

Abstract
Background:
Phragmanthera austroarabica A.G. Mill. and J. A. Nyberg is a semi parasitic plant belonging to family Loranthaceae. It was collected from Saudi Arabia. It is widely used in folk medicine among the kingdom in treatment of various diseases including diabetes mellitus.
Objective:
The total alcoholic extract of P. austroarabica collected from Saudi Arabia was investigated for the chemical structure and prominent biological activity of the main constituents.
Materials and Methods:
Isolation of the active constituents was performed using different chromatographic techniques including column chromatography packed with silica or sephadex and preparative thin layer chromatography. The structures of the isolated compounds were established based on different spectroscopic data as mass spectrum, one-dimensional and two-dimensional nuclear magnetic resonance (correlation spectroscopy, heteronuclear single quantum coherence, and heteronuclear multiple-bond correlation).
Results:
Phytochemical investigation of the plant resulted in isolation of 12 compounds. The isolated compounds were identified as chrysophanic acid, emodin, chrysophanic acid-8-O-glucoside, emodin-8-O-glucoside, pectolinarigenin, quercetin, dillenetin-3-O-glucoside, catechin, catechin-4’-O-gallate, methyl gallate, lupeol and ursolic acid. All the isolated phenolic compounds revealed significant free radical scavenging activities when tested using 2,2-diphenyl-1-picrylhydrazyl reagent.
Conclusion:
The antioxidant activities of the isolated compounds can justify the use of P. austroarabica in traditional medicine for treatment of diabetes and verify its possible application as an antihyperglycemic drug.
Keywords: Antioxidant, Loranthaceae, nuclear magnetic resonance, Phragmanthera
INTRODUCTION
Family Loranthaceae comprises a number of the stem parasites that are commonly known as mistletoes. A number of mistletoes belonging to this family were investigated for chemical constituents and reported to accumulate flavonoids and phenolic compounds.[1,2,3,4] Phragmanthera austroarabica; a plant belonging to family Loranthaceae was collected from Saudi Arabia. It is widely used in folk medicine among the kingdom in treatment of various diseases among them is diabetes mellitus.[5,6] Upon reviewing the literature, it was found that no previous reports on the chemical constituents of P. austroarabica. In the present work, the total extract of the plant was chemically investigated where 12 compounds were isolated and their structures were determined using different spectroscopic techniques [Figure 1]. Additionally, the pure isolated compounds were tested to evaluate their free radical scavenging activities using 2,2-diphenyl-1-picrylhydrazyl (DPPH) reagent.
Figure 1.
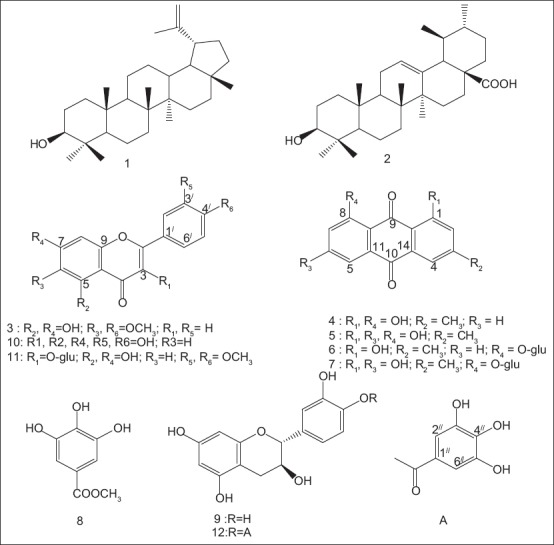
Structures of the isolated compounds
MATERIALS AND METHODS
General experimental procedures
Ultraviolet (UV) spectra were measured on a Hitachi 300 Spectrophotometer. Nuclear magnetic resonance (NMR) spectra were obtained in CD3Cl and CD3OD on Bruker Avance DRX 600 spectrometers at 600 MHz for hydrogen NMR (1H NMR) and 150 MHz for carbon NMR (13C NMR). Mass spectral data were performed on ion trap liquid chromatography/mass spectrum agilent. For column chromatography, silica gel (Merck, 70–230 mesh ASTM) and sephadex LH-20 (pharmacia) were used. Precoated silica gel 60 F-254 plates (Merck) were used for thin-layer chromatography (TLC). Reversed phase-18 (40–63 μm), 6 ml standard tubes (extraction tubes), Damstadt, Germany, were used for purification of the polar compounds. Spots were visualized by exposure to NH3 vapour, UV radiation and P-anisaldehyde/sulfuric acid.
