

SUMMARY
The regulation of protein synthesis contributes to gene expression in both normal physiology and disease, yet kinetic investigations of the human translation mechanism are currently lacking. Using single-molecule fluorescence imaging methods, we have quantified the nature and timing of structural processes in human ribosomes during single-turnover and processive translation reactions. These measurements reveal that functional complexes exhibit dynamic behaviors and thermodynamic stabilities distinct from those observed for bacterial systems. Structurally defined sub-states of pre- and post-translocation complexes were sensitive to specific inhibitors of the eukaryotic ribosome demonstrating the utility of this platform to probe drug mechanism. The application of three-color single-molecule FRET methods further revealed a long-distance allosteric coupling between distal tRNA binding sites within ribosomes bearing three tRNAs, which contributed to the rate of processive translation.
INTRODUCTION
Ribosome-catalyzed protein synthesis is central to faithful gene expression in all organisms. Correspondingly, the translation mechanism has been the subject of intense investigation since before the ribosome's discovery more than fifty years ago (Garrett, 1999). Advances in high-resolution structure-determination, stopped-flow kinetics and single-molecule imaging focused on the individual stages of bacterial protein synthesis - initiation, elongation, termination and recycling – have revealed that each step is regulated by large-scale, coordinated conformational changes within the ribosome and associated translation machinery (Ehrenberg, 2010; Frank, 2012; Green et al., 1997; Munro et al., 2008; 2009; Rodnina, 2013).
The importance of translation control in human physiology is widely recognized (Ruggero, 2013), and mounting evidence implicates aberrant protein synthesis in a number of human disease states (Stumpf and Ruggero, 2011). Emerging methods enabling the first genome-wide investigations of global translation activity and regulation have revealed that individual mRNAs are transited with non-uniform rates (Ingolia et al., 2011). Quantitative analyses suggest that molecular processes within individual ribosomes may constitute a critical and general feature of translation control of gene expression (Munro et al., 2009; Subramaniam et al., 2014), yet little is presently known regarding the nature and timing of conformational events underpinning human ribosome functions.
The human ribosome is a 4.3 MDa, 80S complex composed of a large (60S) and small (40S) subunit assembled from four distinct ribosomal RNA (rRNA) molecules and approximately 80 ribosomal proteins (r-proteins) (Anger et al., 2013; Behrmann et al., 2015). Peripheral to its conserved structural core, human ribosomes contain unique rRNA expansion segments (ES) as well as additional r-proteins making them significantly larger than their prokaryotic counterparts. The functional impact of these structural distinctions is not fully understood.
Global features of the translation cycle are thought to be conserved across species (Budkevich et al., 2014; Kapp and Lorsch, 2004). In eukaryotes, the initiation of protein synthesis pairs the aminoacylated tRNA (aa-tRNA) substrate with the messenger RNA (mRNA) “start” codon within the Peptidyl (P) site of the 80S initiation complex (IC). Protein is made during the elongation cycle, a process in which the evolutionarily conserved GTPases eukaryotic Elongation Factors 1A (eEF1A) and 2 (eEF2) repetitively bind and hydrolyze GTP at the leading edge of the ribosome. eEF1A chaperones aa-tRNA into the aminoacyl (A) site by forming a ternary complex with aa-tRNA and GTP. Proper mRNA codon-tRNA anticodon pairing within the 40S decoding region triggers eEF1A to hydrolyze GTP, allowing entry of the aminoacylated 3’-CCA terminus of aa-tRNA into the peptidyltransferase center (PTC) of the 60S subunit. There, peptide bond formation transfers the nascent polypeptide from P-site tRNA to A-site tRNA, extending its length by one residue. eEF2 operates on this pre-translocation (PRE) complex to promote directional translocation of the tRNA-mRNA module, forming the post-translocation (POST) complex, which bears peptidyl-tRNA in the P site and is competent for further elongation cycles. While providing an essential foundation for understanding the protein synthesis mechanism in mammalian systems, this framework falls short of explaining the prevalence of eukaryote- and prokaryote-specific antibiotics (Wilson et al., 2005) and structural evidence (Budkevich et al., 2011; 2014) indicating that important features of the mammalian protein synthesis mechanism may be functionally divergent.
Towards the goal of gaining quantitative insights into conserved and divergent aspects of the human translation mechanism, we have established the means to interrogate reconstituted human translation reactions in vitro using single-molecule fluorescence resonance energy transfer (smFRET) imaging methods. This approach sheds new light on compositional and conformational processes within the 80S ribosome that directly impact function, mechanisms of drug-mediated inhibition and functional distinctions of the human translation apparatus that contribute to regulation. These data include a direct demonstration of A site-E site allostery, a mechanism that has been debated for almost 30 years (Rheinberger and Nierhaus, 1986; Petropoulos and Greene, 2012). Such findings highlight the potential of single-molecule imaging to offer insights into translation regulation and mis-regulation in humans.
RESULTS
Human 80S ribosomes are translationally active in vitro
Functional ribosome complexes were prepared using fluorescently labeled E. coli tRNA and subunits derived from the actively translating pool of polysomes in HEK293T cells (Figures S1A, S1B; Table S1; Supplemental Information) (Blanchard et al., 2004; Budkevich et al., 2011). Activity assays were performed using in vitro assembled 80S initiation complexes (ICs) programmed with initiator Met-tRNAiMet on a synthetic mRNA following “factor-free” initiation procedures (Supplemental Information).
The efficiency of 80S localization at the single AUG start codon was verified by tracking codon-dependent de-quenching of a Cy3B-labeled Phe-tRNAPhe ternary complex (Burnett et al., 2014) upon aa-tRNA accommodation into the A site (Supplemental Information). The 80S IC concentration-dependence of the de-quenching activity indicated that >75% of ribosomes were properly initiated and functional for tRNA selection (Figure 1A). These results were corroborated by ribosome profiling which showed that ribosome protected fragments tightly clustered around the AUG start codon (Figure S1C; Supplemental Information). 80S ICs were also competent to carry out processive translation reactions comprised of multiple rounds of aa-tRNA selection, peptide bond formation and translocation. As expected, reactions were sensitive to the eukaryotic-specific inhibitors cycloheximide (CHX) and harringtonine (HTN) (Fresno et al., 1977; Garreau de Loubresse et al., 2014) (Figure 1B).
Figure 1. Human ribosomes are translationally active in vitro.
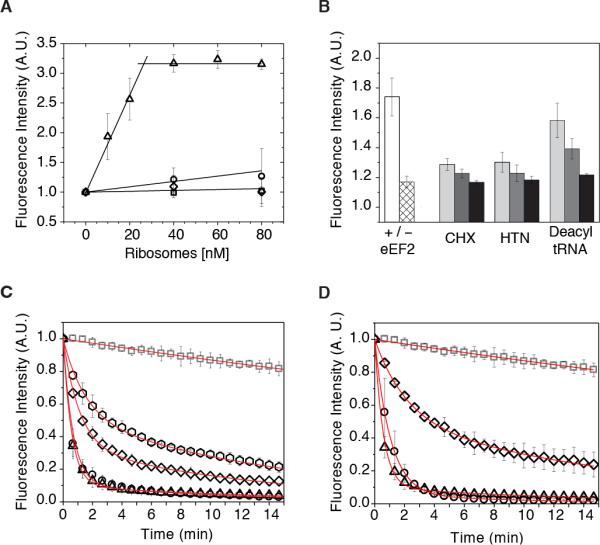
(A) “Fraction-active” assay reporting on the competency of 80S ICs assembled in vitro to accept (Cy3B)Phe-tRNAPhe into the A site. The incorporation of (Cy3B)Phe-tRNAPhe into 80S IC's programmed with Met-tRNAiMet in the P-site and a UUC codon in the A site (triangles); UCU codon in the A site (squares); UUC codon in the A site but lacking P-site tRNA (circles); in the absence of mRNA (diamonds). (B) “Processive translation assay” monitoring (Cy3B)Phe-tRNAPhe ternary complex de-quenching upon mixing with 80S ICs. The left cluster represents biological triplicates in the presence (white bar) and absence (hatched bar) of eEF2. Bar graphs to the right represent biological triplicates of ligand-induced inhibition in the presence of eEF2 and (from left) 5 μM (light gray), 50 μM (dark gray) or 500 μM (black) cycloheximide (CHX); 0.1 μM (light gray), 1 μM (dark gray) or 10 μM (black) harringtonine (HTN); 0.005 μM (light gray), 0.05 μM (dark gray) or 0.5 μM (black) deacylated tRNA. (C) “Puromycin peptidyltransferase assay”: photobleaching control (gray squares); 2 mM PMN (triangles); 2 mM PMN plus 200 μM anisomycin (hexagons), 2 mM PMN plus 20 μM sparsomycin (diamonds), 2 mM PMN plus 200 μM chloramphenicol (circles). (D) PMN assay performed on distinct elongation cycle intermediates: 80S IC photobleaching control (gray squares), 80S IC plus 2 mM PMN (triangles); PRE complex plus 2 mM PMN (diamonds), POST complex plus 2 mM PMN (circles). Data are represented as mean +/− SD. See also Figure S1, Tables S1, S2.
