodendrocytes effectively (Bhat et al., 1996; Lang et al., 2013). For each condition, immunolabeling was examined and quantified in MONs from three to six animals and in two to three adjacent sections from each animal. Primary antibodies were used at a dilution of 1:15 for GFAP, 1:100 for Iba1, and 1:200 for APC. Sections were incubated in primary antibodies together overnight at 4°C, washed, and exposed to appropriate secondary antibodies (1:100 dilution).
Cell counts/stereology and axonal integrity.
Images were obtained with a 40× objective on a fluorescent microscope (Nikon Optiphot-2). NIH ImageJ was used to overlay 5 × 6 grids (200 × 250 μm) onto the images of tissue. Border rules were used so that cells crossing a grid line would be counted in a systematic manner. Cell numbers were quantified by counting every other square in the grid. Cells were only counted if the antibody in question overlaid with the nuclear marker (DAPI) and adhered to the border rules. Pyknotic nuclei were excluded from viable cell counts. Antibody-labeled cell counts were presented as a percentage of the total cell number (including pyknotic nuclei). Structural axon injury assessment was performed using an established composite scoring system and SMI-31 staining for phosphorylated neurofilament in WM (Adams et al., 1983; Tekkök and Goldberg, 2001). Axon integrity was quantified by a number between 0 and 12 (the “axonal integrity score”) according to the following criteria: one to three was “assigned for each of the following parameters: brightness of labeling intensity, preservation of anatomically parallel and linearly organized axonal fibers, continuity in individual fibers, and lack of axonal head and bulb formation” (Adams et al., 1983; Tekkök and Goldberg, 2001). Each tissue section was scored independently by two blinded observers. Statistical significance was determined by Mann–Whitney U test (for nonparametric data) for Figure 4 and by unpaired t test for Figures 5 and 6.
Figure 4.

IPC attenuates axonal degradation after OGD. Axons were fixed and stained for SM-31 (green), an antibody that binds phosphorylated neurofilaments, as well as the nuclear marker DAPI (blue). The 40× objective images in A were obtained with an immunofluorescent microscope as described in Materials and Methods. Higher-magnification images in B are digital zoom. Axonal morphology was quantified systematically as described in Materials and Methods (maximum possible score is 12). IPC pulse alone did not significantly alter axonal morphology, but after OGD, there was a significant increase in the amount of axonal injury. Arrows indicate head/bulb formation in the injured fields after OGD (right panels in A). Axonal integrity after OGD was significantly greater in the Ipsi-OGD (7.5 ± 0.5) versus Contra-OGD (4.8 ± 0.4) MONs; **p < 0.01 (C). Scale bars: A, ∼30 μm; B, ∼8 μm. n = 3 MONs per group.
Figure 5.
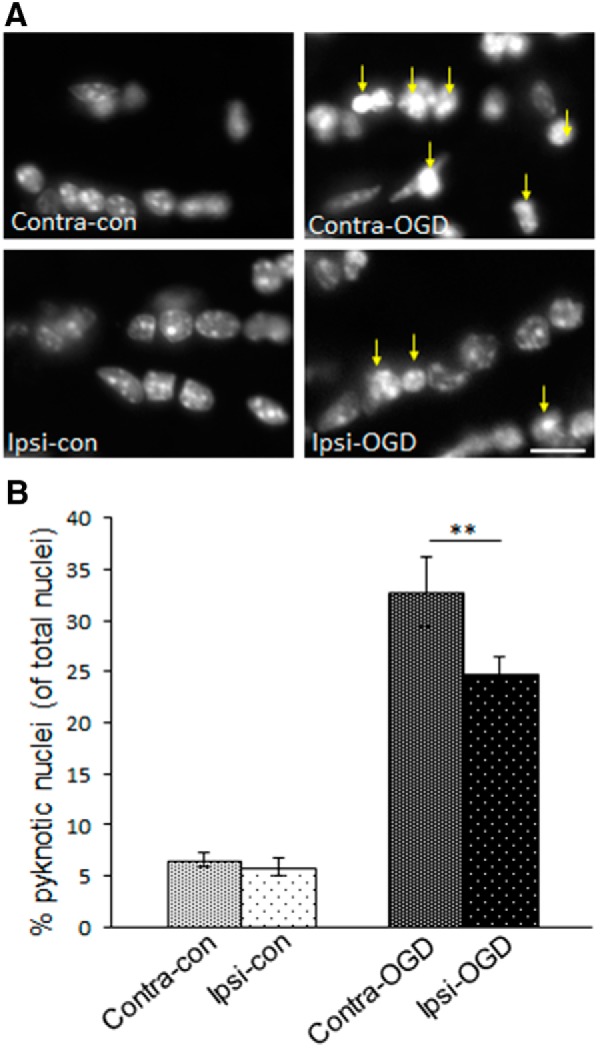
IPC attenuates formation of pyknotic nuclei in MONs after OGD. MONs were stained for the nuclear marker DAPI. The 40× objective images in A were obtained with an immunofluorescent microscope as described in Materials and Methods. The number of pyknotic nuclei were quantified as described in Materials and Methods. IPC pulse alone did not significantly alter the baseline number of pyknotic nuclei (left column in A, quantified in left two bars in B): Contra-con was 7 ± 1% and Ipsi-con was 6 ± 1%. However, after OGD, there was a significant increase in the number of pyknotic nuclei (right column in A, right two bars B) that was more pronounced on the non-preconditioned side: Contra-OGD, 33 ± 3% versus Ipsi-OGD, 25 ± 2%; **p < 0.01. Arrows indicate clumping of chromatin/pyknotic nuclei in the injured fields after OGD (right column in A). Scale bar, ∼15 μm. n = 3 MONs per group.
Figure 6.
