

Abstract
There are now nearly 300 single-gene inborn errors of immunity underlying phenotypes as diverse as infection, malignancy, allergy, auto-immunity, and auto-inflammation. For each of these five categories, a growing variety of phenotypes are ascribed to Primary Immunodeficiency Diseases (PID), making PIDs a rapidly expanding field of medicine. The International Union of Immunological Societies (IUIS) PID expert committee (EC) has published every other year a classification of these disorders into tables, defined by shared pathogenesis and/or clinical consequences. In 2013, the IUIS committee also proposed a more user-friendly, phenotypic classification, based on the selection of key phenotypes at the bedside. We herein propose the revised figures, based on the accompanying 2015 IUIS PID EC classification.
Keywords: Primary immunodeficiencies, classification, IUIS PID expert committee
Introduction
Human Primary Immunodeficiency Diseases (PID) comprise at least 300 genetically-defined single-gene inborn errors of immunity [1]. Long considered as rare diseases, recent studies tend to show that they are more common than generally thought, if only by their rapidly increasing number [2]. They may be even more common, if we consider the emerging monogenic determinants leading to common infectious diseases, such as severe influenza [3]; autoimmune diseases, such as systemic lupus erythematosus [4], and auto-inflammatory diseases, such as Crohn’s disease [5]. The International Union of Immunological Societies (IUIS) PID expert committee has proposed a PID classification [1], which facilitates clinical research and comparative studies world-wide; it is updated every other year to include new disorders or disease-causing genes. This classification is organized in tables, each of which groups PIDs that share a given pathogenesis. As this classification may be cumbersome for use by the clinician at the bedside, the IUIS PID expert committee recently proposed a phenotypic complement to its classification [6]. As the number of PIDs is quickly increasing, and at an even faster pace since the advent of next-generation sequencing, the phenotypic classification from 2013 became outdated and requires revision at the same pace as the classical IUIS classification. Our original phenotypic classification proved successful, which placed it in the 96th percentile for citation rank in Springer journals [7]. Given the success of our user-friendly classification of PIDs, providing a tree-based decision-making process based on the observation of clinical and biological phenotypes, we present here an update of these figures, based on the accompanying 2015 PID classification.
Methodology
We included all diseases included in the 2015 update of the IUIS PID classification [1], keeping the nine major categories unchanged. In addition, we considered other articles proposing a PID classification published recently [8, 9]. An algorithm was assigned to each of the nine main groups of the classification and the same color was used for each group of similar conditions. Disease names are presented in red and genes in bold. In addition, we classed diseases or genes from most common to less common, at the best of our knowledge [10, 11]. These algorithms were first established by a small committee; then validated by one or two experts for each figure.
Results
An update of our classification, validated by the IUIS PID expert committee, is presented in Figs. 1, 2, 3, 4, 5, 6, 7, 8 and 9.
Fig. 1.
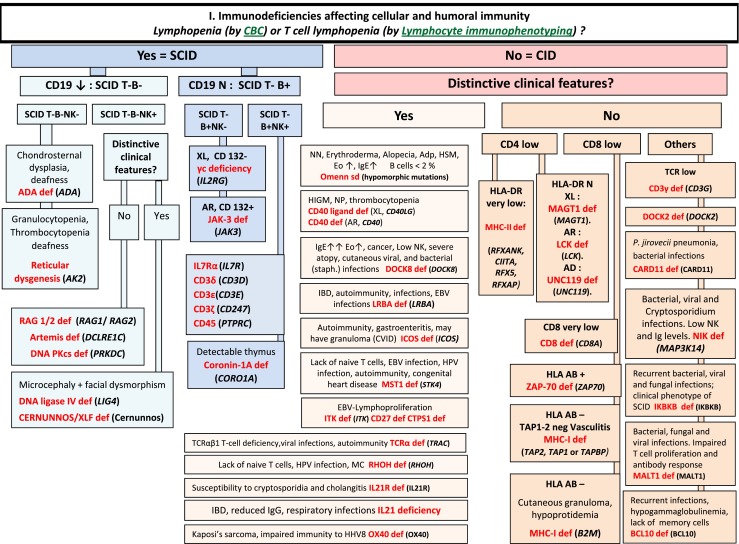
Immunodeficiencies affecting cellular and humoral immunity. ADA Adenosine Deaminase, Adp adenopathy, AR Autosomal Recessive inheritance, CBC Complete Blood Count, CD Cluster of Differentiation, CID Combined Immunodeficiency, EBV Epstein-Barr Virus, EO Eosinophils, HHV8 Human Herpes virus type 8, HIGM Hyper IgM syndrome, HLA Human Leukocyte Antigen, HSM Hepatosplenomegaly, HPV Human papilloma virus, IBD Inflammatory bowel disease, Ig Immunoglobulin, MC Molluscum contagiosum, N Normal, not low, NK Natural Killer, NN Neonatal, NP Neutropenia, SCID Severe Combined ImmunoDeficiency, Staph Staphylococcus sp., TCR T-Cell Receptor, XL X-Linked
Fig. 2.