Surface-immobilized human 80S ribosomes are translationally active
In order to track the behaviors of individual human ribosomes over extended periods, 80S ICs were immobilized via a biotin-streptavidin bridge within passivated microfluidic chambers and imaged using wide-field, prism-based total internal reflection fluorescence (TIRF) microscopy. We first examined the PTC activities of 80S ICs prepared with Cy3(DSP)-Met-tRNAMet in the P site using the inhibitor puromycin (PMN), an analog of the 3’ –CCA end of tRNA that releases nascent polypeptides from translating ribosomes (Blanchard et al., 2004) (Figure S1D; Supplemental Information). Consistent with robust activity, ~90% of the complexes exhibited a rapid loss of the Cy3-Met amino acid upon stopped-flow PMN addition (2 mM) (Figures 1C, 1D and Table S2). The specificity of this reaction was confirmed by its efficient inhibition by both anisomycin, a eukaryotic-specific inhibitor, and sparsomycin, a universal inhibitor (Hansen et al., 2003; Tscherne and Pestka, 1975), and by the reaction's insensitivity to the bacteria-specific inhibitor chloramphenicol (Figure 1C, Table S2).
Competency in single-step, factor-catalyzed aa-tRNA selection and translocation reactions was further verified by assessing the PMN-reactivity of pre- (PRE; A- and P-site bound tRNA) and post- (POST; P- and E-site bound tRNA) translocation complexes. As anticipated by investigations of the bacterial translation apparatus (Blanchard et al., 2004; Green et al., 1998; Moazed and Noller, 1989; Munro et al., 2009; Sharma et al., 2007), the PRE complex, formed by exposure to a cognate ternary complex of eEF1A•GTP•Phe-tRNAPhe (50 nM), exhibited an ~2.5- fold reduction in PMN-reactivity (Figure 1D and Table S2). Consistent with this defect arising from the occlusion of the PMN binding site by A-site bound peptidyl-tRNA, fast PMN reactivity was restored in the POST complex, formed by briefly incubating the PRE complex with eEF2 (2 μM) and GTP (1 mM) (Figure 1D and Table S2). These findings indicated that surface-immobilized human 80S ribosomes were competent for the principal elongation reactions of aatRNA selection and translocation.
tRNA selection on the human 80S ribosome is a multi-step process
To directly monitor the rate and efficiency of aa-tRNA incorporation into surface-immobilized 80S ICs, the process of tRNA selection was imaged under pre-steady state conditions by stopped-flow addition of an eEF1A•GTP•(Cy5)Phe-tRNAPhe ternary complex (20 nM) to ribosomes containing (Cy3)Met-tRNAiMet in the P-site (Figure 2A; Supplemental Information). The binding of ternary complex, marked by the appearance of FRET, exhibited a bimolecular rate constant (~170 μM−1 s−1) consistent with a diffusion-limited macromolecular interaction (Figure S2A) (Blanchard et al., 2004; Rodnina and Wintermeyer, 2001). Ternary complex-ribosome interactions fell into two distinct classes, which in accordance with prior investigations in bacteria (Geggier et al., 2010) were referred to as “non-productive” (Figures S2B-D) and “productive” selection events (Figures 2B, 2C). Idealization of the raw data using hidden Markov modeling (HMM) (Supplemental Information) revealed that productive ternary complex encounters were characterized by a transient low-FRET (~0.25) state that rapidly proceeded to a long-lived, (~500 ms) on-path intermediate-FRET (~0.44) conformation, which then ultimately achieved a high-FRET (~0.69) configuration after what appeared to be repeated attempts to do so (Figure 2B, C left panels). A transition density plot (TDP), which summarizes the specific FRET transitions observed in the idealized data set (McKinney et al., 2006), further illuminated the discrete steps transited along the reaction coordinate, as well as its directionality, as evidenced by the accumulation of specific peaks above the diagonal axis. The microscopically reversible nature of aa-tRNA movements between intermediate- and high-FRET states, as directly seen in individual FRET traces (Figure 2B), was reflected by the peak below the diagonal axis (Figure 2C, right panel).
Figure 2. The human 80S pre-translocation complex undergoes large-scale spontaneous conformational changes.

(A) Schematic representation of aa-tRNA selection depicting the positions of P- and A-site tRNAs site-specifically labeled with Cy3 (green sphere) and Cy5 (red sphere), respectively. aa-tRNA enter the A site in ternary complex with eEF1a (blue) and GTP. (B) Representative pre-steady state single-molecule FRET recording of productive aa-tRNA selection. (C) Population FRET histograms (left panels) and transition density plot (right panel) showing the distribution of FRET states and the frequency of transitions between states, respectively, observed during aa-tRNA selection. Codon recognition (CR), GTPase activated (GA) and fully accommodated (AC) states are indicated (far right). (D) Representative steady-state single-molecule FRET recording of the pre-translocation (PRE) complex. (E) Population FRET histograms (left panels) and transition density plot (right panel) showing the distribution of FRET states and the frequency of transitions between states, respectively, within the PRE complex. Classical (C) and hybrid (H) FRET states are indicated (far right). In FRET histograms and TDPs, FRET state occupancy is indicated by a color heat-map ranging from (least populated) white – blue – green – yellow – orange – red (most populated). See also Figure S2.
From these results, we conclude that the reaction coordinate of aa-tRNA entry into the human ribosome is globally analogous to bacterial systems (Geggier et al., 2010). During initial selection, aa-tRNA rapidly transit a low-FRET, codon-recognition (CR) state to an intermediate-FRET, GTPase-activated (GA) state, an A/T-like configuration where eEF1A-catalyzed GTP hydrolysis occurs. During proofreading, aa-tRNA enters into the PTC, reversibly sampling a high-FRET, fully accommodated (AC) state prior to forming the PRE complex in which A- and P-site tRNAs are “classically” (A/A; P/P) configured (Figures 2A-C). Consistent with these assignments, and analogous to the actions of non-hydrolyzable GTP analogues (Geggier et al., 2010) and hygromycin A (Polikanov et al., 2015) in bacterial systems, the process of tRNA selection on the human ribosome was efficiently blocked within the intermediate-FRET, GA state by GTPγS and harringtonine (Figure S2E). However, as suggested by recent structural investigations (Budkevich et al., 2014), the extended duration of the intermediate-FRET state observed on the uninhibited human ribosome suggests that the nature and timing of aa-tRNA motions during tRNA selection are distinct between bacteria and mammals. This potential divergence in the fidelity mechanism in higher organisms warrants in-depth examinations.