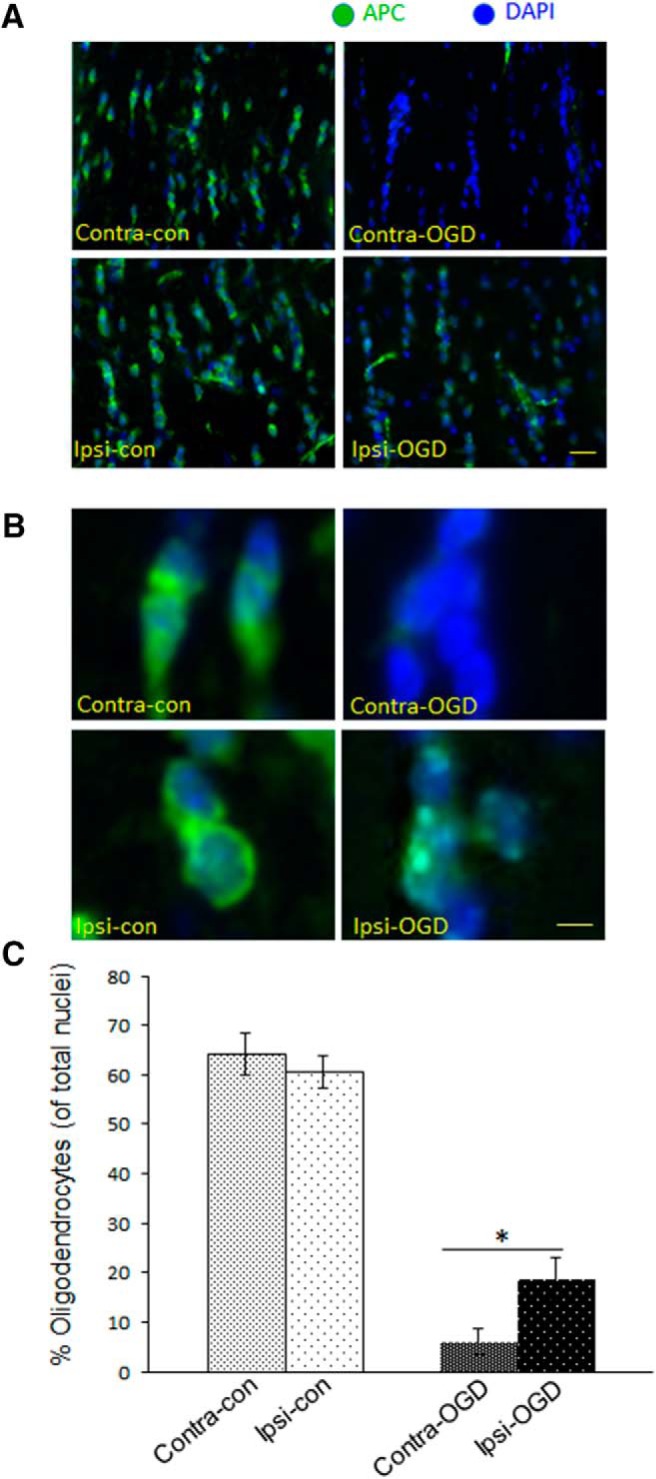
IPC attenuates OGD-induced oligodendrocyte loss in MONs. MONs were stained (green) with APC, an oligodendrocyte marker, and the nuclear marker DAPI (blue). The 40× objective images in A were obtained with an immunofluorescent microscope as described in Materials and Methods. Higher-magnification images in B are digital zoom. The number of APC+ cells were quantified as described in Materials and Methods. IPC pulse alone did not significantly alter the baseline number of oligodendrocytes (left columns in A and B, quantified in left two bars in C): Contra-con was 64 ± 4% and Ipsi-con was 61 ± 3%. However, after OGD, there was a significant decrease in the number of APC+ cells (right columns in A and B, right two bars in C). The number of APC+ cells after OGD was reduced more in the non-preconditioned side: Contra-OGD, 6 ± 3% versus Ipsi-OGD, 19 ± 4%; *p < 0.05. Scale bars: A, 60 μm; B, 15 μm. n = 3 MONs per group.
Glutamate receptor mRNA quantification.
We quantified steady-state levels of AMPA glutamate receptor subtypes/isoforms using qRT-PCR as described previously (Weinstein et al., 2009). In brief, mice were killed 72 h after IPC pulse. For each experiment, three ipsilateral and contralateral MONs were dissected out and pooled separately. MONs were placed in TRIzol LS Reagent (Thermo Fisher Scientific) and homogenized. RNA extraction was performed via the hybrid TRIzol LS-RNeasy Micro kit (Qiagen) method per the protocols of the manufacturer. RNA integrity was analyzed using an Agilent 2100 Bioanalyzer. For the GluR1–GluR4 flip and flop gene expression assay, 150–200 ng of RNA was reverse transcribed using the Transcriptor High Fidelity cDNA Synthesis kit (Roche). In determining gene expression differences between GluR1–GluR4 flip and flop transcript variants, intron-spanning primers were designed such that forward primers were specific to either the flip or flop isoform regions (Sommer et al., 1990), whereas their respective reverse primers recognized sequences unique to individual GluR genes. Singleplex amplification was performed on the StepOnePlus Real-Time PCR System (Applied Biosystems) as described previously (Weinstein et al., 2009). Cycling conditions consisted of the following: (1) 95°C for 10 min; and (2) 50 cycles of 95°C for 10 s, 55°C for 15 s, and 72°C for 30 s. All primer and probe sequences are shown in Table 1. Data were analyzed with StepOne Software version 2.1 (Applied Biosystems). Relative gene expression was calculated using the ΔCt method in which samples were normalized to the cycle threshold (Ct) geometric mean of the housekeeping genes. Numerical data are given as the mean ± SEM of the normalized mean values from each independent experiment. All experiments were performed in triplicate.
Table 1.
Primer/probe sequences for quantification of steady-state levels of GluR 1–4 flip and flop isoforms
| Gene | Isoform | GenBank accession number | Forward primer | Reverse primer | Amplicon size (bp) | UPL probe number | Probe recognition site |
|---|---|---|---|---|---|---|---|
| Genes of interest | |||||||
| GluR1 | Flip | NM_001113325.1 | GCAAGGACTCCGGAAGTAAGG | TCTCTCCACTGCCACTTCCT | 261 | 51 | GGCAGGAG |
| Flop | NM_008165.3 | GGGAGGTGACTCCAAGGACAA | TCTCTCCACTGCCACTTCCT | 257 | |||
| GluR2 | Flip | NM_001083806.1 | GCCAAGGACTCGGGAAGTA | TGGGTTAATATTCTGTGCATTCTTTG | 186 | 67 | TGCTGGAG |
| Flop | NM_013540.2 | GGGGAGGTGATTCCAAGG | TGGGTTAATATTCTGTGCATTCTTTG | 182 | |||
| GluR3 | Flip | NM_016886.3 | GGAATGTGGAGCCAAGGAC | TGGCAGGAGCAGGCTTAAA | 206 | 45 | CTGGGGCT |
| Flop | AB102775.1 | GCGGTGACTCCAAGGACAA | TGGCAGGAGCAGGCTTAAA | 189 | |||
| GluR4 | Flip | NM_019691.4 | AAGGACTCGGGAAGCAAGGA | TCTTATGGCTTCGGAAAAAGTCA | 177 | 37 | CCAGGGCA |
| Flop | NM_001113180.1 | GGGAGGTGACTCCAAGGACAA | TCTTATGGCTTCGGAAAAAGTCA | 175 | |||
| Housekeeping genes | |||||||
| HPRT | N/A | NM_013556.2 | GGAGCGGTAGCACCTCCT | CTGGTTCATCATCGCTAATCAC | 116 | 69 | CTTCCTCC |
| EIF4a2 | Variants 1, 2 | NM_013506.2 | CGATCTACCTACCAATCGTGAA | ACCTTTCCTCCCAAATCGAC | 70 | 53 | TGGCAGAG |
| ATP5b | N/A | NM_016774.3 | GGCACAATGCAGGAAAGG | TCAGCAGGCACATAGATAGCC | 77 | 77 | CCACCACC |
UPL, Universal probe library.
Preparation of primary microglia cultures.
