

Abstract
Two new one-component aluminium-based catalysts for the reaction between epoxides and carbon dioxide have been prepared. The catalysts are composed of aluminium–salen chloride complexes with trialkylammonium groups directly attached to the aromatic rings of the salen ligand. With terminal epoxides, the catalysts induced the formation of cyclic carbonates under mild reaction conditions (25–35 °C; 1–10 bar carbon dioxide pressure). However, with cyclohexene oxide under the same reaction conditions, the same catalysts induced the formation of polycarbonate. The catalysts could be recovered from the reaction mixture and reused.
Keywords: aluminium, carbon dioxide, cyclic carbonate, epoxide, salen
Introduction
Carbon dioxide is a renewable and inexpensive carbon source, so great efforts have been directed at developing novel methods for the valorization of this abundant raw material [1]. One way of achieving this goal is to produce cyclic carbonates or polycarbonates from carbon dioxide and the corresponding epoxides (Scheme 1). Cyclic carbonates are an important class of solvents [2] and starting materials in organic synthesis [3–6].
Scheme 1.

Synthesis of cyclic and polycarbonates.
Although a significant array of catalysts have been developed for the production of cyclic carbonates [7–9] and polycarbonates [10–11] from carbon dioxide and epoxides, the most developed and privileged set of catalysts are based on Lewis acidic metal–salen complexes. In particular, cobalt(III) and chromium(III) complexes were found to be highly efficient for polycarbonate production [12]. Further modification of the salen moiety by the introduction of basic or ammonium salts through alkyl spacers attached to the salen aromatic rings led to the formation of a family of bifunctional catalysts possessing both Lewis acid and nucleophilic catalysis capability (via the anion in the case of catalysts containing ammonium salts), with a concomitant increase in their activity [12–13]. Recently, more environmentally benign aluminium-based complexes, including salen complexes, have been introduced to catalyse cyclic carbonate production [14]. The performance of these catalysts was also greatly improved by the introduction of bifunctional versions of the catalyst system, combining an electrophilic aluminium centre with an ammonium cation/nucleophilic-counteranion combination within the framework of a single catalytic species as reported by North [15], Liu and Darensbourg [14], and Lu [16] (Figure 1).
Figure 1.

Bifunctional aluminium–salen complexes, including those studied in this work.
Unfortunately, the bifunctional derivatives with an alkyl spacer are not very stable at higher temperatures because of the well-known ammonium salt decomposition pathways including: Zaitsev and Hoffman type eliminations [17–19] and retro-Menschutkin reactions [20–23]. We reasoned that the direct introduction of ammonium moieties onto the aromatic rings of the salen ligands (as in structures 1 and 2) would greatly stabilize the whole structure by reducing the number of sp3-hybridized carbon atoms attached to the nitrogen atoms of the ammonium salts and increasing the steric hindrance around the ammonium salts. Herein, we report the synthesis of two aluminium–salen complexes incorporating quaternary ammonium salts directly attached to the salen ligand and their catalytic activities for the coupling of epoxides and carbon dioxide under solvent free conditions. Catalyst recycling experiments are also reported and show the robustness of this system.
Results and Discussion
The preparation of salen ligands 8a and 8b was conducted according to Scheme 2, starting from tert-butylphenol, which was formylated and then nitrated to produce 5-nitro-3-(tert-butyl)salicylaldehyde (5) [24–27]. 5-N,N-Dimethylamino-3-(tert-butyl)salicylaldehyde (6) was prepared directly from 5, using a literature procedure [28–29]. The salen ligand 7 was then obtained in high yield by condensation with (R,R)-cyclohexanediamine, according to a known technique [30]. Ligand 7 was efficiently alkylated under mild reaction conditions using either methyl iodide or benzyl bromide, giving the corresponding positively charged salen ligands 8a and 8b in 95 and 78% yields respectively. The aluminium–salen complexes were prepared by treating 8a and 8b with diethylaluminium chloride (Scheme 3), affording complexes 1 and 2 in 96% and 91% yields respectively. These complexes could be used without any additional purification.
Scheme 2.

Synthesis of salen ligands 8a and 8b.
Scheme 3.

The preparation of aluminum complexes 1, 2 and 10.