Plant material
The plant was collected during March 2011 from Abha, Khamis Mushat at the South of Saudi Arabia; identified by Dr. Nahed Morad, Faculty of Science, King Abdulaziz University. A voucher sample was kept at Department of Natural Products, Faculty of Pharmacy, King Abdulaziz University under the registration code No. 2011-Phaa.
Extraction and isolation
The plant was air dried, finely powdered (2.5 kg) followed by maceration with methyl alcohol (3 mL × 3000 mL) at room temperature. The total alcoholic extract was concentrated under vacuum, fractionated successively with petroleum ether, chloroform and ethyl acetate. Five gram of petroleum ether extract were chromatographed on silica gel column packed in petroleum ether and eluted with a step gradient of petroleum ether-chloroform-methanol. Three main sub fractions were obtained after collecting similar fractions together. Fraction A eluted by 100% chloroform was re-chromatographed on silica gel column using chloroform-methanol gradient elution to obtain finally 10 mg of white powder and 16 mg of another white powder designated as compounds (1) and (2). Fraction B eluted by 2% methanol in chloroform showed two spots that were purified on another silica gel column. This was followed by sephadex using methanol: chloroform (80:20) as an eluting agent to give 7 mg of a yellow powder (compound 3) and 5 mg of yellow residue (compound 4). Fraction C eluted by 4% methanol in chloroform was chromatographed on silica gel column with gradient elution (chloroform: Methanol), followed by crystallization from methanol to afford 8 mg of yellowish-orange crystals of compound 5. Five gram of chloroform extract were chromatographed on silica gel column and eluted with chloroform-methanol gradient. Fractions eluted by 5% methanol in chloroform were chromatographed on silica gel column with gradient elution using chloroform-methanol, followed by purification on sephadex using methanol: chloroform (80:20) as an eluting agent to yield finally 5 mg of a yellow residue designated as compound 6. Fractions eluted by 7% methanol in chloroform were purified by preparative TLC using 5% methanol in chloroform for development to afford 3 mg of an orange residue (compound 7). Seven gram of the ethyl acetate extract were chromatographed on silica gel column and eluted with a step gradient of chloroform-methanol. Fractions eluted by 10% methanol in chloroform were purified on sephadex using methanol for elution followed by further purification on silica gel column to yield finally 55 mg of white needles (compound 8), 60 mg of a creamy powder (compound 9) and 35 mg of a yellow powder (compound 10). Fractions eluted by 20% methanol in chloroform were purified on sephadex and eluted by methanol to yield 5 mg of yellow residue designated as compound 11, while those eluted by 35% methanol in chloroform were purified on reversed phase silica gel extraction tubes using water-acetonitril for gradient elution to give 8 mg of dark orange residue designated as compound 12.
Determination of free radical scavenging activity
Aliquots of different concentrations from the pure compounds (3–12) solutions were pipetted into a series of 5 mL volumetric flasks. To each flask 3 mL of DPPH solution were added, mixed with the solution, volumes were made up with methanol and flasks were allowed to stand in dark at room temperature for 10 min.[7] The absorbance of each of the resulted solutions was measured at 516 nm against similarly treated blank. The free radical scavenging activity was determined for each of the tested pure compounds according to the following equation:
% DPPH radical scavenging = (A0 − A)/A0 × 100
Where A0 is the absorbance of a blank and A is the absorbance of the sample.