Primary microglia were prepared from the cortex of newborn (P4 or P5) IFNAR1fl/fl or IFNAR1fl/fl LysMcre mice as described previously (Giulian and Baker, 1986; Möller et al., 2000). In brief, cortical tissue was freed carefully from blood vessels and meninges, digested with 50 ng/ml DNase, triturated, and washed. Cortical cells were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% heat inactivated fetal bovine serum (FBS; Hyclone) and penicillin/streptomycin (P/S; 50 IU/50 μg/ml; Mediatech/Corning) plus 2 ng/ml granulocyte macrophage colony stimulating factor (GM-CSF; R&D Systems) for 10–21 d (media change every 3–4 d). Microglia were separated from underlying astrocytic monolayer by gentle agitation and spun down (100 × g for 10 min). Cell pellet was resuspended in DMEM/10% FBS with P/S plus 2 ng/ml GM-CSF and plated on BD Primaria culture dishes and plates (Falcon). Non-adherent cells were removed 30–60 min after plating by changing the medium, and adherent microglia were incubated for 24 h in culture medium. We confirmed purity of our primary microglia population by flow cytometry based on their expression of CD11b (>96% of total cell population).
Flow cytometry.
After IFNβ stimulation, primary microglia were detached from 24-well plates in ice-cold 2 mm EDTA/PBS. Cells were then washed with FACS medium (HBSS containing 10% FBS and 10 mm HEPES) and incubated with Fc block (BD Biosciences Pharmingen) for 5 min on ice. Microglia were then stained with DAPI and separately with phycoerythrin-conjugated anti-mouse-IFNAR1 antibody (BioLegend) or isotype control for 60 min on ice. After staining, cells were washed and resuspended in 250 μl of FACS medium. Data acquisition and analysis were performed using a BD FACSCanto II flow cytometer and Flojo (Treestar) software. Numerical data are given as the mean ± SEM of the normalized specific binding median values from each independent experiment. All experiments were performed in triplicate.
Acute isolation of cortical microglia and quantification of ifnar1 expression.
Ex vivo flow cytometric sorting of microglia from mouse brain is a well established method that offers a physiologic window into in vivo microglial biology (Sedgwick et al., 1991; Carson et al., 1998). Microglia were isolated from cortices of naive 12- to 16-week old male mice using our previously published protocol (Su et al., 2014). In brief, cortical tissue was dissociated with mechanical and enzymatic (trypsin) digestion. We used CD11b microbead columns (Miltenyi Biotec) to enrich for myeloid cells and immunostained with a panel of fluorochrome-conjugated antibodies to identify specific innate immune cell populations. Samples were processed for flow cytometry on a BD FACSAria sorter, and CD45dim/F4–80pos/GR-1neg/1A8neg microglia were identified by gating and sorted directly into concentrated TRIzol (Thermo Fisher Scientific). RNA was extracted, and samples were processed for expression of IFNAR1 mRNA via singleplex amplification. Cycling conditions were as follows: (1) 95°C for 10 min; and (2) 40 cycles of 95°C for 10 s and 60°C for 30 s. Primers and probe were custom designed (Integrated DNA Technologies) within ifnar1 exon 10 and 11 coding sequences and overlapping with loxP sites in the IFNAR1fl/fl mouse: forward primer, GAATAGTTGCCCGAGTCCTG; reverse primer, ACGGTCGCTGTAGAAGTAAAG; and probe, TCTGTGAGCTGTACTTCCTGAGGTCC. Relative gene expression was calculated using the ΔΔCt method in which samples were normalized to the Ct geometric mean of three housekeeping genes.
Results
Ischemic preconditioned WM was less susceptible to ischemic injury
To determine whether IPC could induce resistance in WM to a subsequent ischemic insult, we developed a novel experimental WM IPC model with a temporal paradigm based on previous IPC studies (Stenzel-Poore et al., 2003; Zhang et al., 2008). The sequential combination of transient CCAO in vivo, followed 72 h later by ex vivo exposure of the MON to 45 min of OGD (Fig. 2), the latter a well established model of WM injury (Stys et al., 1991; Bakiri et al., 2008; Sozmen et al., 2012; Matute et al., 2013), allowed us to assess directly whether the conditioning ischemia induced WM protection. In all cases, preconditioned MONs (ipsilateral to transient CCAO) showed improved CAP recovery after OGD compared with control MONs (contralateral to transient CCAO; Fig. 3A). The area under the CAP curve, which is proportional to the number of functioning axons (Buchtal and Rosenfalck, 1966; Cummins et al., 1979; Stys et al., 1991), was determined 60 min after the end of the OGD exposure (Fig. 3A, time point b). The MON exposed to IPC (Ipsi-OGD) showed an average CAP recovery to 31 ± 3% of baseline compared with 17 ± 3% in the contralateral MON (Contra-OGD) not exposed to IPC (p < 0.01; Fig. 3B) and 21 ± 4% in the sham-operated (Sham-OGD) MON (p < 0.05; data not shown). There were no significant differences among these groups in the rates of CAP decline at the onset of OGD (Fig. 3A).
Figure 3.
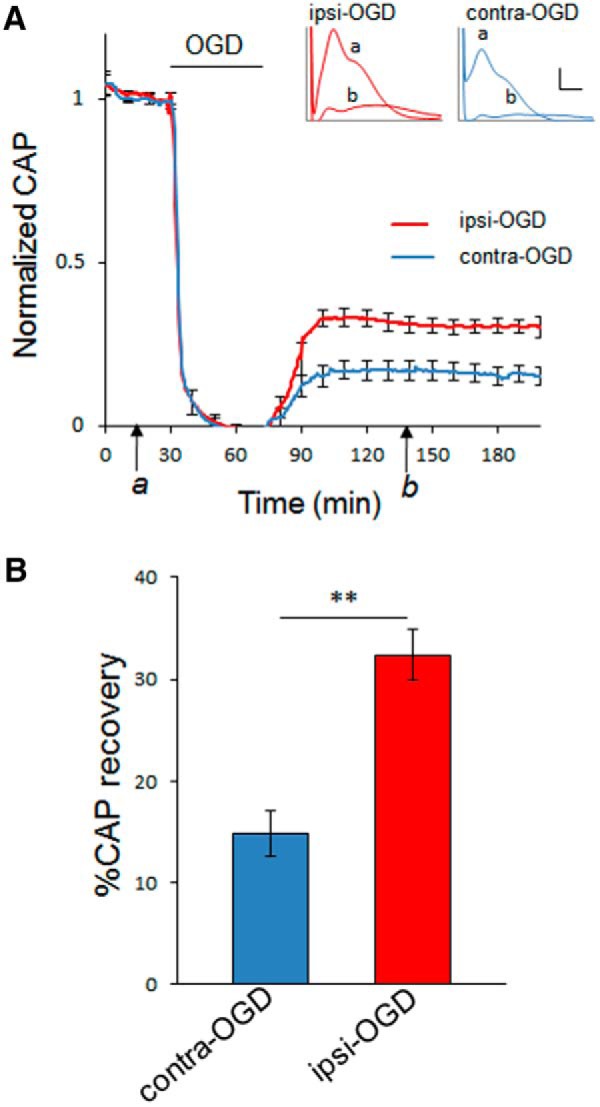
Axon function in WT MONs is improved after IPC. Seventy-two hours after IPC pulse, axon function in MONs from WT mice was assessed. MONs were allowed to equilibrate for 30–60 min to establish a baseline (see time point a). Axons were stimulated every 30 s with a supramaximal pulse; CAPs were quantified. Error bars are shown for every 10 data points. Ischemia was induced by OGD. After 45 min of OGD, normoxic/normoglycemic control conditions were returned for a period of several hours to monitor recovery (see time point b). Insets demonstrate a typical CAP at baseline (a) and during recovery (b) in both ipsilateral preconditioned (red) and contralateral control (blue) MONs. Calibration: insets in A, 1 mV, 0.5 ms. Bar graph showing extent of CAP recovery at 60 min after the end of 45 min OGD pulse (time point b). **p < 0.01. n = 11 MONs per group.
