

Supplemental Digital Content is available in the text.
Key Words: PRAME tumor antigen, AS15 immunostimulant, animal models, cancer immunotherapy, tumor protection
Abstract
The PRAME tumor antigen is a potential target for immunotherapy. We assessed the immunogenicity, the antitumor activity, and the safety and the tolerability of a recombinant PRAME protein (recPRAME) combined with the AS15 immunostimulant (recPRAME+AS15) in preclinical studies in mice and Cynomolgus monkeys. Four groups of 12 CB6F1 mice received 4 injections of phosphate-buffered saline (PBS), recPRAME, AS15, or recPRAME+AS15. Immunized mice were injected with tumor cells expressing PRAME (CT26-PRAME) 2 weeks or 2 months after the last injection. The mean tumor surface was measured twice a week. Two groups of 10 monkeys received 7 injections of saline or recPRAME+AS15. T-cell responses were measured by flow cytometry using intracellular cytokine staining (ICS). In CB6F1 mice, repeated injections of recPRAME+AS15 induced high PRAME-specific antibody titers and mostly CD4+ T cells producing cytokines. This immune response was long-lasting in these animals and was associated with protection against a challenge with PRAME-expressing tumor cells (CT26-PRAME) applied either 2 weeks or 2 months after the last injection; these data indicate the induction of an immune memory. In HLA-A02.01/HLA-DR1 transgenic mice, recPRAME+AS15 induced both CD4+ and CD8+ T-cell responses, indicating that this antigen can be processed by the human leukocyte antigen and is potentially immunogenic in humans. In addition, a repeated-dose toxicity study in monkeys showed that 7 biweekly injections of recPRAME+AS15 were well tolerated, and induced PRAME-specific antibodies and T cells. In conclusion, these preclinical data indicate that repeated injections of the PRAME cancer immunotherapeutic are immunogenic and have an acceptable safety profile.
Although conventional approaches in the therapy of cancer may improve the survival of patients, they are often associated with severe side effects and rarely lead to tumor remission. Moreover, a substantial proportion of cancer patients do not respond to standard anticancer therapies or relapse after treatment.1–3 In addition, certain tumor types such as melanoma, renal cell carcinoma, hepatocellular carcinoma, and gastrointestinal stromal tumors are poorly responsive to standard anticancer therapies.4 Thus, there is a need for alternative, well tolerated and more specific anticancer therapies.
Recent advances in molecular biology and immunology, a greater understanding of the cellular events and pathways driving carcinogenesis, and the discovery of tumor antigens capable of eliciting tumor-specific immune responses have led to the development of various cancer immunotherapeutic approaches including therapeutic anticancer vaccination.5–7
In contrast to chemotherapy, cancer immunotherapy acts on the patient’s immune system, modulating it to recognize and eradicate the tumor.3,8 Antigen-specific cancer immunotherapy aims to harness the natural ability of the immune system to activate tumor-specific T cells against tumor antigens.9–11
Antigen-specific cancer immunotherapies have several potential advantages over conventional chemotherapy, as they target tumor cells specifically, decreasing the risk of damage to normal, healthy tissues, and potentially induce specific, durable, and recall antitumor responses. In addition, the highly specific characteristic of the host immune response minimizes the risk of clinically significant adverse events associated with most chemotherapies used today.3,12–15 However, a limitation of such an approach is the weakness of the immune responses raised by tumor antigens, especially when they are delivered as purified recombinant proteins. To increase the magnitude of the response elicited by protein-based immunotherapies, tumor antigens can be combined with immunostimulants.16–19
The human tumor antigen PRAME (PReferentially expressed Antigen of MElanoma) was originally identified as an antigen recognized by cytotoxic T lymphocytes from a patient with melanoma.20,21 The PRAME antigen is a potential target for immunotherapy as it is expressed in various tumor types including melanoma, non–small cell lung cancer (NSCLC), head and neck carcinomas, breast cancer, and some types of leukemia.20,22–27 It is also expressed in the normal adult testis, and at very low levels in some other normal tissues, such as the ovaries, the kidneys, the endometrium, and the adrenal medulla.20,28 Furthermore, recent studies provided important insights in molecular pathways in which PRAME is implicated, including transcriptional regulation and oncogenesis.29,30 PRAME-specific CD8+ T cells, derived from healthy donors or cancer patients and stimulated in vitro with PRAME peptides, lysed melanoma, renal cell carcinoma, lung carcinoma, and breast and cervical cancer cell lines, suggesting that PRAME is immunogenic.20,28,31 The induction of PRAME-specific T-cell immune responses has been reported ex vivo in studies of melanoma and leukemia in humans.32–34 In addition, PRAME-specific T cells were highly reactive against different PRAME tumor cell lines or freshly isolated metastatic melanoma and primary leukemic cells, but not against nonmalignant cells.35 Thus, PRAME-targeted immunotherapy, able to induce strong T-cell responses against tumors, could potentially provide significant benefit to a large number of cancer patients.