The pre-translocation complex is conformationally metastable
Subsequent to aa-tRNA accommodation at the A site, the PRE complex, imaged under steady state conditions, exhibited predominantly non-classical (lower) FRET states (Figures 2D, 2E) suggesting that the energy of aa-tRNA binding and/or peptide bond formation efficiently drives restructuring of the ribosome such that classical tRNA positions become disfavored (Marshall et al., 2008). Individual FRET traces revealed that the PRE complex spontaneously transits between low- (~0.19), intermediate- (~0.37) and high-FRET (~0.69) states (Figure 2D). These observations suggest that tRNAs within the A and P sites of the human ribosome undergo large-scale (ca. 15-25 Å) rearrangements in relative and absolute positions as a function of time. In the bacterial ribosome, such transitions reflect global remodeling events at the interface of the small and large subunits that are directly implicated in ribosome function, including a 6-9° subunit rotation that directs both coupled and independent movements of A- and P-site tRNAs between classical (A/A, P/P) and hybrid (A/A, P/E; A/P, P/E) positions (Cornish et al., 2008; Dunkle et al., 2011; Feldman et al., 2010; Frank and Agrawal, 2000; Moazed and Noller, 1989; Munro et al., 2007; 2009; Wang et al., 2011).
The generation of a TDP from the idealized FRET data enabled assessment of the prevalence and nature of these conformational processes in the human PRE complex (Figure 2E, right panel). As anticipated for a system in dynamic equilibrium, four clearly distinguished peaks symmetrically distributed with respect to the diagonal axis were observed. (Figures S2F-H). FRET transitions between low- (~0.19) and intermediate-FRET (~0.37) states were the most abundant, whereas intermediate- to high-FRET were less frequent and low- to high-FRET transitions were notably absent (Figure 2E). Thus, in contrast to bacterial PRE complexes (Munro et al. 2007), tRNA movements between classical and hybrid positions appear to preferentially occur through coupled motions on the human ribosome.
Ligands stabilize distinct sub-states of the human PRE complex
To verify the physical basis of the FRET states observed in the PRE complex, steady state experiments were performed in the presence of ligands known to modulate A- and P-site tRNA positions (Budkevich et al., 2011). To fill the E site and promote a classically configured PRE complex, deacylated tRNAArg was titrated into the imaging buffer. As anticipated, the high-FRET (~0.69) classical (A/A; P/P) PRE complex configuration was stabilized in a concentration-dependent fashion (Figure 3A). Similar results were obtained when experiments were performed using mammalian bulk deacylated tRNA (Figure S3A). The apparent EC50 of E-site tRNAArg binding was approximately 10 nM (Figure S3B), an order of magnitude higher affinity than found for rabbit ribosomes (Budkevich et al., 2011). Such disparities may reflect compositional differences in the PRE complexes examined (i.e. tRNA identities) or yet to be identified physical distinctions of the E sites of human and rabbit ribosomes.
Figure 3. Ligand-induced changes of the human 80S pre-translocation complex.

Population FRET histograms showing the impact of (A) deacylated-tRNAArg (B) cycloheximide (CHX) and (C) harringtonine (HTN) on the equilibrium distribution of FRET states in the human 80S PRE complex bearing Cy3- and Cy5-labeled P- and A-site tRNAs, respectively. See also Figure S3.
We next examined the impact of CHX, a potent inhibitor of the translocation mechanism that binds the large subunit E site in a position that physically overlaps with that of the 3’-CCA end of P/E tRNA (Budkevich et al., 2011; Garreau de Loubresse et al., 2014). In line with this mode of binding, CHX stabilized the high-FRET (~0.69) classical configuration of the 80S PRE complex (Figure 3B). Consistent with its inhibitory concentrations in vitro (Figure 1B) and in cells (Ingolia et al., 2011), the apparent EC50 of this impact was approximately 35 μM (Figure S3C), a value again more pronounced than observed for rabbit ribosomes (Budkevich et al., 2011). Collectively, these findings argue that FRET fluctuations in the human PRE complex reflect global conformational processes in the ribosome that direct changes in A- and P-site tRNA positions, and likewise indicate that CHX and deacylated tRNA exert their effects by capturing classical configurations of the PRE complex once they have been spontaneously achieved.
We next investigated the impact of HTN, a PTC inhibitor that binds within the large-subunit A site in a region that overlaps with the 3’-CCA terminus of classically positioned peptidyl-tRNA (Garreau de Loubresse et al., 2014). Consistent with its established binding mode, the supposition that low- and intermediate-FRET states reflect hybrid (P/E) tRNA configurations and the notion that peptidyl-tRNA is dynamic within the A site (Budkevich et al., 2011; Munro et al., 2007; Wang et al., 2011), HTN de-populated both low- and high- FRET states to stabilize an intermediate- (~0.42) FRET configuration (Figure 3C). In line with its inhibitory concentrations in vitro (Figure 1B) and in cells (Fresno et al., 1977; Ingolia et al., 2011), the estimated EC50 was ~1 μM (Figure S3D). These findings establish that HTN can bind with high affinity to the large subunit of the PRE complex to stabilize hybrid tRNA positions by preventing the return of peptidyl-tRNA to a classical (A/A) position. Correspondingly, the intermediate-FRET state stabilized by HTN (~0.42) reflects a ribosome configuration in which the elbow domains of both A- and P-site tRNAs adopt hybrid (A/P; P/E) positions (Budkevich et al., 2011), whereas the intermediate-FRET state observed in the absence of drug (~0.37) likely corresponds to a time-averaged FRET value arising from the rapid exchange of peptidyl-tRNA between classical (A/A) and hybrid (A/P) configurations within the A site.
eEF2 translocates the human ribosome from the hybrid state
To examine the competency of surface-immobilized PRE complexes to directionally translocate, pre-steady state smFRET experiments were performed in the presence of eEF2 (2 μM) and GTP (1 mM). Stopped-flow addition of eEF2•GTP rapidly and efficiently converted PRE complexes to a predominantly high-FRET (~0.82) configuration (Figures 4A, 4B). HTN, which stabilizes hybrid tRNA configurations (Figure 3C), had no observable impact on this process, however formation of this high-FRET configuration was not observed in the presence of saturating concentrations of either CHX (500 μM) or deacylated tRNA (1 μM) (Figure S4A). These findings suggest that the PRE complex is efficiently translocated by eEF2 and that E-site ligands inhibit this process by stabilizing classical tRNA configurations.
Figure 4. eEF2 rapidly catalyzes formation of a stable post-translocation state.

(A) A representative single-molecule FRET trace shows the time between injection of eEF2 and the transition to high FRET upon translocation (gray) and a dynamic POST complex. (B) Population FRET histograms show the distribution of states during translocation. From left: pre translocation complex (PRE); real-time delivery (RTD) of eEF2; post-translocation complex (POST). (C) Translocation monitored by imaging PRE complexes during the stopped-flow delivery of increasing concentrations of eEF2 in the presence of GTP (1 mM) or GDPNP (1 mM) and monitoring the conversion of individual FRET trajectories to a distinct high- (~0.82) FRET state. Curves from top: 5 μM (~ 8 s−1) squares; 250 nM (~ 4 s−1) circles; 50 nM (~ 2 s−1) triangles; 2 μM + GDPNP (~0.4 s−1) diamonds; no eEF2 (no translocation measured) inverted triangles. Data are represented as mean +/− SEM. (D) The KM of eEF2's interaction with the PRE complex was estimated by the 0.5Vmax of tfast (triangles) to be approximately 400 nM. tslow (circles) did not change with eEF2 concentration. Data are represented as mean +/− SD. See also Figure S4.
