

Abstract
Purpose
This review will discuss recent advances in understanding mouse and human pancreatic islet cell development, novel concepts related to β cell dysfunction and improved approaches for replenishing β cells to treat diabetes.
Recent Findings
Considerable knowledge about pancreatic islet development and function has been gained using model systems with subsequent validation in human tissues. Recently, several rodent studies have revealed that differentiated adult islet cells retain remarkable plasticity and can be converted to other islet cell types by perturbing their transcription factor profiles. Furthermore, significant advances have been made in the generation of β-like cells from stem cell populations. Therefore, the generation of functionally mature β cells by the in situ conversion of non-β cell populations or by the directed differentiation of human pluripotent stem cells could represent novel mechanisms for replenishing β cells in diabetic patients.
Summary
The overall conservation between mouse and human pancreatic development, islet physiology and etiology of diabetes encourages the translation of novel β cell replacement therapies to humans. Further deciphering the molecular mechanisms that direct islet cell regeneration, plasticity and function could improve and expand the β cell replacement strategies for treating diabetes.
Keywords: Islet, pancreas, development, endocrine, β cell, diabetes
Introduction
Extensive research using rodent models has characterized many of the essential genes and molecular mechanisms that are important for pancreatic islet cell development and function. Recently, the increased availability of human fetal pancreas has revealed important similarities and differences in mouse versus human islet cell development and morphology. Furthermore, with the advent of high throughput sequencing, mouse studies have facilitated the identification of a number of causative genetic mutations for pancreatic agenesis, perinatal diabetes and mature onset diabetes of the young (MODY) in humans. With these advances, it is an optimal time to evaluate our current knowledge of mouse and human pancreatic islet cell development, the conserved transcriptional program for generating and maintaining functionally mature β cells, and exciting new approaches for repairing or replacing damaged β cells as potential therapies for diabetes.
Pancreatic Specification
The pancreas derives from two distinct segments of the foregut endoderm that can be identified at approximately embryonic day (E) 8.5 in mice by the expression of the pancreatic determination transcription factor (TF) Pdx1 (Pancreatic and duodenal homeobox 1)[1]. The Pdx1+ multipotent pancreatic progenitor cells (MPCs) cells are highly proliferative and give rise to all cell types of the three major pancreatic compartments, including the exocrine, endocrine, and ductal lineages[2]. Between E9.5 and E11, the expansion, rearrangement and morphogenesis of MPCs restructures the endoderm monolayer to multilayered stratified epithelia forming a dorsally and ventrally located pancreatic bud[3]. When cultured ex vivo, as few as eight MPCs isolated from the early pancreatic bud are sufficient to form pancreatic rudiments containing all three pancreatic lineages[4].
Pancreatic specification and maintenance of MPCs requires several TFs, including Pdx1, Ptf1a (Pancreas specific transcription factor 1a), Mnx1 (Motor neuron and pancreas homeobox1), Sox9 (SRY box 9), and HNF1β (Hepatocyte nuclear factor-1 β)[5–10]. Depletion of any of these TFs impairs pancreatic bud formation and leads to varying degrees of pancreatic agenesis. Furthermore, Pdx1 and Ptf1a are sufficient for pancreas specification; ectopic expression of Pdx1 or Ptf1a in specific regions of endoderm can induce ectopic pancreatic bud development[11–13].
Although little is known about the specification of pancreatic endoderm upstream of Pdx1 and Ptf1a, there is evidence that the TFs Foxa1/Foxa2 and Gata4/Gata6 induce pancreatic fate by activating expression of Pdx1 and Ptf1a[14, 15]. Simultaneous loss of Foxa1/Foxa2 or Gata4/Gata6 reduces the expression of Pdx1+ and Ptf1a+, impairs bud development and subsequently leads to pancreatic agenesis. Currently, the mechanism(s) by which the broadly expressed Gata and Foxa TFs induce Pdx1 and Ptf1a specifically in the prospective pancreatic endoderm is unknown. Likely candidates are the presence or absence of additional regulatory factors and the integration of spatiotemporal signals secreted from adjacent structures such as the notochord, mesenchyme and aorta to promote pancreas development [16].
Corresponding morphological and molecular studies of early pancreas development in humans have been much more limited due to the scarcity of human fetal tissues. However, several recent studies have begun to define the similarities and differences between mouse and human pancreas development. Interestingly, pancreatic specification in humans occurs at a relatively later developmental stage – shortly after gut closure has occurred and the gut tube has become separated from the notochord and aorta[17]. Similar to mice, PDX1 can be first be detected in the presumptive dorsal and ventral pancreatic endoderm at 29–31 days post-conception (dpc) in a region of the pancreatic endoderm that is also positive for GATA4 and FOXA2[17]. No information is currently available for GATA6 or PTF1A expression; however, most cases of pancreatic agenesis in humans has been linked to heterozygosity of GATA6 (>56% of cases) and by rare mutations in PTF1a, PDX1, and GATA4[7, 18–25], suggesting there is some degree of conservation in these early developmental processes. In humans, however, GATA4 is not expressed in the early foregut endoderm prior to pancreas specification, which may at least partly explain a greater dependence on GATA6 for pancreatic specification[17].