Histological analysis showed that preconditioned WM was less vulnerable to ischemic injury
Functional axonal impairment can be associated with cytoskeletal changes in axonal structure (Tekkök and Goldberg, 2001). The linear and uniform pattern of phosphorylated neurofilament staining seen in normal axons is lost in proportion to the severity of injury. Ischemia-damaged axons show abnormal patterns of neurofilament staining, including axonal heads and retraction bulbs (Adams et al., 1983; Tekkök and Goldberg, 2001). The IPC protocol used here did not, in and of itself, alter axon morphology as determined by immunofluorescent staining for phosphorylated neurofilament (Fig. 4A,B, quantified in C). After 5 h of ex vivo incubation, MONs ipsilateral and contralateral to CCAO (Ipsi-con and Contra-con, respectively) were indistinguishable from sham control (Sham-con) and showed no evidence of pathological changes in neurofilament structure (Fig. 4). After the ex vivo OGD exposure, axonal integrity was reduced in both the preconditioned (Ipsi-OGD) and control (Contra-OGD) MONs (Fig. 4). However, the axonal integrity score was better preserved in the Ipsi-OGD MON relative to the Contra-OGD MON (68 vs 45% of baseline, respectively; p < 0.01; Fig. 4). Pyknotic nuclei are a reliable marker of apoptotic cell death, and this marker has been used effectively to judge severity of ischemic injury in MONs (Tekkök and Goldberg, 2001). IPC alone did not alter the number of pyknotic nuclei (Fig. 5). Although OGD increased the number of pyknotic cells in MONs both ipsilateral and contralateral to the previous CCAO (Fig. 5), this increase was significantly greater in the Contra-OGD compared with the Ipsi-OGD MON (26 vs 19% of all nuclei, respectively; p < 0.01; Fig. 5B). Together, these data indicate that IPC alone did not induce any demonstrable MON injury, but IPC did offer protection against subsequent ischemic insult.
Immunostaining with cell-specific markers revealed that IPC also reduced loss of oligodendroctye-specific staining (APC+) after OGD (Fig. 6). As with the neurofilament staining and pyknotic nuclei data above, the IPC protocol itself, in the absence of subsequent OGD, did not change the density of APC+ cells (i.e., oligodendrocytes). However, the loss of APC+ cells after ex vivo OGD was less in MONs ipsilateral to the IPC stimulus (69% reduction in Ipsi-OGD versus 91% reduction in Contra-OGD; p < 0.05; Fig. 6C). Neither IPC alone nor subsequent ex vivo OGD induced any changes in the number of astrocytes (GFAP+ cells) or microglia/macrophages (Iba1+ cells) in MONs (data not shown).
Preconditioning did not alter ischemic release of EAAs or AMPA glutamate receptor expression
The mechanisms of ischemic WM injury are distinct from those in GM (Matute et al., 2013). In ischemic WM injury, glutamate is released and causes excitotoxicity via activation of AMPA/kainate glutamate receptors (Tekkök et al., 2007). NMDA-type glutamate receptors are present, but they mediate improved outcome, not damage (Tekkök et al., 2007). Conversely, hypoglycemic WM injury depends on aspartate release and activation of NMDA-type glutamate receptors (Yang et al., 2014). Given the importance of EAA release for WM injury, in general, we hypothesized that IPC might reduce subsequent ischemic injury by decreasing glutamate/aspartate release or alternatively by downregulating glutamate receptor expression. We used HPLC to measure the concentration of glutamate and aspartate released from MONs during OGD. Although we observed the expected increases in EAAs induced by OGD (Tekkök et al., 2007), we did not detect any significant changes in OGD-mediated release between the IPC-exposed MON and the contralateral control optic nerve (Fig. 7A–D). Most AMPA receptors are heterotetrameric, consisting of a dimer of dimers that include the GluR2 subunit, as well as GluR1, GluR3, or GluR4 (Keinänen et al., 1990). The four known AMPA glutamate receptor subunits each have two mRNA isoforms termed flip and flop (Sommer et al., 1990). The physiological function of the channel is determined by both the composition of the subunits of the receptor and flip/flop isoforms that can affect channel open time and possibly injury severity (Keinänen et al., 1990; Sommer et al., 1990). Using qRT-PCR, we quantified steady-state levels of both the flip and flop mRNA isoforms of GluR1–GluR4 in MON 72 h after IPC. We detected expression of the flip isoform of all four AMPA receptor subunits and the flop isoform of GluR1 and GluR2 (Fig. 7E). We did not detect any significant differences in expression of any of the subunits or isoforms between the ipsilateral preconditioned and contralateral control MONs (Fig. 7E).
Figure 7.

IPC did not change the amount of glutamate or aspartate released, nor did it alter AMPA glutamate receptor expression. As expected, OGD for 60 min was associated with a time-dependent increase in glutamate (A) and aspartate (B) release from optic nerve. However, there was no significant difference in release of either glutamate or aspartate from the Ipsi-OGD and Contra-OGD optic nerves at any time point during or after OGD. Samples of extracellular perfusate, ACSF, were collected every 2 min and assessed for amino acid concentrations by HPLC (see Materials and Methods). C and D show data for glutamate and aspartate release, respectively, reorganized into 20 min epochs (mean ± SE), n = 5; release magnitude compared with control/baseline (first 20 min); * over bar indicates a statistically significant release compared with baseline (noted by dashed line). *p < 0.05; **p < 0.01; ***p < 0.001 by ANOVA with Dunnett's post hoc test against 20 min of corresponding basal level release before OGD application. Preconditioned (Ipsi-OGD) and non-preconditioned (Contra-OGD) control displayed no significant differences in either glutamate or aspartate release before, during, or after OGD (Student's t test). Several representative nonsignificant p values are shown. n = 4 MONs per group. In E, the effects of IPC on expression of AMPA GluR mRNA subtypes/isoforms is shown. qRT-PCR on RNA extracted from either ipsilateral or contralateral MONs 72 h after 15 min CCAO, our IPC pulse. Findings are presented as log10 fold change in steady-state levels of each AMPA GluR subtype/isoform mRNA gene of interest (GOI) relative to panel of housekeeping genes (HKG) as described in Materials and Methods. Bar graph shows that six of the eight GluR subtypes/isoforms could be detected by qRT-PCR. IPC did not significantly alter expression of any of these transcripts at the 72 h time point (Student's t test). n = 3 qRT-PCR experiments on pooled samples from a total of 9 MONs per group.