Inspection of individual FRET trajectories revealed that eEF2-induced entry into high-FRET states occurred predominantly via hybrid state PRE complex configurations (Figure 4A). Post-synchronized population FRET histograms confirmed this observation and revealed that the transition into high-FRET states generally occurred within one to two imaging frames (80-120 ms) (Figure S4B). These data indicate that eEF2 rapidly acts on the PRE complex to reduce the distance between E- and P-site tRNAs to resolve into a POST configuration (~0.82 FRET) where tRNAs are compacted (by ~10 Å) compared to the PRE complex (A/A, P/P) (Behrmann et al., 2015).
As expected for a reaction driven by a bimolecular interaction between eEF2 and the ribosome, the time delay (Δttrans) between eEF2•GTP addition and appearance of high (>0.7) FRET states (Figure 4A) decreased in an eEF2 concentration-dependent fashion. This effect was reduced in the presence of the non-hydrolyzable GTP analogue GDPNP (Figure 4C). At each eEF2 concentration, the apparent translocation rate was best approximated as a double exponential process, suggesting the existence of fast (tfast ~73%) and slow (tslow ~27%) populations. Notably, only the rate of the tfast population exhibited an eEF2 concentration-dependence (Figure 4D). At the highest concentration examined (5 μM or 10*KM = ~0.5 μM), the rapid eEF2-induced transition (tfast) occurred at a rate of ~8 s−1, roughly an order of magnitude faster than tslow (~1 s−1). Hence, akin to bacterial systems (Munro et al., 2010b; Wang et al., 2011), we conclude that eEF2•GTP preferentially translocates hybrid state (A/P; P/E) PRE complexes, whereas complexes exhibiting classical tRNA configurations must first spontaneously (ca. 1 s−1) adopt hybrid configurations before translocation can occur.
Although the POST complex exhibited thermodynamic stability (Figure 4B), individual FRET trajectories revealed structural dynamics characterized by intermittent (~1 s−1) fluctuations to lower-FRET configurations (~0.38-0.55) that were transient in nature (~250 ms) (Figures 4A and S4C). While the specific events in the POST complex giving rise to such large changes in inter-tRNA distance (~20-30 Å) are not presently known, these data are consistent with E-site tRNA movements within the human POST complex (Behrmann et al., 2015). Conformational processes anticipated to impact the positioning of E-site tRNA may include reversible motions of the L1-stalk domain away from the subunit body to extended open positions (Munro et al., 2010b; Yamamoto et al., 2014) as well as subunit rotation and rolling (Budkevich et al., 2014).
Binding of aa-tRNA at the A site promotes E-site tRNA release
The apparent stability of the POST complex and the estimated bimolecular rate constant of ternary complex binding (Figure S2A) predicted that the POST complex could be the physiological substrate for tRNA selection. This model is particularly relevant as prior work has suggested the existence of an allosteric mechanism between the A and E sites of the ribosome that contributes to the fidelity of translation (Budkevich et al., 2008; Budkevich et al., 2014; Marquez et al., 2004; Nierhaus, 1990; Rheinberger and Nierhaus, 1986). Correspondingly, we set out to determine whether aa-tRNA binding at the A site influences the positioning, dynamics and stability of E-site tRNA. To do so, ribosomes were driven through two sequential elongation cycles while monitoring the dissociation rate of (Cy3)tRNAiMet from the E site via time-lapse imaging.
As anticipated, both PRE and POST complexes exhibited characteristic FRET signatures (Figure 5A, first and second panel) and notable thermodynamic stability (t1/2 ~35 minutes) (Figure 5B). A PRE2 complex was formed by enzymatic delivery of an eEF1A•GTP•Phe-tRNAPhe ternary complex (50 nM). Following a brief incubation, a small shift towards an even higher-FRET (~0.84) configuration was observed (Figure 5A, third panel), suggesting that the accommodation of aa-tRNA at the A site induces further compaction (≤ 5Å) between E- and P-site tRNAs. Such changes may arise from subunit rolling, which shifts the position of P-site tRNA towards the E site (Budkevich et al., 2014), and/or L1 stalk closure. In notable contrast to the POST complex, the rate of Cy3-labeled E-site tRNA dissociation increased by an order of magnitude (t1/2 ~2.8 minutes) (Figure 5B). As a consequence, the number of molecules exhibiting FRET decreased over time (Figure 5A, right panel). Hence, as initially posited by the model of long-distance, allosteric coupling between the A and E sites (Nierhaus, 1990; Rheinberger and Nierhaus, 1986), these findings demonstrate that formation of peptidyl-tRNA in the A site of the PRE2 complex induces global conformational changes that contribute to the release of deacylated tRNA from the E site.
Figure 5. E-site tRNA dissociates more rapidly from the PRE2 complex.
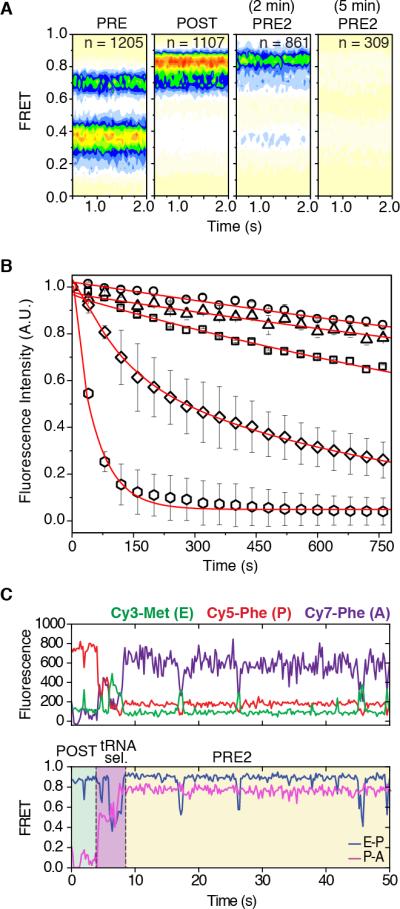
(A) Population FRET histograms show the distribution of states in the PRE complex (first panel), POST complex (second panel), PRE2 complex after 2 min (third panel) and the PRE2 complex after 5 min (last panel). (B) Cy3-tRNAMet dissociation from the E site tracked by the loss of fluorescence via time-lapse imaging. E-site tRNA stability for 80S IC (~0.03 min−1) squares; PRE (~0.02 min−1) circles; POST (~0.02 min−1) triangles; PRE2 (~0.25 min−1) diamonds; PRE2 + eEF2 (~1.2 min−1) hexagons. Data are represented as mean +/− SD. (C) A representative pre-steady state single-molecule FRET trace of productive aa-tRNA selection showing dynamic motions within the ribosome bearing three fluorescently-labeled tRNAs. The area in green indicates the time occupied by a stable POST complex before delivery of Cy7-labeled ternary complex; tRNA selection is highlighted in blue; the PRE2 complex is highlighted in yellow. See also Figure S5.
Three-color smFRET imaging reveals long-distance allostery between the A and E sites
In order to directly track PRE2 complex formation and to examine the changes in dynamics that contribute to E-site tRNA release, pre-steady state three-color smFRET experiments were performed in which a (Cy7)Phe-tRNAPhe ternary complex was enzymatically delivered to the POST complex bearing (Cy3)tRNAiMet and Met-Phe-(Cy5)tRNAPhe in the E- and P-sites, respectively. As observed for tRNA selection on the 80S IC (Figure 2B), inspection of individual FRET trajectories revealed that Cy7-labeled aa-tRNA enters the A site in a step-wise manner in which at least one long-lived (~0.5-1 second) intermediate was evidenced (Figures 5C, S5A and S5B). Notably, Cy3-labeled tRNA remained bound within the E site throughout the selection process. Complete accommodation of aa-tRNA into the A site was marked by high FRET between Cy5 and Cy7 (~0.8 FRET).