CID with associated or syndromic features. These syndromes are generally associated with T-cell immunodeficiency. αFP alpha- fetoprotein, AD Autosomal Dominant inheritance, AR Autosomal Recessive inheritance, CMF Flow cytometry available, EDA Anhidrotic ectodermal dysplasia, EDA-ID Anhidrotic Ectodermal Dysplasia with Immunodeficiency, FILS Facial dysmorphism, immunodeficiency, livedo, and short stature, FISH Fluorescence in situ Hybridization, HSM Hepatosplenomegaly, HSV Herpes simplex virus, Ig Immunoglobulin, VZV Varicella Zoster virus, WAS Wiskott-Aldrich syndrome, XL X-Linked inheritance
Fig. 3.
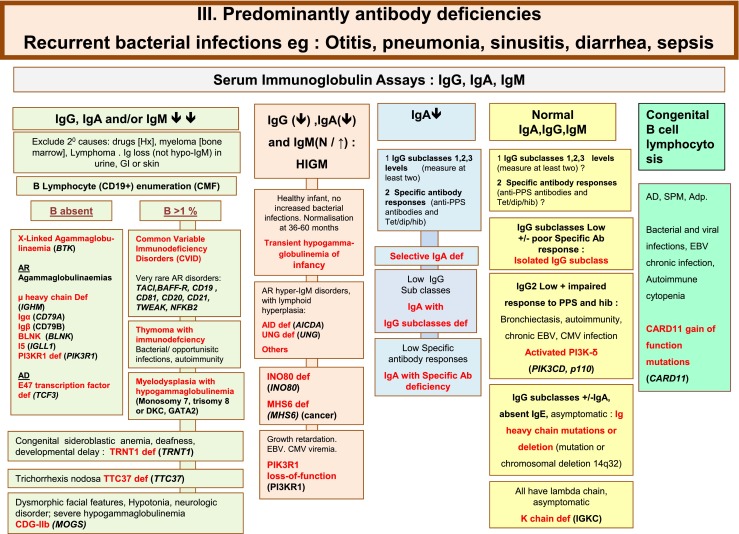
Predominantly Antibody deficiencies. Ab Antibody, Adp adenopathy, Anti PPS Anti- pneumococcus Antibody, AR Autosomal Recessive inheritance, CD Cluster of Differentiation, CDG-IIb Congenital disorder of glycosylation, type IIb, CMV Cytomegalovirus, CT Computed Tomography, EBV Epstein-Barr Virus, Dip Diphtheria, GI Gastrointestinal, Hib Haemophilus influenzae serotype b, Hx medical history, Ig Immunoglobulin, SPM Splenomegaly, subcl subclass, Tet Tetanus, XL X-Linked inheritance
Fig. 4.