Styrene oxide was used as a model substrate to test the catalytic efficiency of complexes 1 and 2 in the coupling reaction with carbon dioxide. Table 1 summarizes the experimental results. It is apparent from the data that both catalysts are effective at promoting the coupling reaction even at room temperature and 1 bar of carbon dioxide pressure (Table 1, entries 1–4 and 10–12). However, complex 2 was a better catalyst, affording 83% conversion of styrene oxide to the corresponding cyclic carbonate after 24 hours, whereas complex 1 gave only 47% conversion under the same conditions (Table 1, entries 4 and 12). Increasing either the temperature of the reaction or the carbon dioxide pressure had positive effects on the catalytic performance (Table 1, entries 8, 9 and 13).
Table 1.
Coupling of CO2 and styrene oxide promoted by complexes 1, 2 and 10.a
| Entry | Catalyst | Catalyst loading (mol %) | Time (h) | Pressure (bar) | Temperature (°C) | Conversion (%) |
| 1 | 1 | 0.2 | 24 | 1 | 25 | 8 |
| 2 | 1 | 1 | 24 | 1 | 25 | 16 |
| 3 | 1 | 2 | 24 | 1 | 25 | 40 |
| 4 | 1 | 2.5 | 24 | 1 | 25 | 47 |
| 5b | 1 | 2.5 | 3 | 1 | 25 | 8 |
| 6b | 1 | 2.5 | 6 | 1 | 25 | 17 |
| 7b | 1 | 2.5 | 24 | 1 | 25 | 72 |
| 8 | 1 | 2.5 | 24 | 10 | 25 | 70 |
| 9 | 1 | 2.5 | 24 | 10 | 35 | 100 |
| 10 | 2 | 2.5 | 3 | 1 | 25 | 14 |
| 11 | 2 | 2.5 | 6 | 1 | 25 | 43 |
| 12 | 2 | 2.5 | 24 | 1 | 25 | 83 |
| 13 | 2 | 2.5 | 24 | 10 | 25 | 100 |
| 14 | 10 | 2.5 | 24 | 1 | 25 | 5 |
| 15c | 10 | 2.5 | 24 | 1 | 25 | 80 |
aIn neat styrene oxide. bOne equivalent of H2O and Et3N were added to the catalyst. cWith 5 mol % tetrabutylammonium iodide as cocatalyst.
Furthermore, the performance of catalyst 1 could be improved by adding one equivalent of water and triethylamine relative to the catalyst loading (Table 1, entries 5–7). Presumably, some of complex 1 was converted into a highly active oxygen-bridged aluminium complex in situ, as shown in Scheme 4. High activities for this type of dinuclear complexes have been reported before [31].
Scheme 4.

Possible formation of a dinuclear complex from 1 by treatment with H2O and Et3N.
In order to prove the bifunctional nature of our catalysts, aluminium complex 10 was prepared (Scheme 3) using 3,5-di-(tert-butyl)salicylaldehyde as a starting material. It was found that this catalyst was almost inactive in the reaction of styrene oxide with carbon dioxide (Table 1, entry 14). After addition of tetrabutylammonium iodide (5 mol %) as a cocatalyst, the conversion was increased to 80% (Table 1, entry 15), which was close to the performance of catalyst 2 (Table 1, entry 12). This supports the hypothesis that complexes 1 and 2 are bifunctional catalysts in which both the aluminium centre and the ammonium halide play important catalytic roles.
After finding the optimal reaction conditions for each catalyst, both complexes 1 and 2 were tested with a range of epoxides. These experiments were carried out without added water to allow direct comparison of the two catalysts and to avoid complicating the reaction system. The results are summarized in Table 2. Both catalysts proved to be efficient for coupling both aromatic and aliphatic substrates. In all cases reported in Table 2, cyclic carbonate, catalyst and unreacted epoxide (for entries 10 and 11) were the only species detected by 1H NMR spectroscopy of the crude reaction product prior to purification by column chromatography. The moderate yield for propylene oxide (Table 2, entry 6) can be explained by volatility of the starting material under the reaction conditions.
Table 2.