Fluctuations in Cy3-Cy5 FRET (ca. Δ0.2-0.4), consistent with transient increases in distance (~10-20 Å) between E- and P-site tRNAs, were present both before and after aa-tRNA accommodation into the A site (Figure 5C). Strikingly, rearrangements of this kind were observed to occur during the tRNA selection process (Figures 5C, S5A and S5B), indicating that compaction of the A site (Budkevich et al., 2014) is sufficient to trigger fluctuations in distance between E- and P-site tRNAs. Based on the measured inter-tRNA distances within the human ribosome (Behrmann et al., 2015), high-FRET configurations observed before and after tRNA selection reflect classical tRNA positions. Thus, the relatively transient lower-FRET states likely arise from conformational process within the ribosome, such as subunit rolling, related L1 stalk motions and/or rotation of the small subunit with respect to the large, that reversibly relax and compact the E site, (Behrmann et al., 2015; Budkevich et al., 2014; Munro et al., 2010a; Yamamoto et al., 2014).
Resolution of the PRE2 complex is rate-limiting to processive translation
Having established the nature and timing of FRET states exhibited during isolated rounds of tRNA selection and translocation, we next sought to track distinct ribosome configurations during processive translation. To do so, pre-steady state three-color FRET experiments were performed wherein a mixture containing ternary complexes of eEF1A•GTP•(Cy5)Phe-tRNAPhe and eEF1A•GTP•(Cy7)Phe-tRNAPhe (10 nM), eEF2 (200 nM) and GTP (1 mM) was stopped-flow delivered to 80S IC's bearing (Cy3)Met-tRNAiMet in the P site (Figure 6A; Supplemental Information). We focused our analysis on the molecules (~50%) in which the first elongation cycle initiated with the appearance of Cy3-Cy5 FRET and the characteristic FRET signatures of PRE complex formation (Figures 2 and 6A, zoom – blue region) followed by translocation to the POST complex (Figures 4 and 6A, zoom – green region). The second elongation cycle, marked by the appearance of Cy5-Cy7 FRET, resulted in formation of a PRE2 complex bearing Cy3-, Cy5- and Cy7-labeled tRNAs (Figures 5C and 6A, zoom – yellow region). Translocation of this PRE2 complex was marked by the loss of FRET as a result of Cy3-labeled tRNA release from the E site.
Figure 6. Stability and dynamics of the human 80S PRE complex bearing three tRNA species.

(A) Fluorescence channels (top panel) and a representative pre-steady state single-molecule FRET trace (mid panel). Zoom (lower panel) demonstrates productive aa-tRNA selection (blue region), translocation to a POST state (green region), and a second round of productive aa-tRNA selection (yellow region), where dynamic motions occur within the ribosome bearing three fluorescently-labeled tRNAs. (B) Mean duration of states during processive translation. Data are represented as mean +/− SD. See also Figure S6.
The temporal progression of individual ribosomes through sequential elongation reactions was assessed by approximating the durations of the FRET signatures of PRE, POST and PRE2 complexes. In line with the notion that intra-ribosomal processes are rate-determining to the elongation process (Munro et al., 2009; Subramaniam et al., 2014), the rate of processive translation was primarily determined by the PRE2 complex lifetime (Figure 6B). This finding indicates that the PRE2 complex predominantly occupies a conformation that is refractory to eEF2-catalyzed translocation, consistent with our supposition that this complex preferentially adopts a classical tRNA configuration.
Nevertheless, the rate of tRNA dissociation from the E site increased in the presence of eEF2 (Figure 5B), suggesting that aa-tRNA restructures the PRE2 complex in a manner that contributes to the translocation mechanism. Given that classical tRNA configurations inhibit translocation (Figure S4A), we speculate that PRE2 complexes bearing A-, P- and E-site tRNAs are capable of transiently achieving hybrid tRNA configurations (A/P; P/E) upon which eEF2 can act. Such states, which necessarily hinge on a substantial repositioning of deacylated tRNA within the E site, may be facilitated by clockwise rotation of the small subunit with respect to the large as well as outward deflections of the L1 stalk to extended-open configurations (Behrmann, et al., 2015; Munro et al., 2010a; 2010b; Yamamoto et al., 2014).
DISCUSSION
Observations that the elongation phase of protein synthesis in both prokaryotic and eukaryotic species occurs with non-uniform rates (Ingolia et al., 2011) predict that dynamic processes intrinsic to the ribosome play a critical role in the regulation of gene expression (Subramaniam et al., 2014). Leveraging strategies first developed for the study of the bacterial translation mechanism (Blanchard et al., 2004), we report here the first quantitative interrogations of the human translation elongation cycle at the single-molecule scale. These efforts provide an essential step toward detailed biophysical examinations of basal ribosome functions as well as the paradigms of translation control underpinning gene expression.
Single-turnover and processive translation reactions provided compelling evidence that human ribosomes exhibit tell-tale signatures of conformational metastability: intrinsic dynamics manifest in distinct functional complexes achieved during the elongation cycle and contribute directly to their regulation. These findings add to a growing body of knowledge that large-scale structural events in the ribosome, which can be achieved by thermal fluctuations, represent an evolutionarily conserved feature of the translation mechanism (Behrmann et al., 2015; Budkevich et al., 2011; 2014; Munro et al., 2008; 2009). While bacterial and human ribosomes both exhibit spontaneous rearrangements in global architecture, the energy landscapes governing these structural changes appear distinct (Figures 2, S2). Such differences influence the energetics of subunit rotation in addition to the nature and timing of tRNA motions within the A, P and E sites. While the functional significance and physical basis of these evolutionary distinctions remains to be fully elucidated, the evidence presented here suggests that mammalian and bacterial elongation cycles may be more dissimilar than previously postulated (Rodnina and Wintermeyer, 2009).
The most divergent aspects of the human ribosome revealed in this work center on the E site, located at the lagging edge of the ribosome (Andersen et al., 2006; Blaha and Nierhaus, 2001; Petropoulos and Green, 2012; Rheinberger and Nierhaus, 1986; Robertson and Wintermeyer, 1987; Rodnina and Wintermeyer, 1992; Wettstein and Noll, 1965). Our insights into these distinctions began with investigations of the PRE complex, which displayed a strong preference for ribosome configurations in which P-site tRNA was positioned in a P/E hybrid state (Figures 2B). Under the same conditions, bacterial ribosomes favored a classical (P/P) state (Figures S2). In the human PRE complex, the P/E hybrid state is notably stable whereas peptidyltRNA fluctuates rapidly between classical and hybrid positions within the A site (Figures 2B, 3C). Consistent with the notion that a rotated ribosome configuration is the preferred substrate for factor-catalyzed translocation (Munro et al., 2010c; Wang et al., 2011), the human PRE complex rapidly translocated (Figure 4). Highlighting the regulatory potential of the E site of the human ribosome, the PRE complex exhibited an exquisite sensitivity to the drug CHX, which inhibits translocation by binding the E site and stabilizing classical tRNA configurations (Figure 3B and S4A). Functional and structural distinctions of the E site were further supported by the notable thermodynamic stability of the POST complex (Figure 5). In bacteria, deacylated tRNA release from the E site occurs at least an order of magnitude more rapidly (Figure S4D) (Chen et al., 2011; Chen et al., 2013; Lill et al., 1986; Robertson and Wintermeyer, 1987). High-affinity interactions between deacylated tRNA and the E site may be a general feature of ribosomes from higher eukaryotes that emerged late in evolution (Anger et al., 2013; Bokov and Steinberg, 2009; Behrmann et al., 2015).