Preconditioning-mediated protection of axonal function was absent in TLR4−/− mice
TLR4 signaling is required for optimal IPC-mediated protection in infarct volume models of IPC (Pradillo et al., 2009). We tested the relevance of this gene product in WM IPC using TLR4−/− mice. As we did for WT mice (see above), we compared CAP recovery after ex vivo OGD in TLR4−/− MONs that were preconditioned (Ipsi-OGD) or not preconditioned (Contra-OGD) by previous transient CCAO. We also quantified CAP recovery from OGD in Sham-OGD control MONs. The IPC protection seen in WT animals (i.e., in MONs ipsilateral to CCAO) was abolished in the TLR4−/− mice (Fig. 8A; Ipsi-OGD CAP recovery, 23 ± 4% and Contra-OGD recovery, 21 ± 5%, p = NS; Sham-OGD CAP recovery, 20 ± 3%, p = NS; data not shown).
Figure 8.

IPC failed to protect WM against ischemic injury in TLR4−/−, IFNAR1−/−, or myeloid cell-specific IFNAR1 knockdown MONs. Seventy-two hours after IPC pulse (15 min CCAO), MONs were removed and exposed to a standardized ischemic insult: 45 min of OGD (see Materials and Methods). Recovery from OGD was determined 60 min after reperfusion with control solution (time point b). For the TLR4−/− mice, recovery of CAP was similar in Contra-OGD (21 ± 5%) and Ipsi-OGD (23 ± 4%; p = NS) MONs (A). For the IFNAR1−/− mice, recovery of CAP was also similar in Contra-OGD (17 ± 5%) and Ipsi-OGD (14 ± 3%; p = NS) MONs (B). n = 8 and 5 MONs per group for TLR4−/− and IFNAR1−/−, respectively. For the IFNAR1fl/fl control mice, recovery of CAP was significantly less in Contra-OGD (25 ± 6%) compared with Ipsi-OGD (45 ± 3%; *p < 0.05) MONs (C, E). For the IFNAR1fl/fl LysMcre mice, recovery of CAP was similar in Contra-OGD (25 ± 4%) and Ipsi-OGD (24 ± 3%; p = NS) MONs (D, F). n = 4 MONs per group for IFNAR1fl/fl control and IFNAR1fl/fl LysMcre, respectively.
Preconditioning-mediated protection of axonal function was absent in IFNAR1−/− mice
IFN signaling (notably IRF3 and IRF7) is required for optimal IPC-mediated protection in infarct volume models of IPC (Stevens et al., 2011). IFNAR1 is the primary receptor mediating cellular signaling for type I IFNs (including all IFNα and IFNβ; Müller et al., 1994). Several recent studies have also shown that IFNAR1 is required for optimal TLR agonist-mediated protection in infarct volume models of ischemic cross-tolerance (Gesuete et al., 2012; Leung et al., 2012). To test the importance of IFNAR1 in IPC-mediated WM protection, we compared CAP recovery after ex vivo OGD in IFNAR1−/− mice whose MONs were preconditioned (Ipsi-OGD) or not preconditioned (Contra-OGD) 72 h earlier by transient CCAO. No IPC protection was detected in IFNAR1−/− mice (Fig. 8B; Ipsi-OGD CAP recovery, 14 ± 3% and Contra-OGD recovery, 17 ± 5%, p = NS).
Preconditioning-mediated protection of axonal function was absent in myeloid cell-targeted IFNAR1 knockdown mice
Microglia, the resident immune cells of the CNS, have been implicated in IPC (Weinstein et al., 2010). Prinz et al. (2008) recently generated a myeloid-selective IFNAR1 knockdown strain, IFNAR1fl/fl LysMcre, that shows a high degree of IFNAR1 knockdown in innate immune cell populations and has been used to specifically implicate microglial/macrophage expression of IFNAR1 in the pathophysiology of experimental autoimmune encephalomyelitis. We used this IFNAR1 knockdown strain to determine whether microglial IFNAR1 signaling plays a role in WM IPC. We first demonstrated that IFNAR1 expression in IFNAR1fl/fl LysMcre mice was “knocked down” by 93 and 70%, respectively, in primary microglia (Fig. 1A) and ex vivo sorted cortical microglia (Fig. 1B). We next determined whether IPC-induced protection from ischemic injury was altered in MONs from IFNAR1fl/fl LysMcre mice compared with MONs from IFNAR1fl/fl mice. Robust protection from ischemia after the standard IPC protocol was present in the IFNAR1fl/fl control line (Fig. 8C,E; Ipsi-OGD CAP recovery, 45 ± 2% and Contra-OGD CAP recovery, 25 ± 6%, p < 0.05) but not in the cell-type-specific IFNAR1fl/fl LysMcre knockdown line (Fig. 8D,F; Ipsi-OGD CAP recovery, 24 ± 3% and Contra-OGD recovery, 25 ± 4%, p = NS).
Discussion
These findings demonstrate for the first time that IPC induces a robust protective effect against subsequent ischemic injury in CNS WM. The relative protective effect of IPC was 1.8-fold according to our physiological data, which represents a proportional 1.8-fold increase in the number of functional axons that survived the ischemic challenge. We also found a significant IPC-induced protective effect in our structural data. Of great interest, IPC-induced protection in WM, as in GM, depended on key immune cell signaling pathways: TLR4 and type 1 IFN. In the absence of either, the powerful protective effect of IPC in WM was abolished. In addition, we used a myeloid cell-targeted IFNAR1 knockdown line to demonstrate that IFNAR1 expression in innate immune cells was critical for IPC-mediated protection in WM. Although the precise downstream mechanism(s) of IPC-induced WM protection remains to be determined, our data indicated that oligodendrocytes were better preserved in the IPC-protected MONs. Thus, IPC-induced protection in WM may be mediated, at least in part, by specific changes that render these cells less vulnerable to ischemic destruction (Fig. 9). Oligodendrocytes are susceptible to glutamate excitotoxicity during ischemia (Salter and Fern, 2005; Alix and Fern, 2009), but we found no differences in the amount of glutamate released from preconditioned and control WM, nor did we find any IPC-induced differences in the expression of AMPA glutamate receptor mRNA subunits/isoforms.
Figure 9.

Hypothesized pathway of IPC in CNS WM. IPC in WM is initiated by a brief (15 min) CCAO ischemic pulse (1). This leads to the extracellular release of DAMP macromolecules from multiple cellular (astrocytes, microglia) and possibly other (axons, myelin sheaths) sources (2). There likely are a variety of DAMP macromolecules involved, but specific examples might include heat shock proteins, peroxiredoxins, high mobility group box 1, and fibronectin. These DAMP macromolecules then bind to TLRs on the cell surface, including TLR4. In WM, TLR4 is found on microglia (3a) and astrocytes (3b). Binding of these receptors induces production of type 1 interferons, including IFNα and IFNβ, which are then secreted from the cells (4). INFα/β can then bind to the type 1 interferon receptor (a heterodimer that includes IFNAR1) on nearby microglia (5). This induces activation of IRFs and the transcription of ISGs in the microglia (6). Secreted ISG products (such as the chemokines CXCL10 and CCL5) are released from the cell (7a), and intracellular ISGs may alter the microglial phenotype (7b). ISG expression ultimately leads to protection of oligodendrocytes and their associated myelin and enhances functional recovery to subsequent ischemic exposure (8).