RESULTS
Comparison of NMR data of compounds 1 and 2 with previously reported data confirmed their identity as lupeol and ursolic acid respectively.[8] Compounds 8, 9 and 10 were identified as methyl gallate, catechin and quercetin respectively by direct comparison using TLC against reference samples previously isolated by the author from Plicosepalus acacia.[4]
Spectroscopic data
Compound 1
White powder, m/z (rel. int.%): 427 [M + 1]+, C30H50O. 13C NMR (150 MHz, CDCl3): δ C 38.2 (C-1), 27.6 (C-2), 79.2 (C-3), 38.1 (C-4), 55.5 (C-5), 18.2 (C-6), 34.5 (C-7), 40.2 (C-8), 50.6 (C-9), 37.4 (C-10), 21.1 (C-11), 25.3 (C-12), 39.0 (C-13), 41.0 (C-14), 27.6 (C-15), 35.8 (C-16), 43.0 (C-17), 48.2 (C-18), 48.5 (C-19), 151.2 (C-20), 30.0 (C-21), 43.2 (C-22), 15.5 (C-23), 28.2 (C-24), 16.2 (C-25), 16.3 (C-26), 14.7 (C-27), 18.5 (C-28), 19.5 (C-29), 109.5 (C-30).
Compound 2
White powder, m/z (rel. int.%): 455 [M - 1]+, C30H48O3. 13C NMR (150 MHz, CDCl3): δC 39.1 (C-1), 28.2 (C-2), 79.2 (C-3), 38.9 (C-4), 56.5 (C-5), 19.9 (C-6), 34.5 (C-7), 38.6 (C-8), 49.4 (C-9), 37.2 (C-10), 25.7 (C-11), 125.9 (C-12), 140.0 (C-13), 40.9 (C-14), 29.9 (C-15), 27.6 (C-16), 42.6 (C-17), 55.5 (C-18), 39.0 (C-19), 38.9 (C-20), 30.7 (C-21), 37.2 (C-22), 29.8 (C-23), 18.5 (C-24), 15.5 (C-25), 16.3 (C-26), 24.0 (C-27), 180.1 (C-28), 16.2 (C-29), 21.1 (C-30).
Compound 3
Yellow powder, m/z (rel. int. %): 315 [M + 1]+, C17H14O6. UV λ max nm (MeOH): 278, 330, λmax nm (MeOH + NaOMe): 277, 296, 370, λ max nm (MeOH + NaOAc): 276, 297, 371, λ max nm (MeOH + AlCl3): 280, 302, 358. 1H NMR (600 MHz, CDCl3): dH 6.91(s, H-3), 6.61 (s, H-8), 7.57 (d, J = 7.8, H-2’ and H-6’), 6.94 (d, J = 7.8, H-3’ and H-5’), 3.79 (s, 6- OCH3), 3.89 (s, 4’- OCH3). 13C NMR (150 MHz, CDCl3): dC 163.5 (C-2), 102.5 (C-3), 182.0 (C-4), 152.3 (C-5), 131.2 (C-6), 157.0 (C-7), 94.2 (C-8), 152.6 (C-9), 109.9 (C-10), 121.3 (C-1’), 131.2 (C-2’, 6’), 115.5 (C-3’, 5’), 158.0 (C-4’), 59.8 (6- OCH3), 55.7 (4’- OCH3).
Compound 4
Yellow residue, m/z (rel. int.%): 253 [M - 1]+, C15H10O4. 1H NMR (600 MHz, CDCl3): δH 7.13 (d, J = 1.2, H-2), 7.66 (d, J = 1.2, H-4), 7.83 (dd, J = 7.8, 1.2, H-5), 7.61 (t, J = 7.8, H-6), 7.31 (dd, J= 8.4, 1.2, H-7), 2.48 (s, 3- CH3), 12.14 (s, 1-OH), 12.04 (s, 8-OH). 13C NMR (150 MHz, CDCl3): dC 162.7 (C-1), 124.4 (C-2), 149.3 (C-3), 121.4 (C-4), 119.9 (C-5), 137.0 (C-6), 124.6 (C-7), 162.4 (C-8), 192.6 (C-9), 182.1 (C-10), 133.3 (C-11), 115.9 (C-12), 113.7 (C-13), 133.6 (C-14), 22.3 (3- CH3).
Compound 5
Yellowish orange crystals, m/z (rel. int.%): 271 [M + 1]+, C15H10O5. 1H NMR (600 MHz, CDCl3): δH 7.09 (br s, H-2), 7.62 (br s, H-4), 7.28 (d, J = 2.1, H-5), 6.67 (d, J = 2.1, H-7), 2.45 (s, 3- CH3), 12.12 (s, 1-OH), 12.29 (s, 8-OH). 13C NMR (150 MHz, CDCl3): dC 162.5 (C-1), 124.6 (C-2), 148.4 (C-3), 121.3 (C-4), 108.8 (C-5), 165.2 (C-6), 108.9 (C-7), 163.4 (C-8), 190.8 (C-9), 182.0 (C-10), 133.1 (C-11), 110.4 (C-12), 113.6 (C-13), 135.3 (C-14), 22.7 (3- CH3).