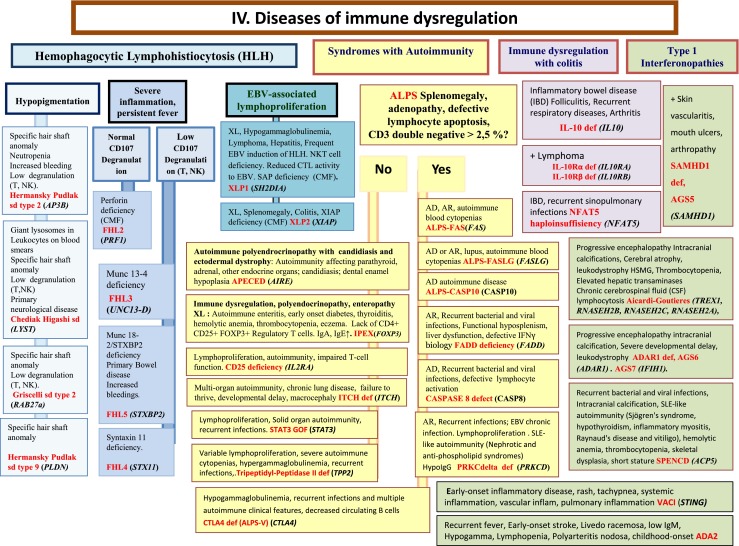
Diseases of Immune Dysregulation. AD Autosomal Dominant inheritance, ALPS Autoimmune lymphoproliferative syndrome, AR Autosomal Recessive inheritance, CD Cluster of Differentiation, CMF Flow cytometry available, CSF Cerebrospinal fluid, CTL Cytotoxic T-Lymphocyte, EBV Epstein-Barr Virus, GOF Gain-of-function, HLH Hemophagocytic lymphohistiocytosis, HSM Hepatosplenomegaly, IBD Inflammatory bowel disease, IFNγ Interferon gamma, Ig Immunoglobulin, IL interleukin, Inflam Inflammation, NK Natural Killer, NKT Natural Killer T cell, T T lymphocyte, XL X-Linked inheritance
Fig. 5.
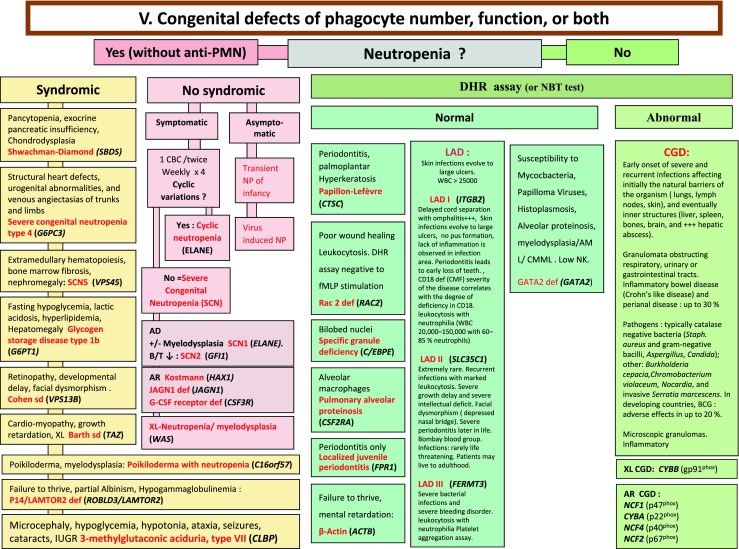
Congenital defects of phagocyte number, function, or both. For DHR assay, the results can distinct XL-CGD from AR-CGD, and gp40phox defect from others AR forms. AD Autosomal Dominant inheritance, AML Acute Myeloid Leukemia, AR Autosomal Recessive inheritance, BCG Bacilli Calmette-Guérin, CBC Complete Blood Count, CD Cluster of Differentiation, CGD Chronic Granulomatous Disease, CMML Chronic MyeloMonocytic Leukemia, DHR DiHydroRhodamine, IUGR Intrauterine growth retard, LAD Leukocyte Adhesion Deficiency, NP Neutropenia, PNN Neutrophils, SCN Severe congenital neutropenia, WBC White Blood Cells, XL X-Linked inheritance
Fig. 6.