Our immunotherapeutic approach is based on the use of a PRAME recombinant protein (recPRAME) combined with the GSK (Rixensart, Belgium) proprietary immunostimulant AS15, to form the PRAME cancer immunotherapeutic (recPRAME+AS15). The PRAME cancer immunotherapeutic is currently in phase I or phase I/II clinical development for the treatment of NSCLC (NCT01159964) and melanoma (NCT01149343), respectively. Given that PRAME expression is not purely restricted to tumor cells, the safety of the potential PRAME-targeted immunotherapy needs to be assessed carefully. Therefore, to support the clinical development of the PRAME cancer immunotherapeutic, we performed nonclinical studies evaluating the immunogenicity and the antitumor activity of recPRAME+AS15 against a PRAME-expressing mouse tumor model. In addition, the safety and the tolerability of recPRAME+AS15 were assessed in Cynomolgus monkeys.
MATERIALS AND METHODS
Experimental Animal Models, Housing, and Husbandry
Mice studies were ethically reviewed and approved by the GSK Belgian ethical Committee for Animal Experimentation. They were carried out in accordance with the European Directive 2010/63/EU36 and the GSK Policy on the Care, Welfare, and Treatment of Animals. GSK facilities are AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) accredited. All efforts were made to minimize suffering: tumors exceeding a maximum allowable size of 17 mm×17 mm, ulceration, tumor necrosis, convulsion, morbidity, and circling behavior were conditions requiring euthanasia by intraperitoneal injection with a barbituric acid derivative (overdose).
The study in monkeys was conducted in an AAALAC-accredited European Contract Research Organization in compliance with the principles of Good Laboratory Practice (GLP), in particular the OECD Principles of Good Laboratory Practice,37 the Directive 2004/10/EC,38 and “Arreté du 14 Mars 2000.”39 Compliance with animal health regulations, in particular the Council directive 86/609/EEC40 and the European Directive 2010/63/EU,36 was also ensured. Serology experiments and the evaluation of T-cell responses in the monkey study were carried out as a post hoc analysis in the GSK facilities.
Female CB6F1 mice (hybrid of C57BL/6 and Balb/C mice), 6–8 weeks old at the study start, with a 20 g approximate weight were kept under specific pathogen-free conditions.
Human leukocyte antigen (HLA)-A02.01/HLA-DR1 transgenic mice for human class I and II HLA were generated by the Pasteur Institute (Paris, France) and were 5 months old at the study start.