Coupling of CO2 and various epoxides promoted by complexes 1 and 2.a
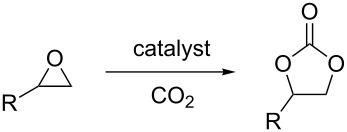 | ||||
| Entry | Catalyst | R | Conversionb (%) | Yieldc (%) |
| 1 | 1 | Ph | 100 | 62 |
| 2 | 1 | CH2OPh | 100 | 78 |
| 3 | 1 | p-ClPh | 100 | 95 |
| 4 | 1 | Bu | 100 | 80 |
| 5 | 1 | Et | 100 | 78 |
| 6 | 1 | Me | 100 | 52 |
| 7 | 1 | CH2Cl | 100 | 82 |
| 8 | 1 | CH2OH | 100 | 85 |
| 9 | 2 | Ph | 100 | 80 |
| 10 | 2 | CH2OPh | 64 | 56 |
| 11 | 2 | p-ClPh | 99 | 84 |
| 12 | 2 | Bu | 100 | 60 |
| 13 | 2 | Et | 100 | 71 |
| 14 | 2 | Me | 100 | 88 |
| 15 | 2 | CH2Cl | 100 | 80 |
| 16 | 2 | CH2OH | 100 | 76 |
aReaction conditions for catalyst 1: solvent free, 10 bar pressure of CO2, 35 °C, 24 h; for catalyst 2: solvent free, 10 bar pressure of CO2, 25 °C, 24 h. bDetermined by 1H NMR spectroscopy of the unpurified product. cAfter purification by column chromatography.
No cyclic carbonate was detected when cyclohexene oxide was used as substrate (Table 3, entries 1–5) and almost no conversion at all was detected in the reaction promoted by complex 1 (Table 3, entries 1 and 2). Catalyst 2 was more active and catalysed the synthesis of the corresponding polycarbonate with 64% conversion at 10 bar (Table 3, entry 4) and 92% at 35 bar carbon dioxide pressure (Table 3, entry 5). Previous reports have indicated that in the presence of a cocatalyst, aluminium–salen complexes can catalyse the formation of either cyclic [32] or polycarbonate [33–34] from cyclohexene oxide, depending on the exact structure of the catalyst and cocatalyst. However, this is the first report of a one-component aluminium–salen-based catalyst for polycyclohexene carbonate synthesis.
Table 3.
Addition CO2 to cyclohexene oxide.a
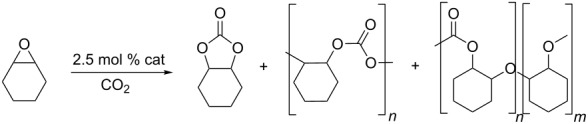 | ||||||
| Entry | Catalyst | Pressure (bar) | Time (h) | Conversion (%) | Polycarbonate (%) | Polyether linkages (%) |
| 1 | 1 | 10 | 24 | 6 | 6 | – |
| 2 | 1 | 10 | 111 | 11 | 11 | – |
| 3 | 2 | 10 | 24 | 8 | 8 | – |
| 4b | 2 | 10 | 96 | 64 | 57 | 5 |
| 5b | 2 | 35 | 96 | 92 | 85 | 7 |
aNeat cyclohexene oxide, temperature: for catalyst 1, 35 °C; for catalyst 2, 25 °C, only traces of cyclic carbonates were detected. bThe ratio of polycarbonate/polyether was determined from the 1H NMR spectrum [11].
MALDI–TOF mass-spectra data (Figure 2) showed that the polycarbonate consisted of a mixture of oligomers with a range of monomer units (n from 4 to 10) with the maximum intensity at n = 6. Both ends of the polymer chain are capped with alcohol groups, suggesting that chain-transfer to adventitious moisture occurred during the polymerisation. GPC data (Figure 3) was consistent with the MALDI–TOF data, showing that most of the polymer has a molecular weight between 300 and 1000 Daltons. This type of low molecular weight polycarbonate–polyol is currently attracting much interest associated with its use in sustainable polyurethanes [35].
Figure 2.

MALDI–TOF spectrum of poly(hexene carbonate) prepared using catalyst 2. The peak at 565 Daltons corresponds to four ring-opened cyclohexene oxide units, three CO2 units, 2 hydrogens (to cap the two terminal oxygens) and a sodium ion. The other peaks are then separated by 142 Daltons corresponding to an additional ring-opened cyclohexene oxide and carbon dioxide.
Figure 3.

GPC trace of poly(cyclohexene carbonate) prepared using catalyst 2. The chromatogram was obtained in THF and is referenced to polystyrene standards.
To show the stability of our catalytic system, catalyst 1 was reused three times. For this purpose the catalyst was precipitated from the reaction mixture by the addition of ether followed by filtration. The catalyst was then dried in vacuo and then reused. The results are summarized in Table 4. As can be seen, there were no significant losses of catalytic activity observed after three catalytic cycles.
Table 4.