Pancreatic Lineage Restriction
During early stages of murine pancreatic bud outgrowth, pancreatic lineage differentiation is primarily limited to a few endocrine glucagon-producing α cells[26]. This early wave of endocrine cell differentiation during murine development, known as the primary transition, is apparently absent in humans, perhaps as a consequence of the relative delay in pancreatic specification[17]. During the secondary transition, which represents the major wave of endocrine cell differentiation, the pancreatic bud drastically reorganizes into an epithelial arboretum with tip and branch structures that are enmeshed in a loose mesenchyme[3]. At this time, the ventral bud descends distally and fuses with the dorsal bud to form a single nascent pancreas. During these complex morphogenetic events, MPCs become lineage restricted and segregate to either Ptf1a+/Gata4+ exocrine progenitors located at the tips of the epithelial plexus or to Sox9+/Nkx6.1+ ductal/endocrine progenitors located in the trunks[27–29]. Shortly after tip/trunk compartmentalization, exocrine progenitors begin to differentiate into acinar cells at the distal end of the tips, while the bi-potent trunk progenitors differentiate into ductal and endocrine precursor cells. Similarly in humans, trip-trunk segregation can be detected by the restriction of GATA4 expression to the tip cells and SOX9 and NKX6.1 to the trunk domain[24].
The specification of MPC towards exocrine or endocrine/ductal bi-potent progenitors involves the mutual repression of Ptf1a and Nkx6 factors[29] and appears to rely on instructive micro-environments established during epithelial polarization and plexus formation[3, 30]. A number of intra-epithelial and mesenchymal signals can instruct MPC proliferation, lineage allocation and differentiation (extensively reviewed in [31]). Notably, Notch signaling can promote both MPC proliferation and trunk bi-potent progenitor specification at the expense of exocrine differentiation[29, 32–37]. Certainly in the future, additional factors that influence MPC fate decisions will be identified, especially those from the developing vasculature that intercalate into the pancreas during the secondary transition[38]. Determining how these signals regulate the Ptf1a/Nkx6 lineage switch during tip trunk compartmentalization will greatly advance the understanding of MPC lineage allocation.
Endocrine islet development
Bi-potent trunk progenitors are directed to an endocrine fate by the transient induction of the TF Neurog3 (Neurogenin3). Neurog3 KO mice fail to develop endocrine cells and display enlarged ducts, suggesting the reallocation of progenitors to the ductal lineage[39–41]. Moreover, ectopic Neurog3 expression in MPCs drives their precocious differentiation into endocrine cells[26, 32, 42]. Shortly after Neurog3 expression, endocrine cells delaminate from the ductal epithelium and aggregate to form nascent islets. Although there is a transient peak in NEUROG3 expression in human fetal pancreas that corresponds to the stage of endocrine lineage commitment, inactivating mutations of NEUROG3 in humans causes relatively mild defects in endocrine islet cell development and function[43–46]. Interestingly, despite the remarkable molecular conservation between zebrafish and murine pancreas development, Neurog3 is also dispensable for endocrine lineage differentiation in zebrafish and, instead, the control of endocrine cell fate requires two basic helix-loop-helix factors, Ascl1b and Neurod1[47]. Since NeuroD1/NEUROD1 is essential for β cell function in mice and humans, NEUROD1 could potentially function redundantly with NEUROG3; however NEUROD1 expression in human pancreas development has yet to be determined[48–50].
In mice, the majority of endocrine cells emerge during the secondary transition as mono-hormonal cells from the Neurog3 endocrine precursor population. The differentiation of specialized mono-hormonal islet cells is controlled by specific combinations of islet TFs that operate as genetic switches by cooperatively inducing islet cell-type specific gene regulatory networks (GRNs) and by repressing alternate islet GRNs (Figure. 1, extensively reviewed in [51, 52]). Although many individual transcription factors have been characterized for their respective roles in islet cell type specification, their cooperative activities are not completely understood. This represents a critical gap in our knowledge since the majority of essential islet TFs, including Pdx1, Nkx2-2, Neurog3, Nkx6.1, Mnx1, Pax6, Isl-1, Glis3, Insm1, Rfx6, and Neurod1 direct the differentiation and specialization for multiple pancreatic cell types[10, 39, 48, 53–63] and extra-pancreatic cell types, such as enteroendocrine cells [48, 61, 64–66] and neuronal cells[67–73]. A further complication is that a single TF may employ distinct mechanisms for regulating different targets in each cell type. For example, in β cells, Nkx2-2 can synergize with Neurog3 to activate NeuroD1, yet it also can interact with the Grg3 co-repressor protein to directly repress Arx, an α cell factor[54, 74]. Furthermore, Neurod1 can promote or impede the development of α cells depending on the presence of Nkx2-2[75]. Therefore the precise mechanisms that control TF target specificity and the downstream GRNs that specify cell type identity are unclear, but are likely to involve unique combinations of TFs forming complexes with chromatin modifying enzymes on the promoters and enhancers of specific targets.
Figure 1. Transcription Factors direct pancreatic islet cell development.