Our prediction that E-site tRNA remains bound to the ribosome during conversion of the POST complex to the PRE2 complex was verified with three-color FRET investigations measuring this process directly via a FRET relay from E to A sites (Figures 5C, 6A). This revealed that entry of aa-tRNA into the A site, located at the leading edge of the ribosome is accompanied by large-scale conformational events within the E site, an effect that correlated with an increased rate of deacylated tRNA release (Figure 5). Given that fluctuations of this kind are triggered during aatRNA selection, these long-distance effects may directly relate to conformational changes specific to mammalian ribosomes, termed subunit rolling, which have been reported to reciprocally open and close the A and E sites (Budkevich et al., 2014). Such findings provide compelling support for an allosteric linkage between the A and E sites of the ribosome and illustrate the potential of single-molecule imaging to provide clarity in systems that exhibit inherent conformational and/or compositional heterogeneities.
Despite the notable decrease in E-site tRNA affinity exhibited by the A site-filled ribosome, translocation of the PRE2 complex was rate-limiting to processive translation (Figure 6B). This finding is consistent with the notion that ribosome complexes bearing deacylated tRNA in the E site exhibit predominantly classical, unrotated configurations (Figure 3A) which are refractory to eEF2 translocation (Figure 4D; Figure S4). We conclude that one or more yet-to-be-identified factors, whose activities may include roles analogous to those of eEF3 in fungi, may be required to facilitate the release of deacylated tRNA from the E site of translating ribosomes (Andersen et al., 2006).
Our observation that eEF2 increased the rate of deacylated tRNA release from the E site (Figure 5B) suggests that dynamic processes within the PRE2 complex contribute to the translocation mechanism (Figures 5C, 6A). In line with this model, FRET changes indicative of 20-30 Å movements between E- and P-site tRNAs were evidenced within the PRE2 complex. (Figures S4B, S4C and 5C). We propose that such motions principally reflect movements of deacylated E site tRNA with respect to a relatively static peptidyl-tRNA within the P site (Figure 7). Given the structure of the 80S E site (Anger et al., 2013; Budkevich et al., 2014; Behrmann et al., 2015) and the mechanism of translocation (Munro et al., 2010c; Ratje et al., 2010; Wang et al., 2011), we speculate that these transient (~250 ms) rearrangements manifest through subunit rolling processes and may include L1-stalk motions away from the 80S particle (Yamamoto et al., 2014) and rotation of the small subunit with respect to the large to enable A- and P-site tRNAs to achieve hybrid configurations. Such configurations, which posit a unique E-site tRNA binding mode, are consistent with earlier suggestions that ribosomes possess multiple E sites with distinct deacylated tRNA affinities (Andersen et al., 2006; Fischer et al., 2010; Robertson and Wintermeyer, 1987; Rodnina and Wintermeyer, 1992). Single-molecule investigations from multiple structural perspectives and single-particle cryo-EM methods will be required to confirm the existence of such fleeting states.
Figure 7. Schematic representation of the translation elongation cycle in human cells.

The first (top) and second (bottom) elongation cycles depicting eEF1A-catalyzed conversion of the 80S IC and POST complexes to PRE and PRE2 complexes, respectively. aa-tRNA entry into the A site proceeds through transient A/T-state intermediates wherein dynamic processes are observed in both the A and E sites (schematized as blurred tRNA positions). A-site tRNA motions are represented as transient excursions of aa-tRNA towards the A site; E-site tRNA motions are represented as transient excursions away from the P site. The spontaneous exchange of PRE and PRE2 complexes between classical (C) and hybrid (H) tRNA positions (schematized as blurred tRNA positions) is influenced by the 40S subunit configuration: unrotated (gray) or rotated (red). eEF2 acts on hybrid state PRE complexes in which both A- and P-site tRNAs adopt hybrid positions (PRE-H and PRE2-H) to catalyze directional substrate translocation, forming the POST complex (POST2 complex not shown). Lines underneath each ribosome complex indicates their relative thermodynamic stabilities: solid (stable) and dashed (transient).
The functional significance of a long-lived, dynamic PRE2 complex may be manifold. The presence of three tRNAs on the elongating ribosome may facilitate translational fidelity and/or processivity (Blaha and Nierhaus, 2001; Marquez et al., 2004) by stabilizing polysomes, so as to protect mRNA from inappropriate degradation (Myasnikov et al., 2014). It has been noted that deacylated tRNAs are not free in the cell during mammalian translation and are instead shuttled from the ribosome to tRNA synthetases and then to eEF1A (Mirande, 2010; Stapulionis and Deutscher, 1995). In this context, the stability of the PRE2 complex observed in vitro may reflect the absence of one or more regulatory factors responsible for facilitating the direct extraction of E-site tRNA from the ribosome.
The high-affinity nature of deacylated tRNA interactions at the E site (Figures 3A, S3A and S3B) could provide a means of sensing growth conditions in the cell, as deacylated tRNA may accumulate in the cytoplasm under starvation conditions (Mirande, 2010; Stapulionis and Deutscher, 1995). Consistent with this notion, low concentrations of exogenous deacylated tRNA (ca. 10-100 nM) inhibited processive translation reactions in vitro (Figures 1B and S1E). Such considerations predict that PRE2 complex stability provides important opportunities for translation control. Further studies must be aimed at exploring these hypotheses in light of the non-uniform, context-dependent rates of protein synthesis exhibited in cells (Ingolia et al., 2011; Subramanian et al., 2014) and the growing list of factors implicated in gene expression control that bind to translating ribosomes (Hsieh et al., 2012; Larsson et al., 2012; Proud, 2014; Ramirez et al., 1991; Reschke et al., 2013; Sattlegger and Hinnebusch, 2005; van Riggelen et al., 2010). We expect single-molecule methods to afford significant advantages in these endeavors by offering the potential to simultaneously track compositional and conformational processes in real time using limited and heterogeneous mixtures of ribosomes and associated factors. This platform also facilitates control over the precise timing of perturbations and the detection of transient interactions that might be missed using traditional methods.
EXPERIMENTAL PROCEDURES
80S initiation complex formation
Purified 40S subunits were mixed with four fold excess of mRNA (Supplemental Information). To this mixture, 1.5-fold excess of fluorescently-labeled or unlabeled Met-tRNAMet was added followed by equimolar amounts of 60S subunits. The mixture and incubated at 37°C for 20 minutes. After the incubation, the MgCl2 concentration of the reaction was raised to 15 mM and the mixture was loaded onto a 10-30% sucrose gradient from which the peak corresponding to 80S complexes was collected and frozen (Supplemental Information).
Single-molecule fluorescence imaging
Complexes were surface-immobilized via the biotin-streptavidin interaction in PEG-passivated quartz chambers. All imaging experiments were performed in HEPES(KOH)-Polymix buffer (pH 7.5) containing 5 mM MgCl2, 50 mM NH4Cl, 2 mM spermidine, 5 mM putrescine, as well as an oxygen scavenging system (2 mM protocatechuic acid (PCA), 50 nM protocatechuate 3,4-dioxygenase (PCD)) together with a cocktail of solution additives (1 mM Trolox, 1 mM cyclooctatetraene, 1 mM nitrobenzyl-alcohol) (Dave et al., 2009) to reduce photobleaching. As previously described (Blanchard et al., 2004), single-molecule fluorescence imaging was performed using a custom prism-based total internal reflection fluorescence (TIRF) microscope. Cy3 fluorophores were illuminated with a 532 nm solid-state laser (LaserQuantum) and fluorescence emission from Cy3, Cy5 (and Cy7 in multicolor experiments) fluorophores was collected using a 60×, 1.27 N.A. Plan-Apo water immersion objective (Nikon) and spectrally separated using a MultiCam-LS device (Cairn) equipped with T635lpxr-UF2 and 740dcxr dichroic mirrors (Chroma) and imaged onto ORCA-Flash 4.0 v2 sCMOS cameras (Hamamatsu). Data were acquired at 15, 40 or 200 ms time resolution using custom software implemented in LabView (National Instruments). See Supplemental Information for additional details.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Drs. Christian Spahn, Tatyana Budkevich and Kaori Yamamoto for purified elongation factors. We also thank the Weill Cornell Genomics and Advanced Bioinformatics Cores for contributions to processing of ribosome profiling data. We thank the Rockefeller University Proteomics Resource Center for mass spectrometry. This work was supported by grants from the NIH (GM 079238) (SCB), the Tri-Institutional Stem Cell Initiative funded by the Starr Foundation (SCB and TCV), the Tri-Institutional Training Program in Chemical Biology (AK) and the German Academic Exchange Service (MFJ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
SCB, AF, RBA and LW designed the project and prepared the manuscript. AF, RBA, LW and BJB prepared reagents. AF and LW performed single-molecule experiments. AF, RBA and MFJ performed tRNA selection experiments. AF, SCB, DST, MFJ and JLA contributed to smFRET data analysis. AF and BJB performed bulk experiments. RAD and AF performed ribosome profiling. Bioinformatics analysis was completed by MMP.