Compound 6
Yellow residue, m/z (rel. int.%): 415 [M - 1]+, C21H20O9. 1H NMR (600 MHz, CD3OD): δH 7.10 (br s, H-2), 7.58 (br s, H-4), 8.20 (dd, J = 2.1, 8.1, H-5), 7.77 (t, J = 8.1, H-6), 7.28 (dd, J = 8.1,2.0, H-7), 2.43 (s, 3- CH3), 11.28 (s, 1-OH), 5.09 (d, J = 7.9, H-1’), 3.07 (m, H-2’), 3.53 (m, H-3’), 3.47 (m, H-4’), 3.52 (m, H-5’), 3.94 (dd, J = 12, 2.4, Ha -6’), 3.73 (dd, J = 12, 4.2, Hb -6’). 13C NMR (150 MHz, CD3OD): dC 163.3 (C-1), 125.7 (C-2), 149.0 (C-3), 121.7 (C-4), 123.8 (C-5), 137.2 (C-6), 125.7 (C-7), 159.7 (C-8), 189.6 (C-9), 183.4 (C-10), 136.7 (C-11), 119.7 (C-12), 115.8 (C-13), 136.0 (C-14), 23.3 (3- CH3), 103.8 (C-1’), 74.9 (C-2’), 77.4 (C-3’), 71.5 (C-4’), 78.2 (C-5’), 62.5 (C-6’).
Compound 7
Orange residue, m/z (rel. int.%): 431 [M - 1]+, C21H20O10. 1H NMR (600 MHz, CD3OD): δH 7.07 (br s, H-2), 7.52 (br s, H-4), 7.30 (d, J = 2.4, H-5), 7.02 (d, J = 2.4, H-7), 2.41 (s, 3- CH3), 12.5 (s, 1-OH), 4.95 (d, J = 7.8, H-1’), 3.64 (m, H-2’), 3.50 (m, H-3’), 3.63 (m, H-4’), 3.52 (m, H-5’), 3.56 (dd, J = 12, 4, Ha -6’), 3.51 (m, Hb -6’). 13C NMR (150 MHz, CD3OD): dC 163.7 (C-1), 125.3 (C-2), 148.5 (C-3), 120.7 (C-4), 112.1 (C-5), 167.0 (C-6), 111.7 (C-7), 163.1 (C-8), 188.7 (C-9), 184.1 (C-10), 138.3 (C-11), 110.1 (C-12), 116.1 (C-13), 134.1 (C-14), 21.9 (3- CH3), 104.2 (C-1’), 73.8 (C-2’), 77.4 (C-3’), 71.1 (C-4’), 78.7 (C-5’), 64.4 (C-6’).
Compound 11
Yellow residue, m/z (rel. int.%): 491 [M - 1]+, C23H24O12. 1H NMR (600 MHz, CD3OD): δH 6.11(d, J = 2.4, H-6), 6.31 (d, J = 2.4, H-8), 7.83 (d, J = 1.8, H-2’), 6.81 (d, J = 8.4, H-5’), 7.49 (dd, J = 8.4, 1.8, H-6’), 3.94 (s, 3’- OCH3), 3.68 (s, 4’- OCH3), 5.31 (d, J = 7.2, H-1’’), 3.44 (m, H-2’’), 3.42 (m, H-3’’), 3.64 (m, H-4’’), 3.21 (m, H-5’’), 3.72 (dd, J = 12, 1.8, Ha -6’’), 3.55 (dd, J = 12, 2.4, Hb -6’’). 13C NMR (150 MHz, CD3OD): dC 148.2 (C-2), 135.1 (C-3), 179.0 (C-4), 163.5 (C-5), 100.0 (C-6), 166.7 (C-7), 94.5 (C-8), 158.7 (C-9), 105.6 (C-10), 122.2 (C-1’), 114.4 (C-2’), 148.0 (C-3’), 151.1 (C-4’), 116.1 (C-5’), 124.9 (C-6’), 56.9 (3’- OCH3), 55.1 (4’- OCH3), 103.8 (C-1’’), 75.7 (C-2’’), 77.8 (C-3’’), 71.4 (C-4’’), 78.0 (C-5’’), 62.7 (C-6’’).