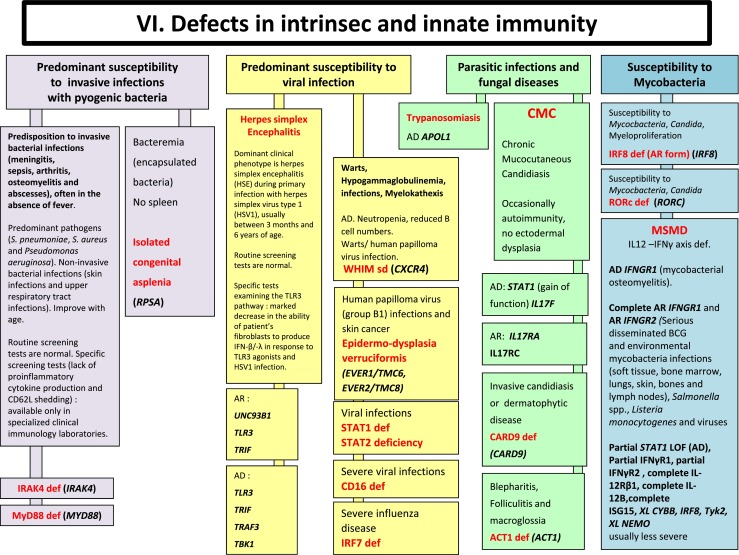
Defects in Intrinsec and Innate Immunity. AD Autosomal Dominant inheritance, AR Autosomal Recessive inheritance, BCG Bacilli Calmette-Guérin, BL B lymphocyte, CMC Chronic mucocutaneous candidiasis, HSV Herpes simplex virus, IFNγ Interferon gamma, Ig Immunoglobulin, IL interleukin, LOF Loss-of-function, MSMD Mendelian Susceptibility to Mycobacterial Disease, PMN Neutrophils, XL X-Linked inheritance
Fig. 7.
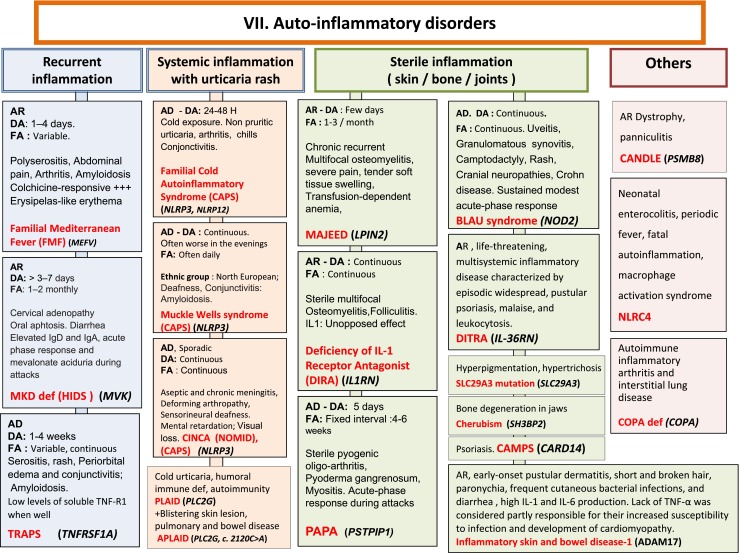
Autoinflammatory Disorders. AD Autosomal Dominant inheritance, AR Autosomal Recessive inheritance, CAMPS CARD14 mediated psoriasis, CANDLE Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome, CAPS Cryopyrin-Associated Periodic syndromes, CINCA Chronic Infantile Neurologic Cutaneous and Articular syndrome, DA Duration of Attacks, DITRA deficiency of interleukin 36 Receptor antagonist, FA Frequency of Attacks, HIDS Hyper IgD syndrome, Ig Immunoglobulin, IL interleukin, MKD Mevalonate Kinase deficiency, MWS Muckle-Wells syndrome, NOMID Neonatal Onset Multisystem Inflammatory Disease, PAPA Pyogenic sterile Arthritis, Pyoderma gangrenosum, Acne syndrome, SPM Splenomegaly, TNF Tumor Necrosis Factor, TRAPS TNF Receptor-Associated Periodic Syndrome
Fig. 8.