Our post-IPC testing protocol was performed on completely isolated MONs, allowing the conclusion that the observed IPC-induced protection was intrinsic to WM. In other words, it was not mediated by nearby GM or by systemic changes that could be communicated by an intact circulation or even diffusion from CSF. Retinal ganglion cells, the cells that give rise to all axons in the optic nerve, are also exposed to ischemia after CCAO (our IPC pulse; Hayreh, 2006). However, it seems unlikely to us that the observed changes in ischemic WM injury were attributable to IPC-stimulated changes in retinal ganglion cells. Although changes initiated in the ganglion cells could theoretically be communicated by axonal transport, it seems improbable that axons thus influenced would be able to provide protection to oligodendrocytes, cells exclusive to WM. Therefore, we currently believe that WM, in and of itself, can mount meaningful protection against ischemic injury after a previous transient ischemic event.
There are several advantages of the combined in vivo/ex vivo experimental IPC model used here. The IPC stimulus and the 72 h “induction period” occurred under normal, in vivo conditions, allowing more direct comparison with IPC-like events in patients. The MON that was contralateral to the transient unilateral CCAO (i.e., the preconditioning event) served as an ideal control for the ipsilateral MON, experiencing IPC, because it eliminated differences attributable to biovariability among individual animals. Finally, the ex vivo MON preparation permitted exclusive assessment of WM ischemic vulnerability after IPC. The test period of WM ischemia could be controlled precisely, and both structural and physiological accompaniments of WM injury could be evaluated quantitatively, enabling unequivocal detection of experimental differences (Buchtal and Rosenfalck, 1966; Cummins et al., 1979; Stys et al., 1991).
The pathophysiology of ischemic WM injury has been studied extensively, but many questions about this destructive cascade remain open (Kettenmann and Ransom, 2013; Fern et al., 2014). One proven tenant is that WM ischemic injury has unique features that distinguish it from GM ischemic pathophysiology. The capacity for recovery from a standardized ischemic insult is greater for WM than for GM (Tekkök and Ransom, 2004). Therefore, in principle, WM offers more opportunity for therapeutic intervention after an ischemic insult than does GM. Ischemia depletes ATP, leading to elevated intracellular [Na+] and cellular depolarization, which in turn cause reversal of Na+-dependent glutamate uptake in astrocytes and a spike in extracellular [glutamate] (Tekkök et al., 2007). Both GM and WM suffer glutamate receptor overexcitation (i.e., “excitotoxicity”) during ischemia, but there is an important difference: ischemic excitotoxicity in GM is mediated by both NMDA- and AMPA/kainate-type glutamate receptors, whereas in WM, only AMPA/kainate-type glutamate receptors mediate injury (Baltan et al., 2008); the situation may be different in perinatal WM (Micu et al., 2006). In fact, NMDA receptor antagonism during WM ischemia reduces recovery (Baltan et al., 2008). Oligodendrocytes are believed to be the primary target of toxic glutamate receptor activation (Baltan et al., 2008), but astrocytes, and even axons, express glutamate receptors and might also suffer. Preceding, or possibly in parallel with, excitotoxicity, WM axons suffer damaging Ca2+ overload during ischemia mediated by collapse of the transmembrane Na+ gradient, causing reverse Na+/Ca2+ exchange (Li et al., 1999; Sánchez-Gómez et al., 2003; Ruiz et al., 2010). Although we do not know whether axonal injury precedes or follows oligodendrocyte injury and associated changes in myelin (Fern et al., 1998), it appears that injury to either adversely affects the other (Waxman et al., 1994).
Because the IPC benefit was not attributable to either of two obvious possibilities—diminished glutamate release during ischemia or reduction in the expression of AMPA glutamate receptor subunits (Fig. 7)—we considered other possibilities. TLR4 signaling is important in stroke pathophysiology (Cao et al., 2007; Caso et al., 2007) and has been implicated directly in IPC-mediated neuroprotection in GM predominant models (Pradillo et al., 2009). Ischemia is presumed to cause release of endogenous TLR4 agonists (including heat shock proteins, high mobility group box 1, and peroxiredoxins), sometimes called “danger-associated molecular pattern” (DAMP) molecules, activating TLR4 on resident microglia, astrocytes or infiltrating macrophages (Weinstein et al., 2010; Shichita et al., 2012). In the case of IPC in GM, this activation attenuates inflammation and enhances immunomodulatory signaling pathways (Stenzel-Poore et al., 2007). We found that IPC-mediated protection in WM was abolished in TLR4−/− mice (Fig. 8A), indicating the presence of at least a partial “common pathway” for IPC in both WM and GM.
Genomic studies on preconditioning in GM have implicated in downstream interferon stimulated gene (ISG) expression in the mechanism of TLR-dependent preconditioning (Stevens et al., 2011). Stimulation of ISG expression depends on activation of IFNAR1, and we found that IPC-mediated protection in WM was eliminated in IFNAR1−/− mice (Fig. 8B), directly implicating IFN signaling in this phenomenon. Although IFNAR1 itself has been implicated previously in models of TLR-agonist-dependent ischemic cross-tolerance (Gesuete et al., 2012; Leung et al., 2012), to our knowledge, this is the first demonstration that IFNAR1 plays a role in any form of IPC.
This observation provides support for the hypothesis that an IPC pathway, with major generic features, operates in both GM and WM. This certainly leaves open the possibility that components of the upstream and downstream IPC cascade may vary significantly between these two vastly different CNS regions. In the CNS, microglia have the highest expression of, and responsiveness to, TLR4 (Holm et al., 2012) and IFNAR1 (Costello and Lynch, 2013), suggesting that these cells are crucial, but perhaps not the only, cellular mediators of IPC. In this regard, it is worth mentioning that microglia are present at approximately the same density in GM and WM (Kettenmann et al., 2011), including within the optic nerve (M.A.H. and B.R.R., unpublished observations). To characterize the cell-type-specific role of microglial IFNAR1 signaling in IPC, we used the IFNAR1fl/fl LysMcre mouse strain (Fig. 8C–F). IPC-mediated protection in WM was eliminated completely in this myeloid cell-targeted IFNAR1 knockdown strain but not in the corresponding IFNAR1fl/fl control line. It is important to note that the LysM promoter is active in both microglia and macrophages so we cannot exclude a role for infiltrating macrophages in this process. Nevertheless, this finding strongly suggests that innate immune cells (either microglia or macrophages), rather than astrocytes, oligodendrocytes, or other WM elements, are the key target for IFN-mediated signaling in WM after an IPC pulse.