The Cynomolgus monkey (Macaca fascicularis) was chosen because of a similar expression pattern of a highly homologous (>94%) PRAME gene (Available at: http://www.ensembl.org/index.html). Moreover, toxicity studies with other adjuvanted vaccines were also conducted in this species, allowing for data comparability. At the study start, monkeys were 25–33 months old, with a mean body weight of 3.3 kg (range: 2.5–4.2 kg) for males and 2.8 kg (range: 2.5–3.0 kg) for females. Each animal was given a complete clinical examination. Monkeys were acclimated to the study conditions for a period of 20 days before the beginning of the treatment period. Housing and dietary conditions were described previously.41
Tumor Models
CT26 is a tumorigenic mouse cell line that expresses major histocompatibility complex class I molecules (MHC I), as determined by fluorescence-activated cell sorting (FACS).42
The CT26-PRAME cell line was generated by stable transfection of the CT26 colon carcinoma parental cell line (ATCC-CRL-2638) with the mammalian expression plasmid, pCDNA3, encoding cDNA for PRAME (Invitrogen, Carlsbad, CA). Individual cell clones expressing PRAME (CT26-PRAME) were selected with geneticin G418 (200 μg/mL). PRAME expression was determined by quantitative real-time polymerase chain reaction and was shown to correspond to 10e3 PRAME mRNA copies/copy of mouse β-actin, which is in the range of the level of PRAME expression in human tumors. CT26-PRAME cells grown in vitro at 37°C with 5% CO2 in Roswell Park Memorial Institute (RPMI) medium with 10% fetal calf serum (FCS), 1% l-glutamine, 1% penicillin-streptomycin, 1% nonessential amino acids (aa), 1% sodium pyruvate, and 0.1% β-mercaptoethanol were trypsinized, washed twice in serum-free medium, and injected in 200 μL RPMI medium subcutaneously in the right flank of CB6F1 mice 14 or 56 days after the last immunization. The product of the 2 main diameters of each tumor was recorded twice a week over 4 weeks.
Antigen Description, Production, and Purification
The recPRAME antigen is a 626-aa recombinant fusion protein produced in Escherichia coli comprising an N-terminal tripeptide (aa 1–3), containing the translator initiator methionine and 2 unrelated aa (aspartic acid and proline), aa residues 20–127 of Haemophilus influenzae protein D (PD) (aa 4–111), the full-length 509-aa-long PRAME sequence (aa 112–620), and a hexahistidine tag (His) (aa 621–626) enabling protein purification.
The manufacturing process of recPRAME purified bulk consisted of the following key steps: (i) antibiotic-free fermentation of the recombinant E. coli cell culture; (ii) disruption of E. coli cells and extraction of inclusion bodies (IB) containing the recPRAME protein; and (iii) purification of recPRAME from the IB pellet. The purification process involved the following steps: (i) extraction of the PD1/3-PRAME-His protein from the IB pellet using the centrifuge system; (ii) carbamidomethylation treatment with iodoacetamide to avoid the formation of disulfide bridges between the cysteine residues; (iii) an immobilized metal ion (Ni2+) affinity chromatography (Ni2+-IMAC) procedure (the interaction between the antigen’s His-tag and immobilized Ni2+ from the resin is responsible for the reversible capture of the antigen on the resin); (iv) hydroxyapatite chromatography of the IMAC eluate to remove E. coli-derived impurities including DNA and endotoxins; (v) ultrafiltration for buffer exchange; and, lastly, (vi) filtration through a 0.45-mm or a 0.22-mm cellulose acetate membrane.
AS15 is an immunostimulant containing 3-O-desacyl-4′-monophosphoryl lipid A produced by GSK, QS-21 Stimulon (Quillaja saponaria Molina, fraction 21; licensed by GSK from Antigenics Inc., a wholly owned subsidiary of Agenus Inc., a Delaware, USA corporation) and a synthetic oligodeoxynucleotide containing unmethylated CG dinucleotides (CpG 7909) in a liposomal formulation. CpG 7909 is a clinical-grade material of CpG 200643 that has been shown to work in both humans and mice and is efficacious in tumor models.44,45
Each dose of recPRAME+AS15 contained either 0.4 or 50 μg of recPRAME for the injection in CB6F1 mice or HLA-A02.01/HLA-DR1 transgenic mice, respectively, and a fixed dose of AS15 (50 μL, 1/10 of a human dose).
Each dose of recPRAME+AS15 for injection in monkeys contained 500 μg of recPRAME and a fixed dose of the AS15 immunostimulant. Each dose corresponded to 1 full human dose and represented an approximately 15–20-fold overexposure of the animals based on a 60–70 kg human and a 3–4 kg monkey.
Control items used were PBS for mice and saline (0.9% NaCl) for monkeys.
Study Objectives
The objectives of the studies in mice included the characterization of immune responses and antitumor effects induced by repeated injections of recPRAME alone or recPRAME+AS15 in CB6F1 mice, and the evaluation of immune responses induced by recPRAME+AS15 in HLA-A02.01/HLA-DR1 transgenic mice.