The catalytic activity of recovered catalyst 1.
 | |
| Cycle | Conversion |
| 1 | 47 |
| 2 | 45 |
| 3 | 43 |
Conclusion
In conclusion, we have developed two new, bifunctional aluminium(salen) catalysts with quaternary ammonium groups directly attached to the aromatic rings. The catalytic system showed high activity in cyclic carbonate formation with a range of substrates. The bifunctional nature of our catalysts was demonstrated by comparing their performance with similar non-modified aluminium complex 10. In contrast to previously reported aluminium–salen complexes, catalysts 1 and 2 produce polycarbonate rather than cyclic carbonate from cyclohexene oxide.
Experimental
Materials
Commercial reagents were used as received unless stated otherwise. Column chromatography was performed using Silica Gel Kieselgel 60 (Merck).
Instrumentation
1H NMR and 13C NMR spectra were recorded on Bruker Avance 300 and Bruker Avance III–400 (operating at 300 and 400 MHz for protons, respectively) spectrometers. Optical rotations were measured on a Perkin–Elmer 341 polarimeter in a 5-cm cell. Melting points were determined in open capillary tubes and are uncorrected. Mass spectra were recorded at the University of York Mass Spectrometry Service Unit using ESI and MALDI ionization methods. GPC was carried out using a set (PSS SDV High) of 3 analytical columns (300 × 8 mm, particle diameter 5 µm) of 1000, 105 and 106 Å pore sizes, plus guard column, supplied by Polymer Standards Service GmbH (PSS) installed in a PSS SECcurity GPCsystem. Elution was with tetrahydrofuran at 1 mL/min with a column temperature of 23 °C and detection by refractive index. 20 µL of a 1 mg/mL sample in THF was injected for each measurement and eluted for 40 minutes. Calibration was carried out in the molecular weight range 400–2 × 106 Da using ReadyCal polystyrene standards supplied by Sigma-Aldrich.
General procedures for compounds (4–10)
3-(tert-Butyl)-2-hydroxybenzaldehyde (4)
Prepared as described in previous work [24]. 1H NMR (400 MHz, CDCl3) δ 11.77 (s, 1H), 9.85 (s, 1H), 7.52 (d, J = 7.7 Hz, 1H), 7.39 (d, J = 7.7 Hz, 1H), 7.00–6.95 (m, 1H), 1.45 (s, 9H).
3-(tert-Butyl)-2-hydroxy-5-nitrobenzaldehyde (5)
Prepared by a modified literature procedure [25]. To a stirred solution of 3-(tert-butyl)-2-hydroxybenzaldehyde (1.0 g, 4.5 mmol) in glacial acetic acid (20 mL) was added 3.3 M nitric acid (4.0 mL). The solution was heated to reflux for 30 minutes. After cooling to room temperature, the solution was poured onto ice. The resulting precipitate was filtered and washed with water, giving 3-(tert-butyl)-2-hydroxy-5-nitrobenzaldehyde (0.9 g, 71%) as a yellow powder. 1H NMR (400 MHz, CDCl3) δ 12.43 (s, 1H), 9.96 (s, 1H), 8.43–8.35 (m, 2H), 1.45 (s, 9H).
3-(tert-Butyl)-5-(dimethylamino)-2-hydroxybenzaldehyde (6)
Prepared by a modified literature procedure [28]. Pd/C (10%, 100 mg) was added to a solution of 3-(tert-butyl)-2-hydroxy-5-nitrobenzaldehyde 5 (200 mg, 0.9 mmol) and 40% aqueous formaldehyde (4.5 mL) in ethanol (20 mL). The reaction mixture was stirred under 1 bar of hydrogen at room temperature for 24 hours. Then, the Pd/C was removed by filtration on celite, the volume of the filtrate was reduced by half under reduced pressure. Distilled water was added to the solution, resulting in formation of an orange precipitate (140 mg, 70%), which was filtered and washed with water (10 mL) and cold ethanol. 1H NMR (400 MHz, CDCl3) δ 11.27 (s, 1H), 9.83 (s, 1H), 7.14 (s, 1H), 6.71 (s, 1H), 2.88 (s, 6H), 1.41 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 197.28, 154.37, 144.05, 138.97, 122.84, 120.34, 114.90, 42.07, 35.14, 29.22; mp 130–131 °C (lit. [28] 134 °C).