Compound 12
Dark orange residue, m/z (rel. int.%): 441 [M - 1]+, C22H18O10. 1H NMR (600 MHz, CD3OD): δ H 4.69 (d, J = 7.8 Hz, H-2), 4.08 (m, H-3), 2.64 (dd, J = 16.2, 7.8 Hz, H-4ax), 2.95 (dd, J = 16.2, 5.4 Hz, H-4eq), 6.22 (d, J = 2.4 Hz, H-6), 6.25 (d, J = 2.4 Hz, H-8), 6.87 (d, J = 2.4 Hz, H-2’), 6.78 (d, J = 7.8 Hz, H-5’), 6.75 (dd, J = 7.8, 2.4 Hz, H-6’), 7.19 (s, H-2’’,6’’);13C NMR (150 MHz, CD3OD): d C 81.6 (C-2), 67.0 (C-3), 27.1 (C-4), 150.3 (C-5), 100.5 (C-6), 155.4 (C-7), 100.6 (C-8), 156.2 (C-9), 105.8 (C-10), 130.5 (C-1’), 113.8 (C-2’), 144.9 (C-3’), 144.8 (C-4’), 114.7 (C-5’), 118.6 (C-6’), 119.3 (C-1’’), 109.1 (C-2’’,6’’), 145.3 (C-3’’,5’’), 139.0 (C-4’’’), 165.7 (C = O).
DISCUSSION
The UV spectrum of compound 3 in different shift reagents indicated a flavone substituted with two hydroxyls at C-5 and C-7. 1H NMR spectrum revealed the presence of two singlets at d 6.91 and 6.61 attributed to H-3 and H-8 respectively. 1H NMR spectrum also indicated the presence of two doublets resonating at δ 7.57, J = 7.8 and 6.94, J = 7.8 each integrated for two equivalent protons confirming an AA’, BB’ system of ring B. Accordingly, the two singlets at d 3.79 and 3.89; attributed to two methoxyls should be situated at C-6 and C-4’ respectively. Both 1H NMR and 13C NMR data were identical to those previously reported for 5,7-dihydroxy, 6,4’- dimethoxy flavone, commonly known as pectolinarigenin.[9]
Different NMR spectra of compounds 4–7 indicated the presence of two aromatic rings and two doubly conjugated carbonyl carbons thus supporting their anthraquinone skeleton.
1HNMR spectrum of compound 4 illustrated the presence of two downfield shifted signals resonating at δ 12.14 and 12.04 supporting the presence of two peri hydroxyl groups. The two signals resonating at δ 192.6 and 182.1 in the 13C NMR spectrum supported the presence of a chelated and nonchelated carbonyls respectively. These findings supported the location of the two peri hydroxyl groups at C-1 and C-8. 1H NMR spectrum illustrated signals at δ 7.13, d, J = 1.2 and 7.66, d, J = 1.2 which were attributed to H-2 and H-4 respectively. Accordingly, the singlet detected at d 2.48 in 1H NMR spectrum is for a methyl group that should be located at C-3. The signals detected at d 7.83, dd, J = 1.2, 7.8; 7.61, t, J = 7.8 and 7.31, dd, J = 1.2, 8.4 are assigned to H-5, H-6 and H-7 respectively. Different NMR data confirmed the structure of 4 to be 1,8-dihydroxy-3-methyl anthraquinones known as chrysophanic acid.
1HNMR data of compound 5 declared the presence of two downfield shifted signals resonating at δ 12.12 and 12.29 for two peri hydroxyl groups locating at C-1 and C-8. This was confirmed from the carbonyl signals detected at δ 190.8 and 182.0 for a chelated and nonchelated carbonyl functionalities. 1H NMR data also revealed the presence of two meta coupled protons resonating at δ 7.09 and 7.62 attributed to H-2 and H-4 respectively, beside other two meta coupled protons at δ 7.28 and 6.67 for H-5 and H-7 respectively. The presence of a methyl group was confirmed from the singlet at δ 2.45 with its corresponding carbon at detected at δ 22.7 which should be located at C-3 as declared from HMBC Therefore, C-6 (δ 165.2) should be substituted with a hydroxyl group. From these data compound 5 is identified as 1,8-dihydroxy-3-methyl-6-hydroxy anthraquinones; known as emodin.