The objectives of the study in monkeys included the evaluation of the immune responses in the context of a repeated-dose toxicity study (CIT35519), using a human dose of recPRAME+AS15.46
The Study Design, Treatment, and Administration
Studies in mice were conducted at GSK (Rixensart, Belgium) between 2008 and 2011. CB6F1 mice were allocated randomly into 4 groups of 12 mice to receive 4 intramuscular (IM) injections of PBS, recPRAME alone, AS15 alone, or recPRAME+AS15 on days 0, 14, 28, and 42. Immune responses were assessed 2 months after the last injection. Eight immunized mice in each group were injected with 10e5 of CT26-PRAME cells 2 weeks (day 56) or 2 months (day 98) after the last injection. Three extra groups of 12 mice were included and were immunized 4 times 2 weeks apart with recPRAME alone, AS15 alone, and recPRAME+AS15, followed by immune response assessment 2 weeks after the last injection.
Another 4 groups of 10 CB6F1 mice were enrolled for tumor specificity experiments. Two groups of mice were immunized with PBS, and 2 with recPRAME+AS15, on days 0 and 14, and then were challenged with either 10e5 of CT26-PRAME cells or 10e5 of CT26-MAGE-A3 cells 2 weeks after the last immunization.
HLA-A02.01/HLA-DR1 transgenic mice were allocated to 2 groups of 5 mice to receive 4 injections of PBS or recPRAME+AS15 on days 0, 14, 28, and 42, and their immune responses were assessed 2 weeks after the last injection.
The repeated-dose toxicity study in monkeys was conducted at the CiToxLAB (Evreux, France) between 2008 and 2009. The study design and the injection schedule were described previously.46 In brief, 20 monkeys (10 male and 10 female) were allocated into 2 groups (by sex) using a manual randomization procedure to receive 7 IM injections (500 μL/injection) of saline (control group) or the full human dose of recPRAME+AS15 (treatment group) on days 1, 15, 29, 43, 57, 71, and 85. At the end of the treatment period (3 d after the last injection), the first 3 animals from each group were killed, whereas the last 2 surviving animals from each group were held for a 28-day treatment-free period before being killed.
In all studies, the IM route was selected to mimic the intended route of administration in human therapeutic use.
The Assessment of Immune Responses
Blood samples for the assessment of the immune responses were taken from CB6F1 mice on days 56 and 98 and from HLA-A02.01/HLA-DR1 mice on day 56. Blood samples from monkeys were taken at a pretreatment time point, and on days 57, 88, and 113.
Antibody Responses
In CB6F1 mice, PRAME-specific antibody levels were measured by an enzyme-linked immunosorbent assay 2 weeks (day 56) or 2 months (day 98) after the fourth injection. Before the addition of sera, the immunoplate was coated with the PRAME antigen overnight at 4°C. After reaction with the sera for 90 minutes at 37°C, a biotinylated sheep whole antibody against mouse immunoglobulins was added for 90 minutes at 37°C. The antigen-antibody complex was revealed by incubation with a streptavidin-biotinylated peroxidase complex for 30 minutes at 37°C. This complex was then revealed by the addition of tetramethyl benzidine for 10 minutes at room temperature; the reaction was stopped with 0.2 M H2SO4. Optical densities were recorded at 450 nm.
In monkeys, full details for the determination of the antibody response are presented elsewhere.46
Geometric mean titers were calculated with 95% confidence intervals.
T-cell Responses
In mice, the frequency of PRAME-specific T cells producing cytokines (at least IFN-γ: IFN-γ single-positive cells or IFN-γ/TNF-α double-positive cells) were detected using ICS and flow cytometry [LSR-II flow cytometer, Becton Dickinson (BD) Biosciences] on individual spleen cells (CB6F1 mice; n=4) or on peripheral blood lymphocytes (1 pool/group of HLA-A02.01/HLA-DR1 transgenic mice). Cells (106 in each well) were restimulated in vitro for 2 hours at 37°C with either medium (background) or a pool of 123 peptides [15-mer peptides with 11 aa overlap (1 μg/mL)] covering the entire sequence of the PRAME protein in the presence of anti-CD49d and anti-CD28 antibodies (BD Biosciences). Cells were then incubated overnight with Brefeldin A (BD Biosciences).