6,6'-((1E,1'E)-((1R,2R)-Cyclohexane-1,2-diylbis(azanylylidene))bis(methanylylidene))bis(2-(tert-butyl)-4-(dimethylamino)phenol) (7)
Prepared by a modified literature procedure [30]. A solution of (1R,2R)-diaminocyclohexane (64 mg, 0.57 mmol) in ethanol (5 mL) was added dropwise to a solution of 6 (250 mg, 1.13 mmol) in ethanol (15 mL). The resulting mixture was refluxed for 4 hours. After cooling to room temperature, distilled water (30 mL) was added. The resulting precipitate was filtered, washed with water and a small amount of cold ethanol to give a yellow powder (250 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 8.23 (s, 2H), 6.88 (d, J = 3.0 Hz, 2H), 6.41 (d, J = 3.0 Hz, 2H), 3.35–3.18 (m, 2H), 2.74 (s, 12H), 2.00–1.90 (m, 4H), 1.79–1.72 (m, 2H), 1.47–1.43 (m, 2H), 1.38 (s, 18H); mp 106–110 °C; [αD] −225 (c 0.01, MeOH); ESIMS m/z: [M + H]+ calcd for C32H48N4O2, 521.38; found, 521.39.
5,5'-((1E,1'E)-((1R,2R)-Cyclohexane-1,2-diylbis(azanylylidene))bis(methanylylidene))bis(3-(tert-butyl)-4-hydroxy-N,N,N-trimethylbenzenaminium iodide) (8a)
To a solution of 7 (100 mg, 0.192 mmol) in dry acetonitrile (5 mL) was added methyl iodide (271 mg, 1.92 mmol). The solution was stirred at room temperature for 24 hours. Diethyl ether (10 mL) was then added to the solution resulting in formation of a precipitate which was filtered and washed with ether to leave a bright yellow powder (146 mg, 95%). 1H NMR (400 MHz, DMSO-d6) δ 8.59 (s, 1H), 7.85 (d, J = 3.2 Hz, 1H), 7.54 (d, J = 3.2 Hz, 1H), 3.58 (s, 1H), 3,49 (s, 9H), 1.88 (d, J = 11.3 Hz, 1H), 1.78 (s, 1H), 1.64 (s, 1H), 1.46 (s, 1H), 1.32 (s, 9H); 13C NMR (75 MHz, CD3OD) δ 164.76, 161.55, 139.89, 137.05, 121.25, 120.27, 118.11, 71.57, 56.59, 35.20, 32.47, 28.07, 23.83; mp 206–208 °C; [αD] −175 (c 0.01, MeOH); ESIMS m/z: [M]2+ calcd for C34H54N4O22+, 275.21; found, 275.21.
5,5'-((1E,1'E)-((1R,2R)-Cyclohexane-1,2-diylbis(azanylylidene))bis(methanylylidene))bis(N-benzyl-3-(tert-butyl)-4-hydroxy-N,N-dimethyl-benzenaminium bromide) (8b)
To a solution of 7 (120 mg, 0.231 mmol) in dry acetonitrile (5 mL) was added benzyl bromide (79 mg, 0.46 mmol). The solution was stirred at room temperature for 24 hours. Diethyl ether (10 mL) was then added to the solution resulting in formation of a precipitate which was filtered and washed with ether to leave a bright yellow powder (165 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 9.00 (s, 1H), 8.80 (d, J = 3.1 Hz, 1H), 7.33–7.27 (m, 1H), 7.19–6.98 (m, 4H), 6.76 (d, J = 3.2 Hz, 1H), 5.51–5.37 (s, 2H), 3.98–3.90 (m, 1H), 3.85 (d, J = 8.0 Hz, 6H), 2.2–2.1 (m, 1H), 2.0–1.9 (m, 1H), 1.6–1.4 (m, 2H), 1.25 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 165.63, 162.37, 139.22, 134.33, 133.01, 130.73, 129.06, 128.87, 123.76, 122.83, 117.57, 72.26, 69.97, 53.21, 35.55, 32.51, 29.37, 29.04, 24.05; mp 140–144 °C; [αD] 107 (c 0.05, MeOH); ESIMS m/z: [M]2+ calcd for C46H62N4O22+, 351.24; found, 351.24.
