

More than any other organs, brain energy demand is entirely dependent on glucose catabolism through the oxidative phosphorylation (OXPHOS). Glucose is the major cerebral energy substrate in the nervous system (NS). Ketone bodies can be utilized as an additional substrate, but in any case, neurons critically depend on oxygen supply. This sounds quite surprising considering that NS contains few mitochondria, which are universally considered the exclusive site of OXPHOS. Several authors have hypothesized that glia may be involved in the energetic support of the axon, supposing an unknown trophic role played by myelin sheath. In fact, the myelin-forming cells, i.e., oligodendrocytes and Schwann cells in central and peripheral NS, respectively, appear fundamental for the maintenance of long-term axonal functional integrity (Lee et al., 2012).
In the last decade, we have focused our attention on myelin sheath, demonstrating that isolated myelin is able to conduct an extramitochondrial OXPHOS, producing ATP through oxygen consumption (Ravera et al., 2009, 2011, 2013b). Glycolytic and Krebs cycle enzymes are functionally expressed in myelin sheath, sustaining the whole glucose catabolism (Ravera et al., 2013a). Moreover, we have recently shown that the sheath layers and the axoplasm are connected by connexins (Ravera et al., 2015a), suggesting a possible way to transfer ATP from sheath to axon. These data appear confirmed by the recent proteome analysis on the Eif2b5 (R132H/R132H) mouse model of vanishing white matter (VWM) disease where the authors postulate a link among VWM and the myelin metabolic function (Gat-Viks et al., 2015).
Considering that some OXPHOS proteins are encoded by mitochondrial DNA, and that the OXPHOS machinery, likely arranged into a “supercomplex” is assembled inside the mitochondrial inner membrane, we have hypothesized that the OXPHOS proteins are transferred from mitochondria to the myelin-forming oligodendrocyte plasma membrane, by a heterologous fusion among mitochondria and endoplasmic reticulum (Ravera et al., 2015a). Therefore, myelin OXPHOS machinery would be the same as expressed in mitochondria.
The OXPHOS is a major cellular source of reactive oxygen species (ROS), both in mitochondria and in myelin, as we have recently demonstrated (Ravera et al., 2015b). ROS production may follow structural damage of the sheath or uncoupling between oxygen consumption and ATP synthesis. Notably, myelin is a lipid-rich membrane with a slow turnover rate; therefore, a sustained oxidative stress may cause lipid peroxidation, worsening the ROS production and sheath damage, in a vicious cycle. Nonetheless, myelin contains some detoxicant enzymes, such as catalase, superoxide dismutase and glutathione peroxidase (Ravera et al., 2015b), which can minimize the oxidative damage in healthy conditions. However, if ROS impairs respiratory chain protein folding or integrity of lipid environment, ATP production decreases causing hypometabolism when a certain threshold of decreased complex activity is reached. This new vision could clarify why the myelin loss not only causes a decrement in the nerve conduction velocity, but also determines axonal degeneration, the main causes of the disabling symptoms in demyelinating diseases (Figure 1). Impaired energy homeostasis has been implied in neurodegenerative disorders. Interestingly, several neurodegenerative diseases are associated with mitochondrial dysfunctions or mutation (Orth and Schapira, 2001).
Figure 1.
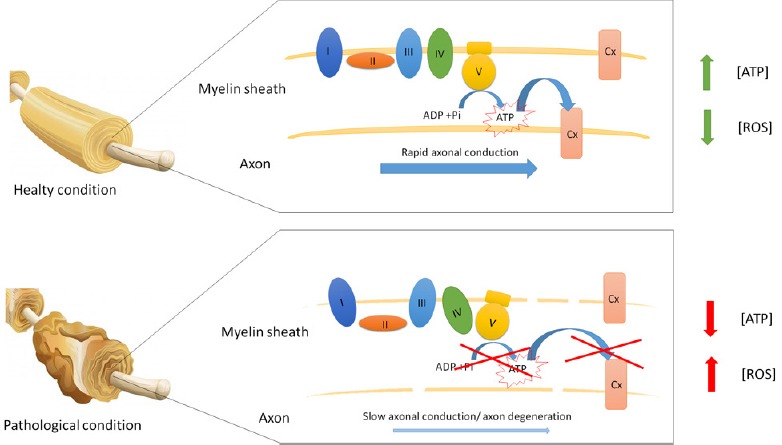
Energy function of myelin sheath in healthy and pathological conditions.
This figure schematizes the oxidative ATP production in myelin. In healthy conditions, myelin produces ATP to support the axon energy demand. By contrast, in pathological conditions, the myelin oxidative phosphorylation is impaired, causing axon degeneration and enhancing the oxidative stress production.