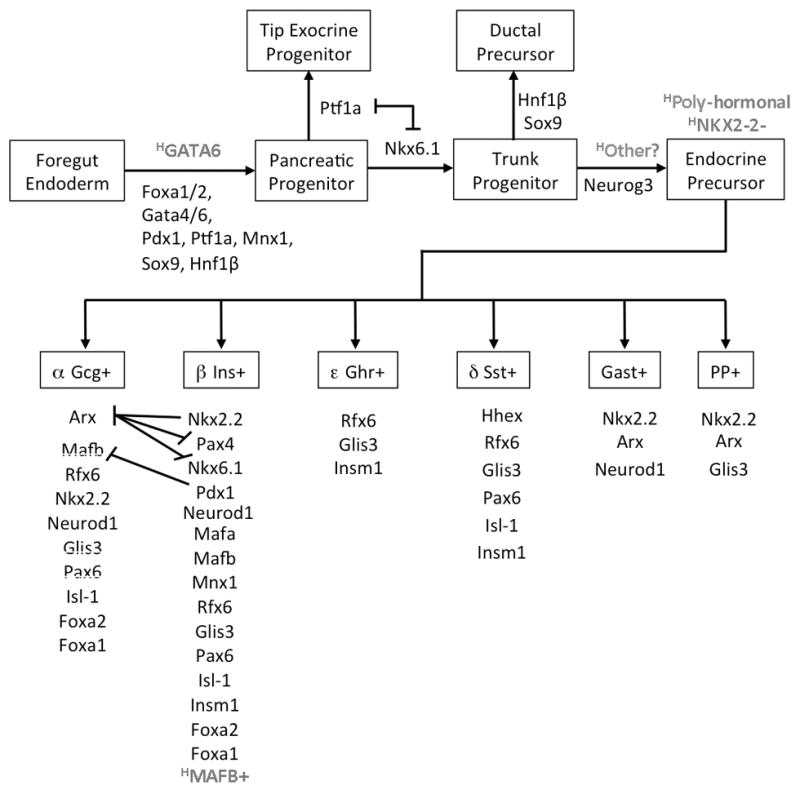
Pancreatic bud development from foregut endoderm depends on the TFs Pdx1, Ptf1a, Mnx1, Foxa1, Foxa2, Gata4 and Gata6. Human pancreas specification has greater dependency on GATA6 possibly due to a lack of GATA4 expression in the foregut endoderm prior to pancreas specification. Lineage restriction of multi-potent pancreatic progenitor populations towards exocrine progenitor cells and ductal/endocrine bi-potent progenitor cells occurs during their segregation to the tips and trunks of the developing pancreatic arboretum respectively and is directed by mutual repression between Nkx6.1 and Ptf1a. Neurog3 is essential for the differentiation of trunk progenitors to hormone producing islet cells in mice but not humans; suggesting functional redundancy by other factors. Specific combinations of functionally conserved TFs are required for Neurog3+ precursors to differentiate into specialized mono-hormonal islet endocrine cells. In humans NKX2-2 is not expressed until endocrine cell differentiation, which may explain why many of the early endocrine cells are poly-hormonal. TFs that differ in human versus mouse by their expression pattern or genetic functions are highlighted in grey with a superscript H to delineate “human”.
The developmental cues and signals that regulate the genetic switches for endocrine cell specification and specialization are also unclear. During early stages of pancreatic bud out growth, the vast majority of endocrine cells are α cells, followed by increased production of β, δ, and ε cells during the secondary transition, and finally the PP cell population during late embryogenesis. The influence of developmental timing on islet cell identity was recently confirmed by the doxycycline controlled ectopic expression of Neurog3 in MPCs (in a Neurog3 null background) at different stages of development[26]. Deciphering the complex transcriptional mechanisms that control islet cell specific GRNs and their regulation by developmental cues will lead to improved strategies for generating functional mono-hormonal β cells from alternative cell sources.
Most TFs analyzed have an expression profile consistent with having a conserved function during human islet development[17, 46, 76]. Moreover, mutations in TFs essential for islet cell development in mice including NEUROD1, NKX2-2, GLIS3, PDX1, RFX6, and MNX1 have been linked to diabetes[50, 77]. Interestingly, the expression pattern of NKX2-2 and MAFB is different in humans and this may explain divergence from mouse islet development[17, 76]. In contrast to mice, a large population of the early endocrine cells in humans is poly-hormonal and the majority of mono-hormonal cell types do not appear until later in development[17, 76, 78]. Interestingly, in humans, NKX2-2 is absent in the early MPCs and is only expressed relatively late during endocrine cell differentiation, corresponding to the appearance of mono-hormonal populations [16]. Given its importance in maintaining islet cell identity in mice[54, 55, 79, 80], NKX2.2 may function to resolve poly-hormonal cells into specialized mono-hormonal cells[17]. In mice, silencing of the TF MafB in the β cell also plays an important role in β cell maturation and identity[81]; however in humans MAFB expression is maintained in β cells indicating that alternative mechanisms may be important for this process [77, 94].
In both mice and humans, all the endocrine cell populations are formed by birth and the full complement of functionally mature endocrine cells aggregate into islet structures shortly after birth. In the adult mouse, 90% of islet cells are β cells that are clustered in the center of the islet and are surrounded by a mantle of the other endocrine islet cell types. In contrast, the human islet has a mosaic distribution of endocrine cells with the proportions of α, δ and β cells reaching 1:1:1 at birth[76, 78]. The relative abundance of α and δ cells in the human islet compared to the mouse islet maybe due to differences in the relative proliferation of these cells to β cells during development [76, 78, 82, 83].
Maintenance of Islet cell identity
The generation of conditional mutations in TFs that are required for islet cell differentiation has revealed that the functional identity of islet cells is not permanently hardwired, but needs to be actively maintained throughout the cell’s lifetime. For example, deletion of the β cell determination TFs Nkx6.1 and Pdx1 in adult β cells leads to their conversion to δ cell-like and α cell-like phenotypes, respectively[81, 84, 85]. β cell function also depends on sustained expression of Neurod1, Rfx6, Pax6, Glis3, Islet1, Foxa1 and Foxa2[49, 86–91]. Similarly, in α cells, deletion of Arx or ectopic expression of Pax4 directs their trans-differentiation to a β cell-like phenotype[92, 93]. In addition to these genetic TF models, sufficient oxygenation of β cells also appears to be required to maintain the functional identity of β cells: culturing islets in hypoxic conditions or disrupting the Vhlh (von Hippel-Lindau) and the Hif1α oxygen sensing pathway alters the expression of differentiation and progenitor markers. Although genetic lineage tracing in human islets is not possible, one study has demonstrated that α cells can also be partially converted to β–like cells when cultured in vitro in the presence of methyltransferase inhibitor[94]. These studies have revealed the existence of a previously unappreciated plasticity in the adult islet that has influenced current ideas about β cell dysfunction and raised the possibility that novel transdifferentiation mechanisms could be used to regenerate or replace β cells in diabetic islets[95].