Aluminium–salen complex (1)
To a solution of 8a (113 mg, 0.14 mmol) in dry acetonitrile (5 mL) under argon was added diethylaluminum chloride (0.14 mL, 1 M solution in hexane). The reaction mixture was heated at reflux for 3 hours. The solvent was evaporated under reduced pressure to give a dark yellow powder (95 mg, 78%) which was used without any additional purification. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (s, 1H), 8.10 (s, 1H), 7.66 (s, 1H), 3.55 (s, 9H), 3.38 (s, 1H), 2.55 (s, 1H), 1.94 (s, 1H), 1.50 (s, 9H), 1.44 (s, 1H), 1.33 (s, 1H); mp >300 °C; [αD] −109.5 (c 0.05, MeOH); ESIMS m/z: [M]2+ calcd for C34H52AlClN4O22+, 305.18; found, 296.19 (substitution of Cl by OH); 303.20 (substitution of Cl by OMe).
Aluminium–salen complex (2)
To a solution of 8b (140 mg, 0.163 mmol) in dry acetonitrile (5 mL) under argon was added diethylaluminum chloride (0.17 mL, 1 M solution in hexane). The reaction mixture was refluxed for 3 hours. Then, the solvent was evaporated under reduced pressure to give a dark yellow powder (136 mg, 91%) which was used without any additional purification. 1H NMR (400 MHz, DMSO-d6) δ 8.33 (s, 1H), 7.83 (d, J = 3.0 Hz, 1H), 7.47 (t, J = 8.6 Hz, 1H), 7.40 (s, 1H), 7.28 (t, J = 7.7 Hz, 2H), 7.02 (d, J = 7.3 Hz, 2H), 4.99 (d, J = 7.5 Hz, 2H), 3.55 (d, J = 7.3 Hz, 6H), 3.35 (s, 1H) 1.89 (s, 2H), 1.48 (d, J = 4.5 Hz, 11H); mp >300 °C; [αD] −83.4 (c 0.01, MeOH); ESIMS m/z: [M]2+ calcd for C46H60AlClN4O22+, 381.21; found, 372.22 (substitution of Cl by OH); 379.20 (substitution of Cl by OMe).
Aluminium–salen complex (10)
Prepared as described in previous work [29]. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (s, 1H), 7.41 (d, J = 2.4 Hz, 1H), 7.36 (d, J = 2.4 Hz, 1H), 2.60 (d, J = 10.7 Hz, 1H), 1.99–1.90 (s, 1H), 1.53 (s, 9H), 1.40–1.20 (m, 3H), 1.29 (s, 9H).
Synthesis of cyclic carbonates
All cyclic carbonate formations were carried out in autoclaves or, in case of 1 bar CO2 reactions, in sample vials with a balloon of CO2 attached to them. In both cases the reactions were magnetically stirred. After completion of the experiment, the reaction mixture was analysed by 1H NMR spectroscopy and passed through a pad of silica to separate the catalyst. In the case of a 100% conversion, CH2Cl2 was used as the eluent, if the conversion was incomplete then column chromatography was used to purify the compounds (SiO2, EtOAc/hexane, 1:3).
Styrene carbonate: 1H NMR (400 MHz, CDCl3) δ 7.44–7.32 (m, 5H), 5.66 (t, J = 8.0 Hz, 1H), 4.82–4.73 (m, 1H), 4.37–4.26 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 155.00, 135.88, 129.80, 129.31, 126.00, 78.11, 71.28.
4-Chlorostyrene carbonate: 1H NMR (400 MHz, CDCl3) δ 7.48–7.25 (m, 4H), 5.65 (t, J = 8.0 Hz, 1H), 4.79 (t, J = 8.2 Hz, 1H), 4.29 (dd, J = 8.6, 7.9 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 154.65, 135.85, 134.35, 129.59, 127.37, 77.34, 71.10.
3-Chloropropylene carbonate: 1H NMR (400 MHz, CDCl3) δ 5.02–4.93 (m, 1H), 4.60–4.53 (m, 1H), 4.37 (dd, J = 8.9, 5.7 Hz, 1H), 3.82–3.67 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 154.49, 74.48, 67.06, 44.03.
3-Phenoxypropylene carbonate: 1H NMR (400 MHz, CDCl3) δ 7.37–6.84 (m, 5H), 5.08–4.94 (m, 1H), 4.63–4.46 (m, 2H), 4.30–4.06 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 157.83, 154.76, 129.78, 122.08, 114.69, 74.20, 66.95, 66.32.
Propylene carbonate: 1H NMR (400 MHz, CDCl3) δ 4.92–4.67 (m, 1H), 4.64–4.38 (m, 1H), 4.07–3.89 (m, 1H), 1.47–1.36 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 155.22, 73.74, 70.78, 19.45.