Compound 6 was found to be closely related to compound 4 except for the presence of one downfield shifted singlet resonating at δ 11.28 attributed to one peri hydroxyl group (instead of two in compound 4). 1H NMR and 13C NMR spectra illustrated signals of a glucose moiety that could be located at C-8. This assumption was confirmed from HMBC through the cross peaks noticed from the anomeric proton of the glucose resonating at δ5.09 to C-8 detected at δ 159.7. Accordingly, compound 6 is identified as chrysophanic acid-8-O-glucoside.
Compound 7 was identified as emodin-8-O-glucoside from 1H NMR, 13C NMR and two-dimensional NMR data which declared the great similarity between compound 7 and 5. The only difference was at C-8 where the perihydroxyl group in compound 5 was replaced by O-glucose in compound 7. This was further proved from long range coupling between H-1’ (δ 4.95) and C-8 (δ163.1) as indicated by HMBC data.
Different NMR data of compound 11 revealed its flavonoid nature. In 1H NMR, the two doublets resonating at δ 6.11, J = 2.4 and 6.31, J = 2.4 were assigned to H-6 and H-8 respectively. Also the signals detected at δ 7.83, d, J = 1.8, 6.81, d, J = 8.4 and 7.49, dd, J = 1.8, 8.4 indicating an ABX system attributed to H-2’, H-5’, H-6’ respectively. 1H NMR spectrum also revealed the presence of two singlets at d 3.94 and 3.68 for two methoxyl groups. In HMBC, cross peaks were noticed from the methoxyl protons (δ 3.94) to C-3’ (δ 148.0) and from the second methoxyl protons (δ 3.68) to C-4’ (δ 151.1). Accordingly, HMBC spectrum confirmed the situation of the two methoxyls at C-3’ and C-4’. Both 1H NMR and 13C NMR data confirmed the presence of a glucose moiety. HMBC spectrum revealed the presence of cross peaks between the anomeric proton resonating at d 5.31 and C-3 detected at d 135.1 thus confirming the location of this glucose moiety at C-3. All the spectral data collectively confirmed the structure of the compound 11 as 5,7-dihydroxy, 3’,4’- dimethoxy flavonol-3-O-glucoside; commonly known as dillenetin-3-O glucoside.
Different 1H NMR data of compound 12 were found to be identical to that of catechin beside the existence of a singlet resonating at δ 7.19. In addition to the carbon signals attributed to catechin, 13C NMR revealed the presence of other five signals at δ 119.3, 109.1, 145.3, 139.0 and 165.7. Investigation of 13C NMR and HSQC spectra proved that those signals are attributed to a galloyl moiety. The galloyl moiety was cited at 4’ based on HMBC correlations where HMBC spectrum revealed the presence of cross peaks between the proton resonating at d 7.19 of the galloyl moiety and C-4’ of the catechin unit detected at δ 144.8. Accordingly, compound 12 was identified as catechin-4’-O-gallate which was previously reported from a number of plants.[10,11]
Generally, phenolics have been considered as powerful antioxidants and many of them proved to be more potent than vitamins C, E and carotenoids.[12,13] All phenolic compounds isolated from P. austroarabica were tested for their free radical scavenging activity using DPPH reagent. Previous investigations on the antioxidant activity of the flavonoid quercetin revealed its powerful effect.[14,15,16] In the present work, quercetin together with other isolated phenolic compounds were examined for their free radical scavenging activity using DPPH reagent at a concentration of 5 μg/ml. The method is based on the reduction of a methanol solution of DPPH in the presence of a hydrogen donating antioxidant due to the formation of the nonradical form DPPH-H.[7] This transformation results in a change in colour from purple to yellow, which was measured spectrophotometrically at 516 nm. The percentage inhibition of the purple colour of the DPPH as a measure of the free radical scavenging activity is shown in Figure 2. Quercetin and catechin-4’-O-gallate revealed the highest activity (89%) followed by methyl gallate (87%). Also; catechin, dillenetin-3-O-glucoside and pectolinarigenin possess high free radical scavenging activities (81, 80 and 77% respectively). Generally, we can notice as illustrated in Figure 2 that all the tested compounds possess antioxidant activities to variable degrees as compared to quercetin the well-known antioxidant flavonoid.[14,15,16]
Figure 2.