The awareness of the existence of an oxidative metabolism in myelin could shed new light on Leber hereditary optic neuropathy (LHON), a disease caused by a mutation affecting some subunits of mitochondrial respiratory complex I (Howell, 1997), a main site of ROS production. Central vision loss in LHON is attributed to progressive retinal ganglion cells (RGC) death. However, how respiratory complex I mutations cause the selective death of RGCs remains to be elucidated, in spite of many pathogenic mechanisms currently proposed, such as mitochondrial network dynamic impairment, DNA maintenance failure or unfolded protein response. A selective RGC loss simply due to a mitochondrial disturbance remains puzzling, as the mitochondria of the other cell types bearing the same mutations, throughout the affected individual, do not suffer any toxic effect. Conceivably, a ROS-damaged inner mitochondrial membrane may not represent a challenge, as mitochondrial turnover is high. By contrast, myelin would be prone to suffer from dysfunction of a mutated complex I, increasing the ROS burden in its lipid-rich spires. This is especially true when the myelin formation is completed, i.e., at 20 years of age for man, as its turnover slows down. Even though axonal loss is yet to be proved to initiate RGC death, it is tempting to presume that progressive ROS accumulation would cause slowing down the ATP production in the sheath, until the axoplasm becomes critically impaired. According to this hypothesis, RGC loss could be explained as a kind of dye-back mechanism. The idea appears consistent with the beneficial effect of treatment of LNOH patients with an analogue of Ubiquinone (Idebenone), recently shown to promote recovery of visual acuity. Quinones, in fact, improve electron transfer efficiency inside respiring membranes.
Impairment of myelin metabolic activity may play a pivotal role also in demyelinating diseases, such as Charcot-Marie-Tooth type 1A (CMT1A) a hereditary dys/demyelinating disease. We have previously evaluated the myelin metabolism in a CMT1A affected rat (CMT1A rat), reporting that the oxygen consumption by myelin was severely impaired in adult affected animals, with respect to controls (Ravera et al., 2013b). By contrast, mitochondria extracted from CMT1A rat Schwann cells did not show any dysfunction. However, an increased mitochondrial density was observed in demyelinated axons (Ravera et al., 2013b), seemingly to supply the impaired energetic support from myelin. The sheath structure is altered in CMT1A, therefore we have supposed that the OXPHOS impairment in CMT1A myelin may depend on the alteration of the lipid environment which houses the complexes (Ravera et al., 2013b).
Another example is Multiple Sclerosis (MS), a chronic neurological autoimmune disease, whose etiology is still debated. Recently, the axon has become the focus of research on MS, as it plays a crucial role in the disease progression. In fact, although damage to myelin sheath by an abnormal immune system is considered the “primum movens” of MS, it is well established that progressive axonal loss is the major cause of the neurological disability. Data strengthen the hypothesis that long-term axonal survival requires trophic support from oligodendrocytes and/or myelin. Several lines of investigation have provided evidence for the involvement of mitochondrial dysfunction and mtDNA deletions in axonal damage and associated neurodegeneration in MS. Evidence shows that chronically demyelinated axons undergo hypometabolism, eventually degenerating (Mao and Reddy, 2010). The hypometabolism and the oxidative stress may also depend by inflammation, due to the production of cytokines and interleukins by the uncontrolled immune system. For this reason, at the present, the principal therapies for MS are devoted to modulate the immune system, i.e., interferon and cortisone, to inhibit the pro-inflammation events. Currently, the axonal energy imbalance is ascribed to the increased Na+/K+ ATPase pump functioning consequent to the redistribution of Na+ channels on the axonal membrane after demyelination. In fact, if myelin is the site of extramitochondrial OXPHOS that supplies axonal functioning, demyelination may cause lack of ATP. Moreover, myelin OXPHOS machinery impairment would also increase ROS production. This in turn would cause oxidative stress damaging the lipid environment of the OXPHOS machinery, increasing the ROS production and the myelin loss. It is worth noting that oxidative stress itself could cause the immune system attack against myelin sheath. In fact, even though MS is considered an autoimmune disease, the real trigger it is still unknown and several factors (i.e., genetic predisposition and environmental factors) are involved in this pathology. Therefore, it is hypothesized that an impaired myelin OXPHOS may induce a lipid structure damage of the sheath, activating the autoimmune response and creating a vicious circle.
An interesting clinical study on patients affected by MS demonstrated that administration of galactose (Gal), an epimer of glucose, improved the performance of patients, reverting the most invalidating symptoms of MS (Hartstein and Ulett, 1957). Authors ascribed the positive effect of Gal to its promoting remyelination. However, Gal may be a good respiring substrate for the sheath, in fact, it is known to be a nutrient able to induce a burst in OXPHOS metabolism. In particular, we have reported that Gal is metabolized by hexose 6 phosphate dehydrogenase (H6PD) (Ravera et al., 2015c), a microsomal enzyme that leads the first two reactions of pentose phosphate pathway (PPP), active in the sheath. In particular, H6PD would oxidize Gal producing NADPH/NADH, which would directly be discharged on complex I. The presence of a microsomal enzyme, as H6PD, in myelin appears confirmative of the hypothesis of the involvement of the ER in the transfer of the OXPHOS machinery from mitochondria to myelin. Notably, H6PD is considered a new gene associated to MS, confirming that this enzyme plays an important role in myelin sheath. The administration of Gal to improve the symptoms in neurodegenerative diseases has been also proposed for Alzheimer's disease. In particular, chronic oral galactose treatment improves cognitive deficits in intracerebroventricular streptozotocin-induced rat models of Alzheimer's dixease.