For cell staining, cell suspensions were washed, and resuspended in 50 μL PBS/1% FCS containing 2% Fc blocking reagent (1/50; 2.4G2). After 10 minutes’ incubation at 4°C, 50 μL of a mixture of anti-CD4-phycoerythrin (1:200 final dilution; BD biosciences) and anti-CD8-peridine chlorophyll-protein complex (1:200 final dilution; BD Biosciences) were added and incubated for 30 minutes at 4°C. After washing in PBS/1% FCS, cells were permeabilized in 200 μL Cytofix-Cytoperm (BD Biosciences) and incubated for 20 minutes at 4°C. Cells were then washed with 1× Perm/Wash solution (BD Biosciences) and resuspended in 50 μL fluorescent rat anti-mouse monoclonal antibodies against IFN-γ (IFNγ-APC) and TNF-fluorescein isothiocyanate (1:50; BD Biosciences) in the Perm/Wash solution. After 2 hours’ incubation at 4°C, cells were washed with Perm/Wash solution, and then with PBS, and resuspended in FACS buffer before FACS analysis. Live cells were gated (FSC/side-scattered light) and acquisition was performed on ∼15,000 CD4+ T cells and ∼10,000 CD8+ T cells for CB6F1 mice. For HLA-A02.01/HLA-DR1 transgenic mice, ∼35,000 CD4+ T cells and ∼5000 CD8+ T cells were acquired. The analyses were performed using the Cellquest or the Diva software (BD Biosciences). The percentage of cells producing at least IFN-γ (single IFN-γ or double IFN-γ/TNF-α-positive cells) in response to antigen stimulation were calculated for the CD4+-gated and CD8+-gated T-cell population by subtracting the response obtained with medium stimulation from the response upon in vitro stimulation with the PRAME peptide pool. The geometric means of CD4+ or CD8+ T cells producing at least IFN-γ are shown.
In monkeys, to focus on the activated CD4+ T cells, the frequency of CD4+CD69+ T cells producing cytokines (at least 1 among IFN-γ, TNF-α, and IL-2) was measured by flow cytometry (LSR-II, BD Biosciences) using ICS in peripheral blood lymphocytes before immunization, and after 7 injections (day 113).
The Assessment of Antitumor Responses
After the immunization of CB6F1 mice and challenge with CT26-PRAME tumor cells, individual tumor growth was recorded twice a week by measuring the product of the 2 main diameters of the tumor during the monitoring phase, starting 7 days after the day of challenge. If a mouse was killed during the study because the tumor size reached the maximum acceptable limit of 289 mm2, the value of the last measurement obtained before sacrifice was carried forward to the next time points. The specificity of tumor protection induced by recPRAME+AS15 was assessed twice a week by measuring the tumor surface in CB6F1 mice immunized on days 0 and 14, and challenged with either CT26-PRAME or CT26 cells genetically engineered to express another tumor antigen, MAGE-A3 (CT26-MAGE-A3) on day 28. Although the tumor protection effects are generally more pronounced after 4 compared with 2 immunizations, as certain protection can already be observed after 2 immunizations, for the specificity of tumor response experiments, mice received only 2 immunizations to gain some time and observe the results faster.
Assessment of the Safety and the Tolerability (Study in Monkeys)
During the study period, each monkey was checked for mortality or signs of morbidity, clinical signs, skin or ophthalmologic reactions, body weight, and food consumption. Rectal temperatures of each animal were recorded and electrocardiography examinations were performed on all animals. Blood samples for the assessment of hematology/biochemistry parameters were taken before the beginning of the treatment period, and on days 2, 4, 15 (before dosing), 84, 86, 88, and 113. A complete microscopic and macroscopic postmortem examination was performed on all monkeys. All assessments were performed as described previously.41
Statistical Analysis
Studies on Mice
A comparison of the percentages of tumor-free mice was performed using the Fisher exact test.47 Pairwise comparisons were adjusted for multiplicity using the Bonferroni method.48 The tumor size measured at the last time point was the main variable and was analyzed using an analysis of variance model with groups as factors. Each group was compared with the recPRAME+AS15 group, and Dunnett’s adjustment for multiple comparisons was performed.48 Serum titers were compared after log-transformation using the same approach, and geometric mean ratios between each group and recPRAME+AS15 were derived.