1,2-Hexylene carbonate: 1H NMR (400 MHz, CDCl3) δ 4.74–4.59 (m, 1H), 4.53–4.43 (m, 1H), 4.02 (dt, J = 9.9, 4.9 Hz, 1H), 1.82–1.57 (m, 2H), 1.46–1.20 (m, 4H), 0.92–0.81 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 155.24, 77.20, 69.51, 33.59, 26.49, 22.31, 13.86.
3-Hydroxypropylene carbonate: 1H NMR (400 MHz, CDCl3) δ 4.90–4.73 (m, 1H), 4.59–4.39 (m, 2H), 3.96 (dt, J = 20.4, 10.2 Hz, 1H), 3.68 (td, J =13.1, 5.5 Hz, 1H), 2.84–2.56 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 155.38, 76.64, 65.85, 61.73.
1,2-Butylene carbonate: 1H NMR (400 MHz, CDCl3) δ 4.71–4.55 (m, 1H), 4.48 (t, J = 8.1 Hz, 1H), 4.04 (dd, J = 8.4, 7.0 Hz, 1H), 1.85–1.63 (m, 2H), 1.03–0.89 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 155.27, 78.16, 69.13, 26.95, 8.52.
Synthesis of polycyclohexene carbonate
Prepared as reported above for the synthesis of cyclic carbonates at 10–35 bar CO2, but without any additional purification of the reaction product. 1H NMR (400 MHz, CDCl3) δ 4.71–4.56 (broad, 2H), 2.21–2.04 (broad, 4H), 1.79–1.62 (broad, 4H).
Acknowledgments
The authors thank the Royal Society for an Anglo–Russian collaboration grant under the international exchange scheme.
This article is part of the Thematic Series "Sustainable catalysis".
Contributor Information
Michael North, Email: Michael.North@york.ac.uk.
Yuri N Belokon, Email: yubel@ineos.ac.ru.
References
- 1.Aresta M, Dibenedetto A, Angelini A. Chem Rev. 2014;114:1709–1742. doi: 10.1021/cr4002758. [DOI] [PubMed] [Google Scholar]
- 2.Schäffner B, Schäffner F, Verevkin S P, Börner A. Chem Rev. 2010;110:4554–4581. doi: 10.1021/cr900393d. [DOI] [PubMed] [Google Scholar]
- 3.Sakakura T, Kohno K. Chem Commun. 2009:1312–1330. doi: 10.1039/b819997c. [DOI] [PubMed] [Google Scholar]
- 4.Takata T, Furusho Y, Murakawa K-i, Endo T, Matsuoka H, Hirasa T, Matsuo J, Sisido M. J Am Chem Soc. 1998;120:4530–4531. doi: 10.1021/ja974337l. [DOI] [Google Scholar]
- 5.Nicolaou K C, Yang Z, Liu J J, Ueno H, Nantermet P G, Guy R K, Claiborne C F, Renaud J, Couladouros E A, Paulvannan K, et al. Nature. 1994;367:630–634. doi: 10.1038/367630a0. [DOI] [PubMed] [Google Scholar]
- 6.Chang H-T, Sharpless K B. Tetrahedron Lett. 1996;37:3219–3222. doi: 10.1016/0040-4039(96)00534-5. [DOI] [Google Scholar]
- 7.He Q, O'Brien J W, Kitselman K A, Tompkins L E, Curtis G C T, Kerton F M. Catal Sci Technol. 2014;4:1513–1528. doi: 10.1039/c3cy00998j. [DOI] [Google Scholar]
- 8.Martín C, Fiorani G, Kleij A W. ACS Catal. 2015;5:1353–1370. doi: 10.1021/cs5018997. [DOI] [Google Scholar]
- 9.Comerford J W, Ingram I D V, North M, Wu X. Green Chem. 2015;17:1966–1987. doi: 10.1039/C4GC01719F. [DOI] [Google Scholar]
- 10.Ikpo N, Flogeras J C, Kerton F M. Dalton Trans. 2013;42:8998–9006. doi: 10.1039/c3dt00049d. [DOI] [PubMed] [Google Scholar]
- 11.Taherimehr M, Pescarmona P P. J Appl Polym Sci. 2014;131:41141. doi: 10.1002/app.41141. [DOI] [Google Scholar]