Loss of β cell identity during the pathogenesis of Type 2 Diabetes
During the pathogenesis of T2D, loss of glycemic control occurs by the deterioration of functional β cells in response to chronic exposure to cellular stressors generated during insulin resistance. Experiments that lineage labeled β cells in several diabetic mouse models revealed that β cell mass is reduced not only due to apoptosis, as previously believed, but also from the transcriptional silencing of insulin and other markers of functionally mature β cells[96]. During the initial stage of the disease, β cells can accommodate the increased demand for insulin by increasing β cell proliferation and insulin production. In particular, the TF Foxo1 is activated by metabolic stress during insulin resistance to enhance β cell function, at least in part, by directly inducing the expression of MafA and Neurod1[96, 97]. However, prolonged metabolic stress impairs Foxo1 activation and eventually causes a subset of β cells to acquire α cell-like phenotypes or express progenitor markers such as Neurog3, Oct4, and Nanog[96, 98]. The reactivation of progenitor markers has been coined “dedifferentiation”, however expression profiling and functional analysis for pluripotency have not yet confirmed whether these former Ins+ β cells have dedifferentiated to a progenitor-like state.
Evaluation of human islets from patients with T2D has also revealed phenotypes consistent with β cell dormancy, which is characterized by diminished expression of insulin and other markers for differentiated β cells, but without significant induction of progenitor markers[98]. Expression analysis of TFs found to be critical for maintaining functionally mature β cells in the mouse revealed diminished expression of TFs PDX1, NKX6.1 and MAFA in human T2D islets[98]. The loss of Pdx1, Nkx6.1 and MafA in the mouse adult β cell leads to many of the same defects observed in T2D islets, suggesting β cell dormancy may be driven by the transcriptional silencing of these TFs during pathogenesis of T2D[81, 84, 85, 99–101]. Recently, several studies have provided mechanistic insight into how β cell transcriptional complexes are inactivated during the pathogenesis for T2D. Most of the SNPs (Single poly-nucleotide polymorphisms) that are associated with T2D are found in non-coding sequences and little is known about how they contribute to β cell dysfunction or dormancy[102, 103]. The genome wide identification of human islet specific enhancers by ChIP-seq analysis has revealed that several of these SNPS are likely to predispose for β cell dormancy by disrupting TF binding sites found in islet specific enhancers[104]. The activity of TFs can also be modified by cellular stressors generated during insulin resistance. Hyperglycemia can lead to the accumulation of highly reactive free radicals that have recently been shown to directly inactivate the β cell TFs MafA and Nkx6.1[98]. Ectopic overexpression of MafA partially prevented β cell dedifferentiation in a mouse model for T2D and significantly improved their pathology[98]. Similarly, administering antioxidants or transgenic overexpression of Gpx-1, an enzyme that reduces free radicals, can prevent the dedifferentiation of β cells in a mouse model for T2D[98, 105, 106]. Better understanding of how cellular stressors generated during insulin resistance inactivate β cell specific transcriptional complexes will likely lead to other novel therapies designed to prevent or reverse β cell dormancy during the pathogenesis of T2D.
Regeneration of β cells in the adult
Adult β cells are mostly quiescent, however a small fraction can be activated to proliferate in response to hyperglycemia from insulin resistance or β cell depletion[107–110]. Compared to mice, human β cells are more resistant to cell cycle entry during development and in response to stress or mitogens[111]. The limited regenerative capacity of β cells has led many researchers to search for alternative approaches to replenish lost or dysfunctional β cells, including taking advantage of the recently discovered islet cell plasticity. It has now been demonstrated that severe β cell depletion (>99%) can lead to a small fraction of either α or δ cells to transdifferentiate into a β cell-like phenotype in adult and adolescent mice respectively[112, 113]. Remarkably, the chemical ablation of β cells combined with treatment of the cytokines CNTF and EGF can convert acinar cells to a β cell-like phenotype, nearly restoring normal glycemic control[114]. Previous experiments suggested ductal cells could transdifferentiate to β cells in response to β cell depletion from partial duct ligation-induced injury[115]. These findings, however have become somewhat controversial in light of new studies using ductal lineage labeling experiments that fail to show duct-derived β cell formation[27, 116].
The ectopic overexpression of β cell TFs can also induce the expression of insulin and other β cell markers in a number of non-islet cell types including liver, exocrine pancreas, gallbladder and mesenchymal stem cells [117–121]. Acinar cells can be converted to a β cell like phenotype in vivo by the adenoviral mediated ectopic overexpression of Pdx1, Neurog3, and MafA[119, 122]. Surprisingly, in a mouse model with ubiquitous transgenic overexpression of the same Pdx1, Neurog3 and MafA combination did not lead to acinar to β cell transdifferentiation, suggesting a permissive effect of either the adenovirus or the nude mouse background used for adenovirus mediated overexpression[119, 123]. The only cells that were converted to a β cell like phenotype by the ubiquitous transgenic overexpression of Pdx1, Neurog3, and MafA were intestinal crypt cells; highlighting their unique potential for reprograming to a β cell fate[123]. Recently both mouse and human intestinal cells could also be converted to a β cell-like phenotype by the depletion of Foxo1 activity[124, 125]. In the future, identifying the factors unique to intestinal cells that allow for their reprogramming to a β cell like fate could be used to directly reprogram other cell types.
Generation of functionally mature β cells from stem cell populations
Previously, protocols based on the extensive knowledge gained from in vivo mouse pancreatic β cell development studies successfully differentiated embryonic stem cells (ESC) to foregut endoderm, pancreatic progenitors and Ins+ cells[126–129]. Although these protocols robustly produced pancreatic endodermal cells, induction of Ins+ cells was inefficient and the majority of Ins+ cells resembled functionally immature β cells that were poly-hormonal and only achieved modest GSIS after 3–4 months of in vivo engraftment[128–131]. However, recent studies by two independent groups have reported significantly improved differentiation protocols that efficiently yield mono-hormonal Ins+ cells with an expression profile and physiology comparable to β cells isolated from human cadaveric islets[132–134]. Within only 2 weeks after transplantation into diabetic mice, the newly improved stem cell derived β cells secreted insulin in response to glucose and partially restored normal glycemic control[132, 134]. This achievement greatly advances the potential for using ESC or IPS cells as a source for β cell replacement therapies for diabetes.