In looking at the CAP recovery levels after OGD, we noted that the IFNAR1fl/fl control mouse strain MONs (Fig. 8C,E) had slightly better recoveries than WT animal MONs (Fig. 3) under control conditions (i.e., without IPC). There could be several explanations: (1) the experiments were performed at different times and might have been subject to slightly different environmental conditions (i.e., food, sleep/wake cycle, etc.); (2) the animals were obtained from different vendors and therefore might be subject to some “genetic drift”; or (3) insertion of the loxP sequences might, themselves, have a subtle effect on recovery from ischemia. However, the important point is that our main experimental findings are not subject to these concerns. In each condition, we compared optic nerves from the same animal, one exposed to IPC, the other not exposed to IPC.
In conclusion, our findings demonstrated the existence of robust IPC-induced protection in WM that depended on the expression of two immune cell signaling molecules/pathways: TLR4 and IFNAR1 (Fig. 9). Furthermore, this is the first in vivo demonstration that cell-type-specific expression of an individual gene (i.e., microglial/macrophage expression of IFNAR1) plays an indispensable role in the IPC phenomenon. These findings support the concept that manipulation of the neuroimmune response may be able to alter the outcome of ischemic brain injury in humans, providing benefit for both WM and GM.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants NS065008 and NS076620 (J.R.W.) and NS115589 (B.R.R.) and the Sidney Gift Fund (B.R.R.). For excellent technical assistance, we thank Katie Ho, Jasdeep Sandhu, Anna Savos, Diana Chao, Nathan Lam, Danielle Hanssen, and Michael Iorga. We thank Dr. Shizuo Akira's laboratory (Research Institute for Microbial Diseases, Osaka University, Osaka, Japan) for permission to use the TLR−/− line through a material transfer agreement (MTA) and Dr. Christopher Wilson (Department of Immunology, University of Washington, Seattle, WA) who physically provided the TLR4−/− line. We also acknowledge that we were kindly granted permission to use the IFNAR1fl/fl mouse line through an MTA with Dr. Ulrich Kalinke's laboratory (Twincore, Centre for Experimental and Clinical Infection Research, Hannover, Germany); these animals, and the IFNAR1−/− line, were kindly provided by Dr. Robert Schreiber (Washington University, St. Louis, MO).
The authors declare no competing financial interests.
References
- Adams JH, Graham DI, Gennarelli TA. Head injury in man and experimental animals: neuropathology. Acta Neurochir Suppl (Wien) 1983;32:15–30. doi: 10.1007/978-3-7091-4147-2_2. [DOI] [PubMed] [Google Scholar]
- Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693. doi: 10.1002/ana.21767. [DOI] [PubMed] [Google Scholar]
- Bahjat FR, Williams-Karnesky RL, Kohama SG, West GA, Doyle KP, Spector MD, Hobbs TR, Stenzel-Poore MP. Proof of concept: pharmacological preconditioning with a Toll-like receptor agonist protects against cerebrovascular injury in a primate model of stroke. J Cereb Blood Flow Metab. 2011;31:1229–1242. doi: 10.1038/jcbfm.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakiri Y, Hamilton NB, Káradóttir R, Attwell D. Testing NMDA receptor block as a therapeutic strategy for reducing ischaemic damage to CNS white matter. Glia. 2008;56:233–240. doi: 10.1002/glia.20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci. 2008;28:1479–1489. doi: 10.1523/JNEUROSCI.5137-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Axt KJ, Fosnaugh JS, Smith KJ, Johnson KA, Hill DE, Kinzler KW, Baraban JM. Expression of the APC tumor suppressor protein in oligodendroglia. Glia. 1996;17:169–174. doi: 10.1002/(SICI)1098-1136(199606)17:2<169::AID-GLIA8>3.0.CO;3B2-Y. [DOI] [PubMed] [Google Scholar]
- Buchtal F, Rosenfalck A. Evoked action potentials and conduction velocity in human sensory nerves. Brain Res. 1966;3:1–122. [Google Scholar]
- Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(SICI)1098-1136(199801)22:1<72::AID-GLIA7>3.0.CO;3B2-A. [DOI] [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- Costello DA, Lynch MA. Toll-like receptor 3 activation modulates hippocampal network excitability, via glial production of interferon-beta. Hippocampus. 2013;23:696–707. doi: 10.1002/hipo.22129. [DOI] [PubMed] [Google Scholar]
- Cummins KL, Perkel DH, Dorfman LJ. Nerve fiber conduction-velocity distributions. I. Estimation based on the single-fiber and compound action potentials. Electroencephalogr Clin Neurophysiol. 1979;46:634–646. doi: 10.1016/0013-4694(79)90101-9. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Raval AP, Lin HW, Dave KR, Della-Morte D, Perez-Pinzon MA. GABA synapses mediate neuroprotection after ischemic and epsilonPKC preconditioning in rat hippocampal slice cultures. J Cereb Blood Flow Metab. 2009;29:375–384. doi: 10.1038/jcbfm.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Barakat DJ, Hernandez E, Shestopalov VI, Ivanov D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol Vis. 2010;16:1907–1912. [PMC free article] [PubMed] [Google Scholar]
- Fernandez DC, Chianelli MS, Rosenstein RE. Involvement of glutamate in retinal protection against ischemia/reperfusion damage induced by post-conditioning. J Neurochem. 2009;111:488–498. doi: 10.1111/j.1471-4159.2009.06334.x. [DOI] [PubMed] [Google Scholar]
- Fern RF, Matute C, Stys PK. White matter injury: Ischemic and nonischemic. Glia. 2014;62:1780–1789. doi: 10.1002/glia.22722. [DOI] [PubMed] [Google Scholar]
- Fern R, Davis P, Waxman SG, Ransom BR. Axon conduction and survival in CNS white matter during energy deprivation: a developmental study. J Neurophysiol. 1998;79:95–105. doi: 10.1152/jn.1998.79.1.95. [DOI] [PubMed] [Google Scholar]
- Fitts DA. Improved stopping rules for the design of efficient small-sample experiments in biomedical and biobehavioral research. Behav Res Methods. 2010;42:3–22. doi: 10.3758/BRM.42.1.3. [DOI] [PubMed] [Google Scholar]
- Gesuete R, Packard AE, Vartanian KB, Conrad VK, Stevens SL, Bahjat FR, Yang T, Stenzel-Poore MP. Poly-ICLC preconditioning protects the blood-brain barrier against ischemic injury in vitro through type I interferon signaling. J Neurochem. 2012;123(Suppl 2):75–85. doi: 10.1111/j.1471-4159.2012.07946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamner MA, Möller T, Ransom BR. Anaerobic function of CNS white matter declines with age. J Cereb Blood Flow Metab. 2011;31:996–1002. doi: 10.1038/jcbfm.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS. Orbital vascular anatomy. Eye (Lond) 2006;20:1130–1144. doi: 10.1038/sj.eye.6702377. [DOI] [PubMed] [Google Scholar]
- Holm TH, Draeby D, Owens T. Microglia are required for astroglial Toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia. 2012;60:630–638. doi: 10.1002/glia.22296. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- Kamphuis E, Junt T, Waibler Z, Forster R, Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- Keinänen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH. A family of AMPA-selective glutamate receptors. Science. 1990;249:556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Ransom BR. Neuroglia. Ed 6. Oxford: Oxford UP; 2013. [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kirino T, Nakagomi T, Kanemitsu H, Tamura A. Ischemic tolerance. Adv Neurol. 1996;71:505–511. [PubMed] [Google Scholar]