Nonparametric comparisons between the percentages of CD4+ T cells were performed after a rank transformation. Dunnett’s adjustment was also applied.
For the study on HLA-A02.01/HLA-DR1 transgenic mice, the analysis of the tumor size measured at the last time point was performed using an analysis of variance model; comparisons of the recPRAME+AS15 with the other groups were adjusted using the Dunnett method.
Study on Monkeys
The percentages of CD4+/CD69+ T cells producing cytokines were compared between the monkey groups in an analysis of covariance model with group as the factor; the percentage of CD4+/CD69+ T cells producing cytokines without stimulation was used as a covariate in the model.
RESULTS
Studies on Mice
Immune Responses
Two weeks after the fourth injection, recPRAME-specific antibody levels were significantly higher in CB6F1 mice immunized with recPRAME+AS15, compared with mice injected with PBS, recPRAME alone, or the AS15 immunostimulant alone; 2 months after the fourth injection, these levels remained significantly higher in mice from the recPRAME+AS15 group compared with the levels in the recPRAME and the AS15 groups (Fig. 1). Similarly, 2 weeks after the fourth injection, the percentage of T cells (mostly CD4+ T cells) producing at least IFN-γ (IFN-γ single-positive cells and IFN-γ/TNF-α double-positive cells) was also significantly higher in CB6F1 mice immunized with recPRAME+AS15 compared with the other groups, and remained significantly higher 2 months after injection (Fig. 2). Of note, this response was slightly lower than the one measured 2 weeks after the last immunization, suggesting that the cellular response decreased with time.
FIGURE 1.
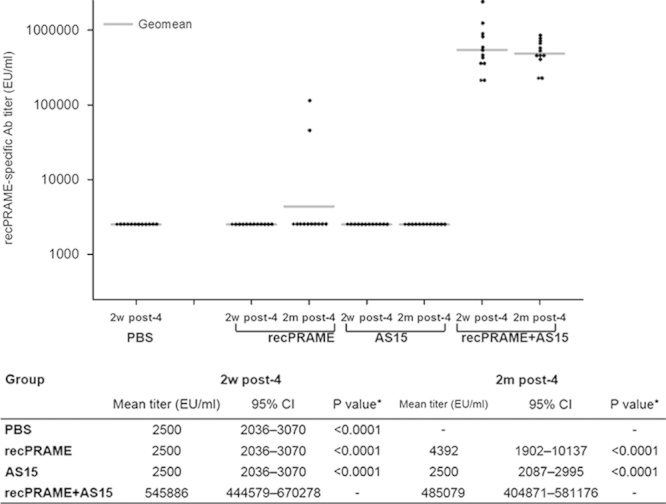
recPRAME-specific antibody responses in CB6F1 mice injected with PBS, recPRAME protein alone, AS15 immunostimulant alone, or recPRAME+AS15 at 2 weeks and 2 months after the fourth injection. Ab indicates antibody; AS15, mice injected with the AS15 immunostimulant alone; EU, enzyme-linked immunosorbent assay units; PBS, mice injected with phosphate-buffered saline; recPRAME, mice injected with the recombinant PRAME protein alone; recPRAME+AS15, mice injected with recPRAME formulated with the AS15 immunostimulant; 95% CI, 95% confidence interval (lower limit–upper limit). The dots represent individual antibody titers. *Comparison with recPRAME+AS15 group.
FIGURE 2.