- 12.Decortes A, Castilla A M, Kleij A W. Angew Chem, Int Ed. 2010;49:9822–9837. doi: 10.1002/anie.201002087. [DOI] [PubMed] [Google Scholar]
- 13.North M, Pasquale R, Young C. Green Chem. 2010;12:1514–1539. doi: 10.1039/c0gc00065e. [DOI] [Google Scholar]
- 14.Tian D, Liu B, Gan Q, Li H, Darensbourg D J. ACS Catal. 2012;2:2029–2035. doi: 10.1021/cs300462r. [DOI] [Google Scholar]
- 15.Meléndez J, North M, Villuendas P. Chem Commun. 2009:2577–2579. doi: 10.1039/b900180h. [DOI] [PubMed] [Google Scholar]
- 16.Ren W-M, Liu Y, Lu X-B. J Org Chem. 2014;79:9771–9777. doi: 10.1021/jo501926p. [DOI] [PubMed] [Google Scholar]
- 17.Hanhart W, Ingold C K. J Chem Soc. 1927:997–1020. doi: 10.1039/jr9270000997. [DOI] [Google Scholar]
- 18.Hughes E D, Ingold C K, Patel C S. J Chem Soc. 1933:526–530. doi: 10.1039/jr9330000526. [DOI] [Google Scholar]
- 19.de la Zerda J, Neumann R, Sasson Y. J Chem Soc, Perkin Trans 2. 1986:823–826. doi: 10.1039/p29860000823. [DOI] [Google Scholar]
- 20.Hofman A W. Proc R Soc London. 1859;10:594–596. doi: 10.1098/rspl.1859.0121. [DOI] [Google Scholar]
- 21.Collie N, Schryver S B. J Chem Soc, Trans. 1890;57:767–782. doi: 10.1039/ct8905700767. [DOI] [Google Scholar]
- 22.Zaki A, Fahim H. J Chem Soc. 1942:270–272. doi: 10.1039/jr9420000270. [DOI] [Google Scholar]
- 23.Gordon J E. J Org Chem. 1965;30:2760–2763. doi: 10.1021/jo01019a060. [DOI] [Google Scholar]
- 24.Gisch N, Balzarini J, Meier C. J Med Chem. 2007;50:1658–1667. doi: 10.1021/jm0613267. [DOI] [PubMed] [Google Scholar]
- 25.Braun M, Fleischer R, Mai B, Schneider M-A, Lachenicht S. Adv Synth Catal. 2004;346:474–482. doi: 10.1002/adsc.200303178. [DOI] [Google Scholar]
- 26.Chen C-T, Kao J-Q, Salunke S B, Lin Y-H. Org Lett. 2011;13:26–29. doi: 10.1021/ol1024053. [DOI] [PubMed] [Google Scholar]
- 27.Sun M, Mu Y, Wu Q, Gao W, Ye L. New J Chem. 2010;34:2979–2987. doi: 10.1039/c0nj00439a. [DOI] [Google Scholar]
- 28.Waibel M, Hasserodt J. Tetrahedron Lett. 2009;50:2767–2769. doi: 10.1016/j.tetlet.2009.03.139. [DOI] [Google Scholar]
- 29.Sigman M S, Jacobsen E N. J Am Chem Soc. 1998;120:5315–5316. doi: 10.1021/ja980299+. [DOI] [Google Scholar]
- 30.Chiang L, Kochem A, Jarjayes O, Dunn T J, Vezin H, Sakaguchi M, Ogura T, Orio M, Shimazaki Y, Thomas F, et al. Chem – Eur J. 2012;18:14117–14127. doi: 10.1002/chem.201201410. [DOI] [PubMed] [Google Scholar]
- 31.North M. ARKIVOC. 2012;(i):610–628. [Google Scholar]
- 32.Beattie C, North M, Villuendas P, Young C. J Org Chem. 2013;78:419–426. doi: 10.1021/jo302317w. [DOI] [PubMed] [Google Scholar]
- 33.Darensbourg D J, Billodeaux D R. Inorg Chem. 2005;44:1433–1442. doi: 10.1021/ic048508g. [DOI] [PubMed] [Google Scholar]
- 34.Sugimoto H, H. Ohtsuka H, Inoue S. J Polym Sci, Part A: Polym Chem. 2005;43:4172–4186. doi: 10.1002/pola.20894. [DOI] [Google Scholar]
- 35.Chapman A M, Keyworth C, Kember M R, Lennox A J J, Williams C K. ACS Catal. 2015;5:1581–1588. doi: 10.1021/cs501798s. [DOI] [Google Scholar]