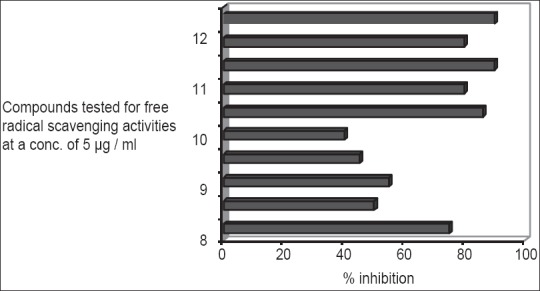
Free radical scavenging activity of investigated isolated compounds (5 μg/ml) using 2,2-diphenyl-1-picrylhydrazyl reagent
Moreover, previous investigation of the known isolated compounds revealed that many of them possess potent antiinflammatory activities. As examples for those compounds; lupeol,[17] ursolic acid,[18] pectolinarigenin,[19] emodin,[20] quercetin,[21] catechin[22] and methyl gallate.[23] These findings can justify the folk use of the plant in treatment of diabetes as it is well known the relation between both antioxidant and antiinflammatory activities from one side and the hypoglycemic activity (especially in type II diabetes) from the other side.[24]
CONCLUSION
Chemical investigation of P. austroarabica resulted in the isolation of 12 compounds. The isolated compounds revealed antioxidant and anti-inflammatory activities. Since both these activities are key factors in treatment of diabetes mellitus (type II), the results of the study supported the traditional use of the plant as a hypoglycemic agent. Further pharmacological studies should be carried out to consider this plant in drug discovery.
Footnotes
Source of Support: Nil
Conflicts of Interest: None declared.
REFERENCES
- 1.Lin J, Lin YT. Flavonoids from the leaves of Loranthus kaoi (Chao) Kiu. J Food Drug Anal. 1999;7:185–90. [Google Scholar]
- 2.Kim YK, Kim YS, Choi SU, Ryu SY. Isolation of flavonol rhamnosides from Loranthus tanakae and cytotoxic effect of them on human tumor cell lines. Arch Pharm Res. 2004;27:44–7. doi: 10.1007/BF02980044. [DOI] [PubMed] [Google Scholar]
- 3.Al-Taweel AM, Perveen S, Fawzy GA, Alqasoumi SI, El Tahir KE. New flavane gallates isolated from the leaves of Plicosepalus curviflorus and their hypoglycemic activity. Fitoterapia. 2012;83:1610–5. doi: 10.1016/j.fitote.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Badr JM, Shaala LA, Youssef DT. A new polyhydroxylated flavanocoumarin from Plicosepalus acacia with significant free radical scavenging and antimicrobial activity. Phytochem Lett. 2013;6:113–7. [Google Scholar]
- 5.Ogunmefun OT, Fasola TR, Saba AB, Oridupa OA. The ethnobotanical, phytochemical and mineral analyses of Phragmanthera incana (Klotzsch), a species of mistletoe growing on three plant hosts in South-Western Nigeria. Int J Biomed Sci. 2013;9:33–40. [PMC free article] [PubMed] [Google Scholar]
- 6.Din N, Dibong SD, Mpondo EM, Priso RJ, Kwin NF, Ngoye A. Inventory and identification of plants used in the treatment of diabetes in Douala Town (Cameroon) Eur J Med Plants. 2011;3:60–73. [Google Scholar]
- 7.Williams BW, Cuverlier ME, Berset C. Use of free radical method to evaluate antioxidant activity. Food Sci Technol. 1995;28:25–30. [Google Scholar]
- 8.Goad LJ, Akihisa T. London, UK: Blackie Academic and Professional; 1992. Analysis of Sterols. [Google Scholar]
- 9.Juang FC, Chen YF, Lin FM, Huang KF. Constituents from the leaves of Lantana camara (IV) J Chin Med. 2005;16:149–55. [Google Scholar]
- 10.Tanaka T, Kataoka M, Tsuboi N, Kouno I. New monoterpene glycoside esters and phenolic constituents of Paeoniae radix, and increase of water solubility of proanthocyanidins in the presence of paeoniflorin. Chem Pharm Bull (Tokyo) 2000;48:201–7. doi: 10.1248/cpb.48.201. [DOI] [PubMed] [Google Scholar]