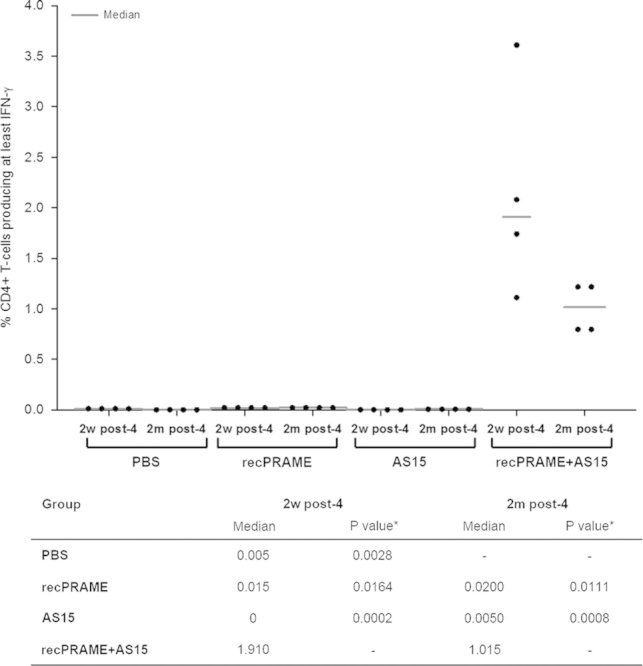
The percentage of CD4+ T cells producing cytokines in CB6F1 mice injected with PBS, recPRAME protein alone, AS15 immunostimulant alone, or recPRAME+AS15 at 2 weeks or 2 months after the fourth injection. AS15 indicates mice injected with the AS15 immunostimulant alone; PBS, mice injected with phosphate-buffered saline; recPRAME, mice injected with the recombinant PRAME protein alone; recPRAME+AS15, mice injected with recPRAME formulated with the AS15 immunostimulant. The dots represent individual percentages of T cells producing cytokines. *Comparison with recPRAME+AS15 group.
Because the PRAME-specific CD8+ T-cell response could not be measured in the mouse strain used, the immunogenicity of recPRAME+AS15 was also evaluated in HLA-A02.01/HLA-DR1 transgenic mice. These mice have the advantage to present antigens in the context of the most common human HLA classes I and II. In these mice, in contrast to CB6F1 mice, both CD4+ and CD8+ T-cell responses could be measured 2 weeks after the fourth injection. The percentages of CD4+ and CD8+ T cells producing cytokines were substantially higher in the recPRAME+AS15 group, as compared with the PBS group (Fig. 3).
FIGURE 3.
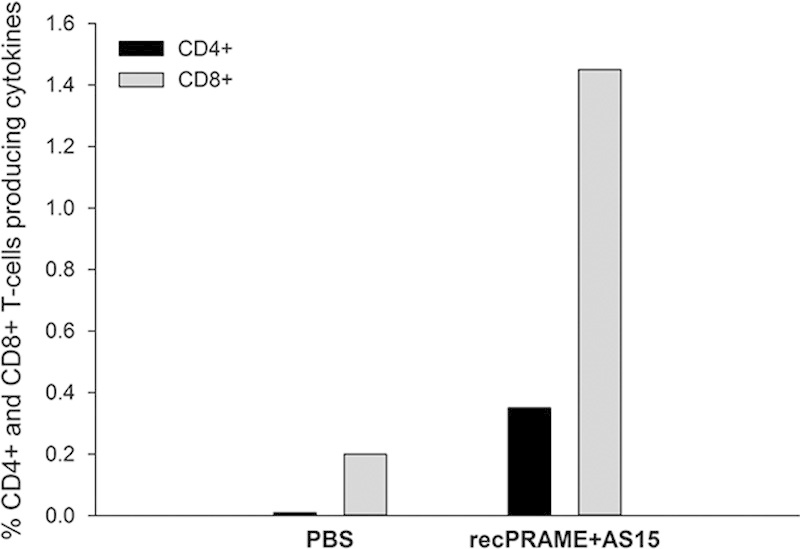
The percentage of CD4+ and CD8+ T cells producing cytokines in HLA-A02.01/HLA-DR1 transgenic mice injected with phosphate-buffered saline (PBS) or recPRAME+AS15 at 2 weeks after the fourth injection. Data are expressed as the pool of 5 mice per group. Percentages of CD4+ and CD8+ in the PBS group: 0.01 and 0.20, respectively; CD4+ and CD8+ in the recPRAME+AS15 group: 0.35 and 1.45, respectively. PBS indicates mice injected with PBS; recPRAME+AS15, mice injected with recPRAME formulated with the AS15 immunostimulant. Each bar represents a pool of 5 mice.