In conclusion, we believe that the proposed metabolic function for myelin is pivotal in the energy support of the axon and that its alterations may promote neurodegeneration. This vision can change the concept of the etiopathogenesis of demyelinating diseases as mainly caused by axonal hypometabolism. This may allow envisaging new therapies focalized to maintenance or restoration of the extramitochondrial OXPHOS functionality in myelin.
This study was supported by: Grant from the “Fondazione Giuseppe Levi–Accademia Nazionale dei Lincei” for the research project entitled: “Produzione extramitocondriale di ATP in mielina: localizzazione dei complessi della catena respiratoria e possible ruolo nella degenerazione assonale in Sclerosi Multipla”, No. Borsa “Giuseppe Levi”_2013; Grant from the ‘‘Compagnia di San Paolo’’- Neuroscience Program, for the research project entitled: ‘‘Energetic metabolism in myelinated axon: a new trophic role of myelin sheath’’, No. 2008.1142.
References
- 1.Gat-Viks I, Geiger T, Barbi M, Raini G, Elroy-Stein O. Proteomics-level analysis of myelin formation and regeneration in a mouse model for Vanishing White Matter disease. J Neurochem. 2015;134:513–526. doi: 10.1111/jnc.13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartstein J, Ulett GA. Galactose treatment of multiple sclerosis: a preliminary report. Dis Nerv Syst. 1957;18:255–258. [PubMed] [Google Scholar]
- 3.Howell N. Leber hereditary optic neuropathy: mitochondrial mutations and degeneration of the optic nerve. Vis Res. 1997;37:3495–3507. doi: 10.1016/S0042-6989(96)00167-8. [DOI] [PubMed] [Google Scholar]
- 4.Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao P, Reddy PH. Is multiple sclerosis a mitochondrial disease? Biochim Biophys Acta. 2010;1802:66–79. doi: 10.1016/j.bbadis.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001;106:27–36. doi: 10.1002/ajmg.1425. [DOI] [PubMed] [Google Scholar]
- 7.Ravera S, Panfoli I, Calzia D, Aluigi MG, Bianchini P, Diaspro A, Mancardi G, Morelli A. Evidence for aerobic ATP synthesis in isolated myelin vesicles. Int J Biochem Cell Biol. 2009;41:1581–1591. doi: 10.1016/j.biocel.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Ravera S, Panfoli I, Aluigi MG, Calzia D, Morelli A. Characterization of Myelin Sheath F(o)F(1)-ATP synthase and its regulation by IF(1) Cell Biochem Biophys. 2011;59:63–70. doi: 10.1007/s12013-010-9112-1. [DOI] [PubMed] [Google Scholar]
- 9.Ravera S, Bartolucci M, Calzia D, Aluigi MG, Ramoino P, Morelli A, Panfoli I. Tricarboxylic acid cycle-sustained oxidative phosphorylation in isolated myelin vesicles. Biochimie. 2013a;95:1991–1998. doi: 10.1016/j.biochi.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Ravera S, Nobbio L, Visigalli D, Bartolucci M, Calzia D, Fiorese F, Mancardi G, Schenone A, Morelli A, Panfoli I. Oxydative phosphorylation in sciatic nerve myelin and its impairment in a model of dysmyelinating peripheral neuropathy. J Neurochem. 2013b;126:82–92. doi: 10.1111/jnc.12253. [DOI] [PubMed] [Google Scholar]
- 11.Ravera S, Bartolucci M, Adriano E, Garbati P, Ferrando S, Ramoino P, Calzia D, Morelli A, Balestrino M, Panfoli I. Support of nerve conduction by respiring myelin sheath: role of connexons. Mol Neurobiol. 2015a doi: 10.1007/s12035-015-9216-0. doi: 10.1007/s12035-015-9216-0. [DOI] [PubMed] [Google Scholar]
- 12.Ravera S, Bartolucci M, Cuccarolo P, Litamè E, Illarcio M, Calzia D, Degan P, Morelli A, Panfoli I. Oxidative stress in myelin sheath: The other face of the extramitochondrial oxidative phosphorylation ability. Free Radic Res. 2015b:1–36. doi: 10.3109/10715762.2015.1050962. [DOI] [PubMed] [Google Scholar]
- 13.Ravera S, Bartolucci M, Calzia D, Morelli A, Panfoli I. Galactose and hexose 6-phosphate dehydrogenase support the myelin metabolic role. Indian J Res. 2015c;4:21–24. [Google Scholar]