- 11.Lee MW, Morimoto SM, Nonaka GN, Nishioka IN. Tannins and related compounds. 111. Flavan-3-ol gallates and proanthocyanidins from Pithecellobium lobatum. Phytochemistry. 1992;31:2117–20. [Google Scholar]
- 12.Rice-Evans CA, Miller NJ, Bolwell PG, Bramley PM, Pridham JB. The relative antioxidant activities of plant-derived polyphenolic flavonoids. Free Radic Res. 1995;22:375–83. doi: 10.3109/10715769509145649. [DOI] [PubMed] [Google Scholar]
- 13.Rice-Evans CA, Miller NJ, Paganga G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med. 1996;20:933–56. doi: 10.1016/0891-5849(95)02227-9. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Swarts SG, Yin L, Liu C, Tian Y, Cao Y, et al. Antioxidant properties of quercetin. Adv Exp Med Biol. 2011;701:283–9. doi: 10.1007/978-1-4419-7756-4_38. [DOI] [PubMed] [Google Scholar]
- 15.Geetha T, Malhotra V, Chopra K, Kaur IP. Antimutagenic and antioxidant/prooxidant activity of quercetin. Indian J Exp Biol. 2005;43:61–7. [PubMed] [Google Scholar]
- 16.Coşkun O, Kanter M, Armutçu F, Çetin K, Kaybolmaz B, Yazgan O. Protective effects of quercetin, a flavonoid antioxidant, in absolute ethanol-induced acute gastric ulcer. Eur J Gen Med. 2004;1:37–42. [Google Scholar]
- 17.Saleem M. Lupeol, a novel anti-inflammatory and anticancer dietary triterpene. Cancer Lett. 2009;285:109–15. doi: 10.1016/j.canlet.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vasconcelos MA, Royo VA, Ferreira DS, Crotti AE, Andrade e Silva ML, Carvalho JC, et al. In vivo analgesic and anti-inflammatory activities of ursolic acid and oleanoic acid from Miconia albicans (Melastomataceae) Z Naturforsch C. 2006;61:477–82. doi: 10.1515/znc-2006-7-803. [DOI] [PubMed] [Google Scholar]
- 19.Lim H, Son KH, Chang HW, Bae K, Kang SS, Kim HP. Anti-inflammatory activity of pectolinarigenin and pectolinarin isolated from Cirsium chanroenicum. Biol Pharm Bull. 2008;31:2063–7. doi: 10.1248/bpb.31.2063. [DOI] [PubMed] [Google Scholar]
- 20.Chang CH, Lin CC, Yang JJ, Namba T, Hattori M. Anti-inflammatory effects of emodin from ventilago leiocarpa. Am J Chin Med. 1996;24:139–42. doi: 10.1142/S0192415X96000189. [DOI] [PubMed] [Google Scholar]
- 21.Boots AW, Wilms LC, Swennen EL, Kleinjans JC, Bast A, Haenen GR. In vitro and ex vivo anti-inflammatory activity of quercetin in healthy volunteers. Nutrition. 2008;24:703–10. doi: 10.1016/j.nut.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 22.Babu PV, Liu D. Green tea catechins and cardiovascular health: An update. Curr Med Chem. 2008;15:1840–50. doi: 10.2174/092986708785132979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SJ, Jin M, Lee E, Moon TC, Quan Z, Yang JH, et al. Effects of methyl gallate on arachidonic acid metabolizing enzymes: Cyclooxygenase-2 and 5-lipoxygenase in mouse bone marrow-derived mast cells. Arch Pharm Res. 2006;29:874–8. doi: 10.1007/BF02973908. [DOI] [PubMed] [Google Scholar]
- 24.Ceriello A, Testa R. Antioxidant anti-inflammatory treatment in type 2 diabetes. Diabetes Care. 2009;32:S232–6. doi: 10.2337/dc09-S316. [DOI] [PMC free article] [PubMed] [Google Scholar]