Conclusions
The generation of functionally mature β cells by the differentiation of stem cells or the in situ conversion of other islet cell types, acinar and intestinal cells are major breakthroughs towards replacing dysfunctional β cells in diabetic patients. Moreover, elucidating the etiology of β cell dysfunction during the pathogenesis of T2D has revealed many new potential therapies for preventing or repairing dysfunctional β cells. These breakthroughs were possible due to insight from over 25 years of basic and clinical research aimed at understanding the molecular mechanisms that control key events during islet cell development and diabetes-related dysfunction. This review has highlighted current knowledge and gaps in the understanding of the mechanisms that control pancreatic specification, subsequent lineage restriction and differentiation of MPCs to functionally mature islet cells. Further elucidation of the signaling and transcriptional mechanisms that direct cell type specific GRNs during the development of functionally mature islet cells will enhance the efficiency of β cell differentiation, transitioning from transgenic to pharmacological approaches for reprogramming, and for diversifying the sources of β cell differentiation. The overall conservation of the mechanisms that direct the development of functional β cells in mice and humans reinforces the idea that these strategies may translate to treating patients with diabetes.
Key points.
Many of the key transcription factors (TFs) that regulate pancreatic islet cell development are conserved between mice and humans, however divergent expression patterns and/or functions of some factors may explain phenotypic differences between murine and human islets.
The functional maturity and identity of β cells are actively maintained by TFs that are important for islet development.
Inactivation of β cell TFs during the pathogenesis of T2D can lead to β cell dormancy and/or dysfunction.
Functionally mature β cells can be generated by the differentiation of embryonic stem cells or the in situ trans-differentiation of non-β cell populations, such as α cells, acinar cells and intestinal cells.
Acknowledgments
We thank members of the Sussel lab for intellectual input and critical reading of the manuscript. We also apologize to the many authors whose outstanding research and/or original publications we could not cite or discuss due to space limitations.
Financial Support and sponsorship
NIH R01 DK082590 (LS)
NIH R01 DK087711 (LS)
NYSTEM CO2811 (LS)
T32 DK007328 (AR)
F32 DK103506 (AR)
Footnotes
None of the authors declare a conflict of interest.
References and recommended reading
- 1.Jorgensen MC, et al. An illustrated review of early pancreas development in the mouse. Endocr Rev. 2007;28(6):685–705. doi: 10.1210/er.2007-0016. [DOI] [PubMed] [Google Scholar]
- 2.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 3.Villasenor A, et al. Epithelial dynamics of pancreatic branching morphogenesis. Development. 2010;137(24):4295–305. doi: 10.1242/dev.052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greggio C, et al. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development. 2013;140(21):4452–62. doi: 10.1242/dev.096628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawaguchi Y, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–34. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 6.Offield MF, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–95. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 7.Stoffers DA, et al. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106–10. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 8.Seymour PA, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104(6):1865–70. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haumaitre C, et al. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci U S A. 2005;102(5):1490–5. doi: 10.1073/pnas.0405776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, et al. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23(1):67–70. doi: 10.1038/12669. [DOI] [PubMed] [Google Scholar]
- 11.Grapin-Botton A, Majithia AR, Melton DA. Key events of pancreas formation are triggered in gut endoderm by ectopic expression of pancreatic regulatory genes. Genes Dev. 2001;15(4):444–54. doi: 10.1101/gad.846001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Afelik S, Chen Y, Pieler T. Combined ectopic expression of Pdx1 and Ptf1a/p48 results in the stable conversion of posterior endoderm into endocrine and exocrine pancreatic tissue. Genes Dev. 2006;20(11):1441–6. doi: 10.1101/gad.378706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *13.Willet SG, et al. Dominant and context-specific control of endodermal organ allocation by Ptf1a. Development. 2014;141(22):4385–94. doi: 10.1242/dev.114165. In this study, ectopic pan-endodermal Ptf1a expression redirected tissues adjacent to the pancreatic bud, including glandular stomach, rostral duodenum and biliary hepatic duct to pancreatic fates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xuan S, et al. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest. 2012;122(10):3516–28. doi: 10.1172/JCI63352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao N, et al. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008;22(24):3435–48. doi: 10.1101/gad.1752608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCracken KW, Wells JM. Molecular pathways controlling pancreas induction. Semin Cell Dev Biol. 2012;23(6):656–62. doi: 10.1016/j.semcdb.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings RE, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62(10):3514–22. doi: 10.2337/db12-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwitzgebel VM, et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab. 2003;88(9):4398–406. doi: 10.1210/jc.2003-030046. [DOI] [PubMed] [Google Scholar]
- 19.Sellick GS, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301–5. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- 20.D’Amato E, et al. Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabet Med. 2010;27(10):1195–200. doi: 10.1111/j.1464-5491.2010.03046.x. [DOI] [PubMed] [Google Scholar]
- 21.Thomas IH, et al. Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF-1 Pro63fsX60 mutation. Pediatr Diabetes. 2009;10(7):492–6. doi: 10.1111/j.1399-5448.2009.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *22.Weedon MN, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61–4. doi: 10.1038/ng.2826. This study used whole genome sequence analysis to identify homozygous recessive mutations found in six individuals with pancreatic agenesis and used ChIP analysis to map the mutations to a Ptf1a enhancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.Shaw-Smith C, et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes. 2014;63(8):2888–94. doi: 10.2337/db14-0061. This study found heterozygous mutations in GATA4 in five patients with variable diabetes and pancreatic agenesis. A patient heterozygous for a missense mutation for GATA4 that impairs its DNA binding and transactivation activity was found to cause complete pancreatic agenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Franco E, et al. GATA6 mutations cause a broad phenotypic spectrum of diabetes from pancreatic agenesis to adult-onset diabetes without exocrine insufficiency. Diabetes. 