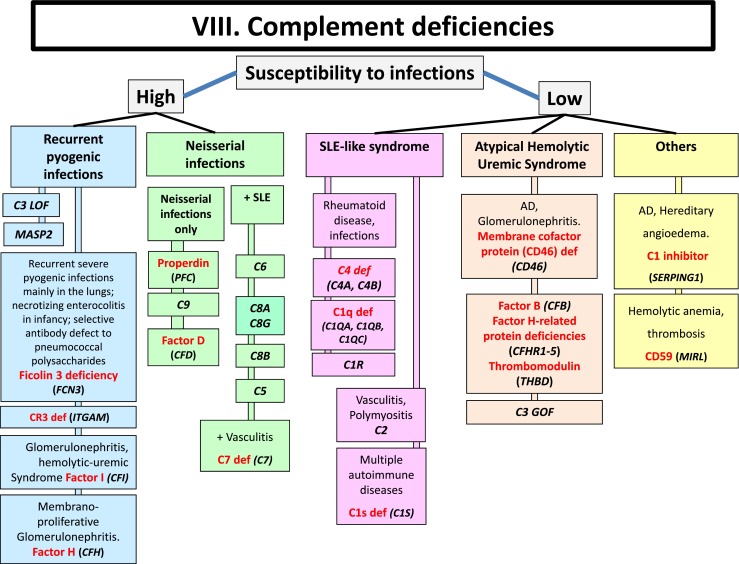
Complement deficiencies. AD Autosomal Dominant inheritance, GOF Gain-of-function, LOF Loss-of-function, LAD Leukocyte Adhesion Deficiency, SLE Systemic Lupus Erythematosus
Fig. 9.

Phenocopies of primary immunodeficiencies. Ab Antibody, ALPS Autoimmune lymphoproliferative syndrome, CMC Chronic mucocutaneous candidiasis, CID Combined Immunodeficiency, HUS Hemolytic uremic syndrome, IFNγ Interferon gamma, IL Interleukin, MSMD Mendelian Susceptibility to Mycobacteria Disease, VZV Varicella Zoster virus
Discussion
Since our 2013 study, 70 new diseases have been included in the 2015 classification. Four disorders have been removed, as the reports concerning associated immunodeficiency or genetic base were not confirmed. We also eliminated duplication of a disease in more than one figure and profoundly revised some figures, following the 2015 IUIS classification.
Conclusion
The IUIS PID expert committee developed this phenotypic classification in order to help clinicians at the bedside to diagnose PIDs but also to promote collaboration with national and international research centers. Needless to say, the expert committee encourages the development of other types of PID classification. Indeed, given the success encountered by the two current IUIS classifications, others classifications are likely to be useful and complementary.
Abbreviations
- αFP
Alpha- fetoprotein
- Ab
Antibody
- AD
Autosomal dominant inheritance
- ADA
Adenosine deaminase
- Adp
Adenopathy
- ALPS
Autoimmune lymphoproliferative syndrome
- AML
Acute myeloid leukemia
- Anti PPS
Anti- pneumococcus antibody
- AR
Autosomal recessive inheritance
- BCG
Bacilli Calmette-Guerin
- BL
B lymphocyte
- CAMPS
CARD14 mediated psoriasis
- CANDLE
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome
- CAPS
Cryopyrin-associated periodic syndromes
- CBC
Complete blood count
- CD
Cluster of differentiation
- CDG-IIb
Congenital disorder of glycosylation, type IIb
- CGD
Chronic granulomatous disease
- CID
Combined immunodeficiency
- CINCA
Chronic infantile neurologic cutaneous and articular syndrome
- CMC
Chronic mucocutaneous candidiasis
- CMF
Flow cytometry available
- CMV
Cytomegalovirus
- CMML
Chronic myelomonocytic leukemia
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- CT
Computed tomography
- CTL
Cytotoxic T-lymphocyte
- DA
Duration of attacks
- Def
Deficiency
- DHR
DiHydroRhodamine
- Dip
Diphtheria
- DITRA
Deficiency of interleukin 36 receptor antagonist
- EBV
Epstein-Barr virus
- EDA
Anhidrotic ectodermal dysplasia
- EDA-ID
Anhidrotic ectodermal dysplasia with immunodeficiency
- EO
Eosinophils
- FA
Frequency of attacks
- FCAS
Familial cold autoinflammatory syndrome
- FILS
Facial dysmorphism, immunodeficiency, livedo, and short stature
- FISH
Fluorescence in situ hybridization
- GI
Gastrointestinal
- GOF
Gain-of-function
- HHV8
Human herpes virus type 8
- Hib
Haemophilus influenzae serotype b
- HIDS
Hyper IgD syndrome
- HIES
Hyper IgE syndrome
- HIGM
Hyper Ig M syndrome
- HLA
Human leukocyte antigen
- HLH
Hemophagocytic lymphohistiocytosis
- HPV
Human papilloma virus
- HSM
Hepatosplenomegaly
- HSV
Herpes simplex virus
- HUS
Hemolytic uremic syndrome
- Hx
Medical history
- IBD
Inflammatory bowel disease
- IFNγ
Interferon gamma
- Ig
Immunoglobulin
- IL
Interleukin
- IUGR
Intrauterine growth retard
- LAD