REFERENCES
- Andersen CBF, Becker T, Blau M, Anand M, Halic M, Balar B, Mielke T, Boesen T, Pedersen JS, Spahn CMT, et al. Structure of eEF3 and the mechanism of transfer RNA release from the E-site. Nature. 2006;443:663–668. doi: 10.1038/nature05126. [DOI] [PubMed] [Google Scholar]
- Anger AM, Armache J-P, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- Behrmann E, Loerke J, Budkevich TV, Yamamoto K, Schmidt A, Penczek PA, Vos MR, Bürger J, Mielke T, Scheerer P, et al. Structural snapshots of actively translating human ribosomes. Cell. 2015;161:845–857. doi: 10.1016/j.cell.2015.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaha G, Nierhaus KH. Features and functions of the ribosomal E site. Cold Spring Harb. Symp. Quant. Biol. 2001;66:135–146. doi: 10.1101/sqb.2001.66.135. [DOI] [PubMed] [Google Scholar]
- Blanchard SC, Kim HD, Gonzalez RL, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokov K, Steinberg SV. A hierarchical model for evolution of 23S ribosomal RNA. Nature. 2009;457:977–980. doi: 10.1038/nature07749. [DOI] [PubMed] [Google Scholar]
- Budkevich TV, El'skaya AV, Nierhaus KH. Features of 80S mammalian ribosome and its subunits. Nucleic Acids Research. 2008;36:4736–4744. doi: 10.1093/nar/gkn424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budkevich TV, Giesebrecht J, Behrmann E, Loerke J, Ramrath DJF, Mielke T, Ismer J, Hildebrand PW, Tung C-S, Nierhaus KH, et al. Regulation of the mammalian elongation cycle by subunit rolling: a eukaryotic-specific ribosome rearrangement. Cell. 2014;158:121–131. doi: 10.1016/j.cell.2014.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budkevich T, Giesebrecht J, Altman RB, Munro JB, Mielke T, Nierhaus KH, Blanchard SC, Spahn CMT. Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol. Cell. 2011;44:214–224. doi: 10.1016/j.molcel.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett BJ, Altman RB, Ferguson A, Wasserman MR, Zhou Z, Blanchard SC. Direct Evidence of an Elongation Factor-Tu/Ts·GTP·Aminoacyl-tRNA Quaternary Complex. J. Biol. Chem. 2014;289:23917–23927. doi: 10.1074/jbc.M114.583385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Stevens B, Kaur J, Smilansky Z, Cooperman BS, Goldman YE. Allosteric vs. spontaneous exit-site (E-site) tRNA dissociation early in protein synthesis. Proc. Natl. Acad. Sci. U.S.A. 2011;108:16980–16985. doi: 10.1073/pnas.1106999108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Petrov A, Tsai A, O'Leary SE, Puglisi JD. Coordinated conformational and compositional dynamics drive ribosome translocation. Nat. Struct. Mol. Biol. 2013;20:718–727. doi: 10.1038/nsmb.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish PV, Ermolenko DN, Noller HF, Ha T. Spontaneous intersubunit rotation in single ribosomes. Mol. Cell. 2008;30:578–588. doi: 10.1016/j.molcel.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave R, Terry DS, Munro JB, Blanchard SC. Mitigating unwanted photophysical processes for improved single-molecule fluorescence imaging. Biophys. J. 2009;96:2371–2381. doi: 10.1016/j.bpj.2008.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkle JA, Wang L, Feldman MB, Pulk A, Chen VB, Kapral GJ, Noeske J, Richardson JS, Blanchard SC, Cate JHD. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg M. Protein synthesis: Translocation in slow motion. Nature. 2010;466:325–326. doi: 10.1038/466325a. [DOI] [PubMed] [Google Scholar]
- Feldman MB, Terry DS, Altman RB, Blanchard SC. Aminoglycoside activity observed on single pre-translocation ribosome complexes. Nature Chemical Biology. 2010;6:54–62. doi: 10.1038/nchembio.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466:329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- Frank J, Agrawal RK. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 2000;406:318–322. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- Frank J. Intermediate states during mRNA-tRNA translocation. Curr. Opin. Struct. Biol. 2012;22:778–785. doi: 10.1016/j.sbi.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresno M, Jiménez A, Vázquez D. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem. 1977;72:323–330. doi: 10.1111/j.1432-1033.1977.tb11256.x. [DOI] [PubMed] [Google Scholar]
- Garreau de Loubresse NG, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014:1–17. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Garrett R. Mechanics of the ribosome. Nature. 1999;400:811–812. doi: 10.1038/23573. [DOI] [PubMed] [Google Scholar]
- Geggier P, Dave R, Feldman MB, Terry DS, Altman RB, Munro JB, Blanchard SC. Conformational sampling of aminoacyl-tRNA during selection on the bacterial ribosome. J. Mol. Biol. 2010;399:576–595. doi: 10.1016/j.jmb.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R, Switzer C, Noller HF. Ribosome-catalyzed peptide-bond formation with an A-site substrate covalently linked to 23S ribosomal RNA. Science. 1998;280:286–289. doi: 10.1126/science.280.5361.286. [DOI] [PubMed] [Google Scholar]
- Green R, Noller HF. Ribosomes and translation. Annu. Rev. Biochem. 1997;66:679–716. doi: 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003;330:1061–1075. doi: 10.1016/s0022-2836(03)00668-5. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp LD, Lorsch JR. The molecular mechanics of eukaryotic translation. Annu. Rev. Biochem. 2004;73:657–704. doi: 10.1146/annurev.biochem.73.030403.080419. [DOI] [PubMed] [Google Scholar]
- Khatter H, Myasnikov AG, Mastio L, Billas IML, Birck C, Stella S, Klaholz BP. Purification, characterization and crystallization of the human 80S ribosome. Nucleic Acids Research. 2014;42:e49. doi: 10.1093/nar/gkt1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O, Morita M, Topisirovic I, Alain T, Blouin M-J, Pollak M, Sonenberg N. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proc. Natl. Acad. Sci. U.S.a. 2012;109:8977–8982. doi: 10.1073/pnas.1201689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Robertson JM, Wintermeyer W. Affinities of tRNA binding sites of ribosomes from Escherichia coli. Biochemistry. 1986;25:3245–3255. doi: 10.1021/bi00359a025. [DOI] [PubMed] [Google Scholar]
- Marquez V, Wilson DN, Tate WP, Triana-Alonso F, Nierhaus KH. Maintaining the ribosomal reading frame: the influence of the E site during translational regulation of release factor 2. Cell. 2004;118:45–55. doi: 10.1016/j.cell.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Marshall RA, Dorywalska M, Puglisi JD. Irreversible chemical steps control intersubunit dynamics during translation. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15364–15369. doi: 10.1073/pnas.0805299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney SA, Joo C, Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirande M. Processivity of translation in the eukaryote cell: role of aminoacyl-tRNA synthetases. FEBS Lett. 2010;584:443–447. doi: 10.1016/j.febslet.2009.11.027. [DOI] [PubMed] [Google Scholar]
- Moazed D, Noller HF. Intermediate states in the movement of transfer RNA in the ribosome. Nature. 1989;342:142–148. doi: 10.1038/342142a0. [DOI] [PubMed] [Google Scholar]