2013;62(3):993–7. doi: 10.2337/db12-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lango Allen H, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44(1):20–2. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johansson KA, et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell. 2007;12(3):457–65. doi: 10.1016/j.devcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Solar M, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17(6):849–60. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Q, et al. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13(1):103–14. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Schaffer AE, et al. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell. 2010;18(6):1022–9. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kesavan G, et al. Cdc42-mediated tubulogenesis controls cell specification. Cell. 2009;139(4):791–801. doi: 10.1016/j.cell.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 31.Serup P. Signaling pathways regulating murine pancreatic development. Semin Cell Dev Biol. 2012;23(6):663–72. doi: 10.1016/j.semcdb.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Apelqvist A, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–81. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 33.Esni F, et al. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131(17):4213–24. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- 34.Hald J, et al. Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev Biol. 2003;260(2):426–37. doi: 10.1016/s0012-1606(03)00326-9. [DOI] [PubMed] [Google Scholar]
- 35.Jensen J, et al. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24(1):36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 36.Murtaugh LC, et al. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920–5. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cleveland MH, et al. Exocrine ontogenies: on the development of pancreatic acinar, ductal and centroacinar cells. Semin Cell Dev Biol. 2012;23(6):711–9. doi: 10.1016/j.semcdb.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villasenor A, Cleaver O. Crosstalk between the developing pancreas and its blood vessels: an evolving dialog. Semin Cell Dev Biol. 2012;23(6):685–92. doi: 10.1016/j.semcdb.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gradwohl G, et al. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–11. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, et al. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol. 2010;339(1):26–37. doi: 10.1016/j.ydbio.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magenheim J, et al. Ngn3(+) endocrine progenitor cells control the fate and morphogenesis of pancreatic ductal epithelium. Dev Biol. 2011;359(1):26–36. doi: 10.1016/j.ydbio.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwitzgebel VM, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127(16):3533–42. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- *43.Rubio-Cabezas O, et al. Neurogenin 3 is important but not essential for pancreatic islet development in humans. Diabetologia. 2014;57(11):2421–4. doi: 10.1007/s00125-014-3349-y. This paper reports four patients with malabsorptive diarrhea that have homozygous missense mutations in NEUROG3. Three of these patents had neonatal diabetes. This paper also summarizes clinical data about previously reported patients who have homozygous NEUROG3 mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubio-Cabezas O, et al. Permanent Neonatal Diabetes and Enteric Anendocrinosis Associated With Biallelic Mutations in NEUROG3. Diabetes. 2011;60(4):1349–53. doi: 10.2337/db10-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N Engl J Med. 2006;355(3):270–80. doi: 10.1056/NEJMoa054288. [DOI] [PubMed] [Google Scholar]
- 46.Salisbury RJ, et al. The window period of NEUROGENIN3 during human gestation. Islets. 2014;6(3):e954436. doi: 10.4161/19382014.2014.954436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flasse LC, et al. Ascl1b and Neurod1, instead of Neurog3, control pancreatic endocrine cell fate in zebrafish. BMC Biol. 2013;11:78. doi: 10.1186/1741-7007-11-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naya FJ, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11(18):2323–34. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu C, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010;11(4):298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malecki MT, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23(3):323–8. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 51.Oliver-Krasinski JM, Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22(15):1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240(3):530–65. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 53.Fujitani Y, et al. Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev. 2006;20(2):253–66. doi: 10.1101/gad.1360106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Papizan JB, et al. Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev. 2011;25(21):2291–305. doi: 10.1101/gad.173039.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sussel L, et al. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. 1998;125(12):2213–21. doi: 10.1242/dev.125.12.2213. [DOI] [PubMed] [Google Scholar]
- 56.Sander M, et al. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11(13):1662–73. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- 57.St-Onge L, et al. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387(6631):406–9. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- 58.Du A, et al. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes. 2009;58(9):2059–69. doi: 10.2337/db08-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang HS, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol. 2009;29(24):6366–79. doi: 10.1128/MCB.01259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe N, et al. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. 2009;583(12):2108–13. doi: 10.1016/j.febslet.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 61.Gierl MS, et al. The zinc-finger factor Insm1 (IA-1) is essential for the development of pancreatic beta cells and intestinal endocrine cells. Genes Dev. 2006;20(17):2465–78. doi: 10.1101/gad.381806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith SB, et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature. 2010;463(7282):775–80. doi: 10.1038/nature08748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Henseleit KD, et al. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development. 2005;132(13):3139–49. doi: 10.1242/dev.01875. [DOI] [PubMed] [Google Scholar]
- 64.Desai S, et al. Nkx2.2 regulates cell fate choice in the enteroendocrine cell lineages of the intestine. Dev Biol. 2008;313(1):58–66. doi: 10.1016/j.ydbio.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jenny M, et al. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. EMBO J. 2002;21(23):6338–47. doi: 10.1093/emboj/cdf649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Larsson LI, et al. Pax 4 and 6 regulate gastrointestinal endocrine cell development. Mech Dev. 1998;79(1–2):153–9. doi: 10.1016/s0925-4773(98)00182-8. [DOI] [PubMed] [Google Scholar]
- 67.Briscoe J, et al. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. 1999;398(6728):622–7. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- 68.Arber S, et al. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23(4):659–74. doi: 10.1016/s0896-6273(01)80026-x. [DOI] [PubMed] [Google Scholar]