- Lang J, Maeda Y, Bannerman P, Xu J, Horiuchi M, Pleasure D, Guo F. Adenomatous polyposis coli regulates oligodendroglial development. J Neurosci. 2013;33:3113–3130. doi: 10.1523/JNEUROSCI.3467-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PY, Stevens SL, Packard AE, Lessov NS, Yang T, Conrad VK, van den Dungen NN, Simon RP, Stenzel-Poore MP. Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke. 2012;43:1383–1389. doi: 10.1161/STROKEAHA.111.641522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. (1–9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Domercq M, Pérez-Samartin A, Ransom BR. Protecting white matter from stroke injury. Stroke. 2013;44:1204–1211. doi: 10.1161/STROKEAHA.112.658328. [DOI] [PubMed] [Google Scholar]
- Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, Antel JP, Moore CS. Roles of microglia in brain development, tissue maintenance and repair. Brain. 2015;138:1138–1159. doi: 10.1093/brain/awv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Mohr JP. Stroke: pathophysiology, diagnosis, and management. New York: Churchill Livingstone; 2004. [Google Scholar]
- Möller T, Hanisch UK, Ransom BR. Thrombin-induced activation of cultured rodent microglia. J Neurochem. 2000;75:1539–1547. doi: 10.1046/j.1471-4159.2000.0751539.x. [DOI] [PubMed] [Google Scholar]
- Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G. Do transient ischemic attacks have a neuroprotective effect? Neurology. 2000;54:2089–2094. doi: 10.1212/WNL.54.11.2089. [DOI] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Packard AE, Hedges JC, Bahjat FR, Stevens SL, Conlin MJ, Salazar AM, Stenzel-Poore MP. Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. J Cereb Blood Flow Metab. 2012;32:242–247. doi: 10.1038/jcbfm.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradillo JM, Fernández-López D, García-Yébenes I, Sobrado M, Hurtado O, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem. 2009;109:287–294. doi: 10.1111/j.1471-4159.2009.05972.x. [DOI] [PubMed] [Google Scholar]
- Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J, Merkler D, Detje C, Gutcher I, Mages J, Lang R, Martin R, Gold R, Becher B, Brück W, Kalinke U. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity. 2008;28:675–686. doi: 10.1016/j.immuni.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Matute C, Alberdi E. Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis. 2010;1:e54. doi: 10.1038/cddis.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gómez MV, Alberdi E, Ibarretxe G, Torre I, Matute C. Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci. 2003;23:9519–9528. doi: 10.1523/JNEUROSCI.23-29-09519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shichita T, Ago T, Kamouchi M, Kitazono T, Yoshimura A, Ooboshi H. Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J Neurochem. 2012;123(Suppl 2):29–38. doi: 10.1111/j.1471-4159.2012.07941.x. [DOI] [PubMed] [Google Scholar]
- Sommer B, Keinänen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- Sozmen EG, Hinman JD, Carmichael ST. Models that matter: white matter stroke models. Neurotherapeutics. 2012;9:349–358. doi: 10.1007/s13311-012-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAIR. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999;30:2752–2758. doi: 10.1161/01.STR.30.12.2752. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Simon RP. Genomics of preconditioning. Stroke. 2004;35:2683–2686. doi: 10.1161/01.STR.0000143735.89281.bb. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, King JS, Simon RP. Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke. 2007;38:680–685. doi: 10.1161/01.STR.0000251444.56487.4c. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP, Stenzel-Poore MP. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci. 2011;31:8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Ransom BR, Waxman SG. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 1991;546:18–32. doi: 10.1016/0006-8993(91)91154-S. [DOI] [PubMed] [Google Scholar]
- Su W, Hopkins S, Nesser NK, Sopher B, Silvestroni A, Ammanuel S, Jayadev S, Möller T, Weinstein J, Garden GA. The p53 transcription factor modulates microglia behavior through microRNA-dependent regulation of c-Maf. J Immunol. 2014;192:358–366. doi: 10.4049/jimmunol.1301397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Goldberg MP. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Ransom BR. Anoxia effects on CNS function and survival: regional differences. Neurochem Res. 2004;29:2163–2169. doi: 10.1007/s11064-004-6890-0. [DOI] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab. 2007;27:1540–1552. doi: 10.1038/sj.jcbfm.9600455. [DOI] [PubMed] [Google Scholar]
- Tian D, Dmitrieva RI, Doris PA, Crary JF, Sondhi R, Sacktor TC, Bergold PJ. Protein kinase M zeta regulation of Na/K ATPase: a persistent neuroprotective mechanism of ischemic preconditioning in hippocampal slice cultures. Brain Res. 2008;1213:127–139. doi: 10.1016/j.brainres.2008.03.046. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Black JA, Ransom BR, Stys PK. Anoxic injury of rat optic nerve: ultrastructural evidence for coupling between Na+ influx and Ca(2+)-mediated injury in myelinated CNS axons. Brain Res. 1994;644:197–204. doi: 10.1016/0006-8993(94)91680-2. [DOI] [PubMed] [Google Scholar]
- Wegener S, Gottschalk B, Jovanovic V, Knab R, Fiebach JB, Schellinger PD, Kucinski T, Jungehülsing GJ, Brunecker P, Müller B, Banasik A, Amberger N, Wernecke KD, Siebler M, Röther J, Villringer A, Weih M. Transient ischemic attacks before ischemic stroke: preconditioning the human brain? A multicenter magnetic resonance imaging study. Stroke. 2004;35:616–621. doi: 10.1161/01.STR.0000115767.17923.6A. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Zhang M, Kutlubaev M, Lee R, Bishop C, Andersen H, Hanisch UK, Möller T. Thrombin-induced regulation of CD95(Fas) expression in the N9 microglial cell line: evidence for involvement of proteinase-activated receptor(1) and extracellular signal-regulated kinase 1/2. Neurochem Res. 2009;34:445–452. doi: 10.1007/s11064-008-9803-9. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Koerner IP, Möller T. Microglia in ischemic brain injury. Future Neurol. 2010;5:227–246. doi: 10.2217/fnl.10.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Hamner MA, Brown AM, Evans RD, Ye ZC, Chen S, Ransom BR. Novel hypoglycemic injury mechanism: N-methyl-d-aspartate receptor-mediated white matter damage. Ann Neurol. 2014;75:492–507. doi: 10.1002/ana.24050. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang ZJ, Klaus JA, Koehler RC, Huang J. Delayed tolerance with repetitive transient focal ischemic preconditioning in the mouse. Stroke. 2008;39:967–974. doi: 10.1161/STROKEAHA.107.497412. [DOI] [PMC free article] [PubMed] [Google Scholar]