- Munro JB, Altman RB, O'Connor N, Blanchard SC. Identification of two distinct hybrid state intermediates on the ribosome. Mol. Cell. 2007;25:505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung C-S, Cate JHD, Sanbonmatsu KY, Blanchard SC. Spontaneous formation of the unlocked state of the ribosome is a multistep process. Proc. Natl. Acad. Sci. U.S.a. 2010a;107:709–714. doi: 10.1073/pnas.0908597107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Altman RB, Tung C-S, Sanbonmatsu KY, Blanchard SC. A fast dynamic mode of the EF-G-bound ribosome. EMBO J. 2010b;29:770–781. doi: 10.1038/emboj.2009.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Sanbonmatsu KY, Spahn CMT, Blanchard SC. Navigating the ribosome's metastable energy landscape. Trends in Biochemical Sciences. 2009;34:390–400. doi: 10.1016/j.tibs.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Vaiana A, Sanbonmatsu KY, Blanchard SC. A new view of protein synthesis: mapping the free energy landscape of the ribosome using single-molecule FRET. Biopolymers. 2008;89:565–577. doi: 10.1002/bip.20961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro JB, Wasserman MR, Altman RB, Wang L, Blanchard SC. Correlated conformational events in EF-G and the ribosome regulate translocation. Nat. Struct. Mol. Biol. 2010c;17:1470–1477. doi: 10.1038/nsmb.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myasnikov AG, Afonina ZA, Menetret J-F, Shirokov VA, Spirin AS, Klaholz BP. The molecular structure of the left-handed supra-molecular helix of eukaryotic polyribosomes. Nat. Commun. 2014;5:1–8. doi: 10.1038/ncomms6294. [DOI] [PubMed] [Google Scholar]
- Nierhaus KH. The allosteric three-site model for the ribosomal elongation cycle: features and future. Biochemistry. 1990;29:4997–5008. doi: 10.1021/bi00473a001. [DOI] [PubMed] [Google Scholar]
- Petropoulos AD, Green R. Further in Vitro Exploration Fails to Support the Allosteric Three-site Model. J. Biol. Chem. 2012;287:11642–11648. doi: 10.1074/jbc.C111.330068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polikanov YS, Starosta AL, Juette MF, Altman RB, Terry DS, Lu W, Burnett BJ, Dinos G, Reynolds KA, Blanchard SC, et al. Distinct tRNA Accommodation Intermediates Observed on the Ribosome with the Antibiotics Hygromycin A and A201A. Mol. Cell. 2015;58:832–844. doi: 10.1016/j.molcel.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. Control of the translational machinery by amino acids. Am. J. Clin. Nutr. 2014;99:231S–236S. doi: 10.3945/ajcn.113.066753. [DOI] [PubMed] [Google Scholar]
- Ramirez M, Wek RC, Hinnebusch AG. Ribosome association of GCN2 protein kinase, a translational activator of the GCN4 gene of Saccharomyces cerevisiae. Mol. Cell. Biol. 1991;11:3027–3036. doi: 10.1128/mcb.11.6.3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratje AH, Loerke J, Mikolajka A, Brünner M, Hildebrand PW, Starosta AL, Dönhöfer A, Connell SR, Fucini P, Mielke T, et al. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature. 2010;468:713–716. doi: 10.1038/nature09547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reschke M, Clohessy JG, Seitzer N, Goldstein DP, Breitkopf SB, Schmolze DB, Ala U, Asara JM, Beck AH, Pandolfi PP. Characterization and Analysis of the Composition and Dynamics of the Mammalian Riboproteome. Cell Reports. 2013;4:1276–1287. doi: 10.1016/j.celrep.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheinberger HJ, Nierhaus KH. Allosteric interactions between the ribosomal transfer RNA-binding sites A and E. J. Biol. Chem. 1986;261:9133–9139. [PubMed] [Google Scholar]
- Robertson JM, Wintermeyer W. Mechanism of ribosomal translocation. tRNA binds transiently to an exit site before leaving the ribosome during translocation. J. Mol. Biol. 1987;196:525–540. doi: 10.1016/0022-2836(87)90030-1. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W. Two tRNA-binding sites in addition to A and P sites on eukaryotic ribosomes. J. Mol. Biol. 1992;228:450–459. doi: 10.1016/0022-2836(92)90834-7. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W. Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms. Annu. Rev. Biochem. 2001;70:415–435. doi: 10.1146/annurev.biochem.70.1.415. [DOI] [PubMed] [Google Scholar]
- Rodnina MV. The ribosome as a versatile catalyst: reactions at the peptidyl transferase center. Curr. Opin. Struct. Biol. 2013;23:595–602. doi: 10.1016/j.sbi.2013.04.012. [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W. Recent mechanistic insights into eukaryotic ribosomes. Curr. Opin. Cell Biol. 2009;21:435–443. doi: 10.1016/j.ceb.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Ruggero D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013;5 doi: 10.1101/cshperspect.a012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattlegger E, Hinnebusch AG. Polyribosome binding by GCN1 is required for full activation of eukaryotic translation initiation factor 2{alpha} kinase GCN2 during amino acid starvation. J. Biol. Chem. 2005;280:16514–16521. doi: 10.1074/jbc.M414566200. [DOI] [PubMed] [Google Scholar]
- Sharma D, Cukras AR, Rogers EJ, Southworth DR, Green R. Mutational analysis of S12 protein and implications for the accuracy of decoding by the ribosome. J. Mol. Biol. 2007;374:1065–1076. doi: 10.1016/j.jmb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn CM, Beckmann R, Eswar N, Penczek PA, Sali A, Blobel G, Frank J. Structure of the 80S ribosome from Saccharomyces cerevisiae--tRNA-ribosome and subunit-subunit interactions. Cell. 2001;107:373–386. doi: 10.1016/s0092-8674(01)00539-6. [DOI] [PubMed] [Google Scholar]
- Stapulionis R, Deutscher MP. A channeled tRNA cycle during mammalian protein synthesis. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7158–7161. doi: 10.1073/pnas.92.16.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr. Opin. Genet. Dev. 2011;21:474–483. doi: 10.1016/j.gde.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Zid BM, O'Shea EK. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell. 2014;159:1200–1211. doi: 10.1016/j.cell.2014.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne JS, Pestka S. Inhibition of protein synthesis in intact HeLa cells. Antimicrob. Agents Chemother. 1975;8:479–487. doi: 10.1128/aac.8.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer. 2010;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- Wang L, Altman RB, Blanchard SC. Insights into the molecular determinants of EF-G catalyzed translocation. RNA. 2011;17:2189–2200. doi: 10.1261/rna.029033.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein FO, Noll H. Binding of transfer ribonucleic acid to ribosomes engaged in protein synthesis: number and properties of ribosomal binding sites. J. Mol. Biol. 1965;11:35–53. doi: 10.1016/s0022-2836(65)80169-3. [DOI] [PubMed] [Google Scholar]
- Wilson DN, Harms JM, Nierhaus KH, Schlünzen F, Fucini P. Species-specific antibiotic-ribosome interactions: implications for drug development. Biol. Chem. 2005;386 doi: 10.1515/BC.2005.141. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Unbehaun A, Loerke J, Behrmann E, Collier M, Bürger J, Mielke T, Spahn CMT. Structure of the mammalian 80S initiation complex with initiation factor 5B on HCV-IRES RNA. Nat. Struct. Mol. Biol. 2014;21:721–727. doi: 10.1038/nsmb.2859. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.