- 69.Toresson H, Potter SS, Campbell K. Genetic control of dorsal-ventral identity in the telencephalon: opposing roles for Pax6 and Gsh2. Development. 2000;127(20):4361–71. doi: 10.1242/dev.127.20.4361. [DOI] [PubMed] [Google Scholar]
- 70.Pfaff SL, et al. Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 1996;84(2):309–20. doi: 10.1016/s0092-8674(00)80985-x. [DOI] [PubMed] [Google Scholar]
- 71.Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13(13):1647–52. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wildner H, et al. Insm1 (IA-1) is a crucial component of the transcriptional network that controls differentiation of the sympatho-adrenal lineage. Development. 2008;135(3):473–81. doi: 10.1242/dev.011783. [DOI] [PubMed] [Google Scholar]
- 73.Sander M, et al. Ventral neural patterning by Nkx homeobox genes: Nkx6.1 controls somatic motor neuron and ventral interneuron fates. Genes Dev. 2000;14(17):2134–9. doi: 10.1101/gad.820400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anderson KR, et al. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J Biol Chem. 2009;284(45):31236–48. doi: 10.1074/jbc.M109.048694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mastracci TL, et al. Regulation of Neurod1 contributes to the lineage potential of Neurogenin3+ endocrine precursor cells in the pancreas. PLoS Genet. 2013;9(2):e1003278. doi: 10.1371/journal.pgen.1003278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riedel MJ, et al. Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia. 2012;55(2):372–81. doi: 10.1007/s00125-011-2344-9. [DOI] [PubMed] [Google Scholar]
- *77.Flanagan SE, et al. Analysis of transcription factors key for mouse pancreatic development establishes NKX2-2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 2014;19(1):146–54. doi: 10.1016/j.cmet.2013.11.021. This study identifies recessive homozygous mutations for factors known to be important for β cell function and development in 121 of 147 consanguineous patients with neonatal diabetes. Eleven of these patients had mutations in TFs essential for pancreas and islet cell development including novel mutations in NKX2-2 and MNX1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Piper K, et al. Beta cell differentiation during early human pancreas development. J Endocrinol. 2004;181(1):11–23. doi: 10.1677/joe.0.1810011. [DOI] [PubMed] [Google Scholar]
- 79.Arnes L, et al. Generation of Nkx2.2:lacZ mice using recombination-mediated cassette exchange technology. Genesis. 2012;50(8):612–24. doi: 10.1002/dvg.22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balderes DA, Magnuson MA, Sussel L. Nkx2.2:Cre knock-in mouse line: a novel tool for pancreas- and CNS-specific gene deletion. Genesis. 2013;51(12):844–51. doi: 10.1002/dvg.22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *81.Gao T, et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014;19(2):259–71. doi: 10.1016/j.cmet.2013.12.002. In this study, depletion of Pdx1 in the adult β cell leads to their conversion to an α cell-like phenotype. Pdx1 was shown to maintain β cell identity at least in part by repressing MafB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jeon J, et al. Endocrine cell clustering during human pancreas development. J Histochem Cytochem. 2009;57(9):811–24. doi: 10.1369/jhc.2009.953307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gregg BE, et al. Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97(9):3197–206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaffer AE, et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet. 2013;9(1):e1003274. doi: 10.1371/journal.pgen.1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013;4(6):1262–75. doi: 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *86.Chandra V, et al. RFX6 regulates insulin secretion by modulating Ca2+ homeostasis in human beta cells. Cell Rep. 2014;9(6):2206–18. doi: 10.1016/j.celrep.2014.11.010. This paper demonstrates Rfx6 is required in the adult mouse β cell to maintain their functional identity by maintaining the expression of factors important for β cell function, including insulin and voltage gated Ca2+ channels, as well as repressing “disallowed” factors known to impair glucose stimulated insulin secretion. [DOI] [PubMed] [Google Scholar]
- *87.Piccand J, et al. Rfx6 maintains the functional identity of adult pancreatic beta cells. Cell Rep. 2014;9(6):2219–32. doi: 10.1016/j.celrep.2014.11.033. This paper demonstrates a conserved function for RFX6 that is essential for human β cell function by maintaining the expression of GSIS factors including Insulin and Ca+2 voltage gated channels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hart AW, et al. The developmental regulator Pax6 is essential for maintenance of islet cell function in the adult mouse pancreas. PLoS One. 2013;8(1):e54173. doi: 10.1371/journal.pone.0054173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang Y, Chang BH, Chan L. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol Med. 2013;5(1):92–104. doi: 10.1002/emmm.201201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *90.Ediger BN, et al. Islet-1 Is essential for pancreatic beta-cell function. Diabetes. 2014;63(12):4206–17. doi: 10.2337/db14-0096. The TF Isl1 is shown to be required to maintain the functional identity of adult β cells by maintaining the expression of Pdx1, MafA, insulin and other factors important for β cell function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gao N, et al. Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab. 2007;6(4):267–79. doi: 10.1016/j.cmet.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 92.Courtney M, et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013;9(10):e1003934. doi: 10.1371/journal.pgen.1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Al-Hasani K, et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell. 2013;26(1):86–100. doi: 10.1016/j.devcel.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 94.Bramswig NC, et al. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Invest. 2013;123(3):1275–84. doi: 10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ziv O, Glaser B, Dor Y. The plastic pancreas. Dev Cell. 2013;26(1):3–7. doi: 10.1016/j.devcel.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 96.Talchai C, et al. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150(6):1223–34. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kitamura YI, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2(3):153–63. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 98.Guo S, et al. Inactivation of specific beta cell transcription factors in type 2 diabetes. J Clin Invest. 2013 doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ahlgren U, et al. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12(12):1763–8. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Artner I, et al. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59(10):2530–9. doi: 10.2337/db10-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang C, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25(12):4969–76. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scott RA, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44(9):991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **104.Pasquali L, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46(2):136–43. doi: 10.1038/ng.2870. Chip analysis for critical islet TFs using human islet chromatin identified islet specific enhancers. Causative roles for several diabetes-associated SNPs were discovered since they mapped to islet specific enhancers and were found to disrupt TF binding. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harmon JS, et al. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology. 2009;150(11):4855–62. doi: 10.1210/en.2009-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaneto H, et al. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes. 1999;48(12):2398–406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