Leukocyte adhesion deficiency
- LOF
Loss-of-function
- MC
Molluscum contagiosum
- MKD
Mevalonate kinase deficiency
- MSMD
Mendelian susceptibility to mycobacterial disease
- MWS
Muckle-wells syndrome
- N
Normal, not low
- NK
Natural killer
- NKT
Natural killer T cell
- NN
Neonatal
- NOMID
Neonatal onset multisystem inflammatory disease
- NP
Neutropenia
- PAPA
Pyogenic sterile arthritis, pyoderma gangrenosum, acne syndrome
- PMN
Neutrophils
- SCID
Severe combined immuno deficiency
- Sd
Syndrome
- SLE
Systemic lupus erythematosus
- SPM
Splenomegaly
- Staph
Staphylococcus sp.
- subcl
Subclass
- TCR
T-cell receptor
- Tet
Tetanus
- T
T lymphocyte
- TNF
Tumor necrosis factor
- TRAPS
TNF receptor-associated periodic syndrome
- VZV
Varicella zoster virus
- WBC
White blood cells
- XL
X-linked
References
- 1.IUIS classification (to be precised) 2015.
- 2.Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, et al. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013;33(1):1–7. doi: 10.1007/s10875-012-9751-7. [DOI] [PubMed] [Google Scholar]
- 3.Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science. 2015;348(6233):448–53. doi: 10.1126/science.aaa1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Troedson C, Wong M, Dalby-Payne J, Wilson M, Dexter M, Rice GI, et al. Systemic lupus erythematosus due to C1q deficiency with progressive encephalopathy, intracranial calcification and acquired moyamoya cerebral vasculopathy. Lupus. 2013;22(6):639–43. doi: 10.1177/0961203313486950. [DOI] [PubMed] [Google Scholar]
- 5.Sewell GW, Rahman FZ, Levine AP, Jostins L, Smith PJ, Walker AP, et al. Defective tumor necrosis factor release from Crohn’s disease macrophages in response to Toll-like receptor activation: relationship to phenotype and genome-wide association susceptibility loci. Inflamm Bowel Dis. 2012;18(11):2120–7. doi: 10.1002/ibd.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bousfiha AA, Jeddane L, Ailal F, Al Herz W, Conley ME, Cunningham-Rundles C, et al. A phenotypic approach for IUIS PID classification and diagnosis: guidelines for clinicians at the bedside. J Clin Immunol. 2013;33(6):1078–87. doi: 10.1007/s10875-013-9901-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Springer Science. In: Citations Springer. 2015. http://citations.springer.com/item?doi=10.1007/s10875-013-9901-6. Accessed 20 Jul 2015.
- 8.Ochs HD, Hagin D. Primary immunodeficiency disorders: general classification, new molecular insights, and practical approach to diagnosis and treatment. Ann Allergy Asthma Immunol. 2014;112(6):489–95. doi: 10.1016/j.anai.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Federici S, Martini A, Gattorno M. The central role of anti-IL1 blockade in the treatment of monogenic and multi-factorial autoinflammatory diseases. Front Immunol. 2013;4:351. doi: 10.3389/fimmu.2013.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Modell V, Knaus M, Modell F, Roifman C, Orange J, Notarangelo LD. Global overview of primary immunodeficiencies: a report from Jeffrey Modell Centers worldwide focused on diagnosis, treatment, and discovery. Immunol Res. 2014;60(1):132–44. doi: 10.1007/s12026-014-8498-z. [DOI] [PubMed] [Google Scholar]
- 11.Online Mendelian Inheritance in Man (OMIM). An Online Catalog of Human Genes and Genetic Disorders. In: Online Mendelian Inheritance in Man. http://omim.org/ Accessed 20 Jul 2015.