- 107.Dor Y, et al. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429(6987):41–6. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 108.Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest. 2007;117(9):2553–61. doi: 10.1172/JCI32959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Porat S, et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab. 2011;13(4):440–9. doi: 10.1016/j.cmet.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tschen SI, et al. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58(6):1312–20. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *111.Wang P, et al. Diabetes mellitus-advances and challenges in human beta-cell proliferation. Nat Rev Endocrinol. 2015 doi: 10.1038/nrendo.2015.9. Extensive review about human and rodent β cell proliferation. [DOI] [PubMed] [Google Scholar]
- **112.Chera S, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature. 2014;514(7523):503–7. doi: 10.1038/nature13633. Complete β cell ablation in adolescent (but not adult) mice leads to the conversion of δ cells to a β cell like phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thorel F, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464(7292):1149–54. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **114.Baeyens L, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32(1):76–83. doi: 10.1038/nbt.2747. In situ conversion of acinar cells to β cells following chemical induced β cell ablation and treatment with the cytokines. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Xu X, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132(2):197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 116.Kopp JL, et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138(4):653–65. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kojima H, et al. NeuroD-betacellulin gene therapy induces islet neogenesis in the liver and reverses diabetes in mice. Nat Med. 2003;9(5):596–603. doi: 10.1038/nm867. [DOI] [PubMed] [Google Scholar]
- 118.Hickey RD, et al. Generation of islet-like cells from mouse gall bladder by direct ex vivo reprogramming. Stem Cell Res. 2013;11(1):503–15. doi: 10.1016/j.scr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhou Q, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ferber S, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6(5):568–72. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- 121.Guo QS, et al. Combined transfection of the three transcriptional factors, PDX-1, NeuroD1, and MafA, causes differentiation of bone marrow mesenchymal stem cells into insulin-producing cells. Exp Diabetes Res. 2012;2012:672013. doi: 10.1155/2012/672013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **122.Li W, et al. Long-term persistence and development of induced pancreatic beta cells generated by lineage conversion of acinar cells. Nat Biotechnol. 2014;32(12):1223–30. doi: 10.1038/nbt.3082. This study significantly improves the efficiency for the in situ reprograming of acinar cells to a β cell phenotype by using an adenovirus for polycistronic overexpression of Pdx1, Neurog3 and MafA. The reprogrammed β cells from this protocol form islet clusters that are maintained for at least 13 months and can sustain normal glycemic control in diabetic mice. [DOI] [PubMed] [Google Scholar]
- **123.Chen YJ, et al. De novo formation of insulin-producing “neo-beta cell islets” from intestinal crypts. Cell Rep. 2014;6(6):1046–58. doi: 10.1016/j.celrep.2014.02.013. In this paper a transgenic mouse with ubiquitous ectopic expression of the TFs Pdx1, Ngn3, and MafA induced formation of islet cell clusters at the base of intestinal crypts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **124.Bouchi R, et al. FOXO1 inhibition yields functional insulin-producing cells in human gut organoid cultures. Nat Commun. 2014;5:4242. doi: 10.1038/ncomms5242. Insulin-producing cells were generated in gut organoids that were differentiated from human iPS cells by perturbing the TF Foxo1 with siRNAs or chemical inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Talchai C, et al. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat Genet. 2012;44(4):406–12. S1. doi: 10.1038/ng.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.D’Amour KA, et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005;23(12):1534–41. doi: 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- 127.D’Amour KA, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24(11):1392–401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- 128.Kroon E, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26(4):443–52. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- 129.Rezania A, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61(8):2016–29. doi: 10.2337/db11-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *130.Bruin JE, et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014;12(1):194–208. doi: 10.1016/j.scr.2013.10.003. Many of the insulin+ cells differentiated from human ES cells were found to be poly-hormonal with defects in glucose stimulated insulin secretion due at least in part from reduced expression of the glucose transporter GLUT1 and potassium channel subunit SUR1. [DOI] [PubMed] [Google Scholar]
- *131.Hrvatin S, et al. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc Natl Acad Sci U S A. 2014;111(8):3038–43. doi: 10.1073/pnas.1400709111. Transcriptome analysis revealed ES cell-derived insulin + cells have a transcriptional profile more similar to immature fetal β cells compared to functionally mature adult β cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **132.Rezania A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32(11):1121–33. doi: 10.1038/nbt.3033. Human ES cells are successfully differentiated into mono-hormonal insulin+ β cells that are capable of partially restoring normal glycemic control in diabetic mice just two weeks after transplantation. While the β cells derived from ES cells express important markers for functionally mature β cells, physiological analysis in culture revealed significant impairment of glucose stimulated insulin secretion compared to cadaveric β cells. [DOI] [PubMed] [Google Scholar]
- 133.Rezania A, et al. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells. 2013;31(11):2432–42. doi: 10.1002/stem.1489. [DOI] [PubMed] [Google Scholar]
- **134.Pagliuca FW, et al. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159(2):428–39. doi: 10.1016/j.cell.2014.09.040. In this paper, human ES cells are successfully differentiated into mono-hormonal β cells that secrete insulin in response to glucose and can restore normal glycemic control in diabetic mice shortly after their transplantation. Global gene expression analysis indicated ES cell-derived β cells had a transcriptome similar to mature human β cells, but with some notable exceptions. [DOI] [PMC free article] [PubMed] [Google Scholar]