In addition, CD8+ T-cell responses were also evaluated in an experiment with outbred mice (CD1). Two weeks after the 2 injections of recPRAME+AS15, CD8+ T cells producing at least IFN-γ were induced in 50% of the mice (Supplementary Fig. S1, Supplemental Digital Content 1, http://links.lww.com/JIT/A395).
Antitumor Responses and Long-term Protection
To assess the capacity of recPRAME+AS15 to induce an immune memory, tumor-free mice immunized with recPRAME+AS15 were challenged with PRAME-expressing tumor cells (CT26-PRAME cells), either 2 weeks (Fig. 4A) or 2 months (Fig. 4B) after the last injection. At both time points of assessment, most mice were protected and remained tumor free (Figs. 4A, B). In contrast, mice immunized with PBS, recPRAME alone, or AS15 alone, and subjected to a similar tumor challenge, had significantly higher mean tumor sizes compared with mice injected with recPRAME+AS15 at 2 weeks after the challenge, with no tumor-free mice detected in these groups (Fig. 4A). At 2 months after the tumor challenge with CT26-PRAME cells, all mice that received recPRAME+AS15 were tumor free, whereas only 3/8 mice were tumor free after the repeated injections of recPRAME alone, and none were tumor free after injections of PBS or AS15 (Fig. 4B). Mean tumor sizes in mice injected with PBS or AS15 were higher compared with mice injected with recPRAME alone (Fig. 4B). These data suggest that long-term antitumor immunity induced by recPRAME+AS15 injections was still able to protect the mice against the tumor challenge at 2 months after the last immunization.
FIGURE 4.
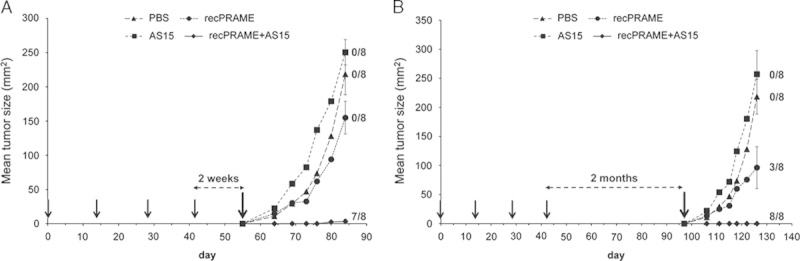
Tumor protection expressed as the tumor size in CB6F1 mice after 4 injections of phosphate-buffered saline (PBS), recPRAME protein alone, AS15 immunostimulant alone, or recPRAME+AS15, and challenged with CT26-PRAME tumor cells 2 weeks (A) or 2 months (B) after the last injection. Mean tumor sizes at the last time point in each group: (A) recPRAME+AS15, 3.3 mm2; PBS, 218.4 mm2; recPRAME, 154.9 mm2; AS15, 250.5 mm2; (B) recPRAME+AS15, 0.0 mm2; PBS, 218.4 mm2; recPRAME, 96.3 mm2; AS15, 257.1 mm2. Statistically significant differences in mean tumor sizes were observed between groups: (A) recPRAME+AS15 versus AS15, P<0.0001; recPRAME+AS15 versus PBS, P=0.0005; recPRAME+AS15 versus recPRAME, P=0.0010; (B) recPRAME+AS15 versus AS15, P=0.0004; recPRAME+AS15 versus PBS, P<0.0001; recPRAME+AS15 versus recPRAME, P=0.1053. Black arrows represent the injection time points; the last black arrows (in A and B) indicate tumor challenge time points. Data are expressed as the mean tumor size of 8 mice per group. Error bars represent SEs. The numbers of tumor-free mice among the 8 mice included in the analysis per group at the last time point of assessment are indicated. AS15 indicates mice injected with AS15 immunostimulant alone; PBS, mice injected with PBS; recPRAME, mice injected with recombinant PRAME protein alone; recPRAME+AS15, mice injected with recPRAME formulated with AS15 immunostimulant.
In addition, although not a part of the experiments presented in this manuscript, the role of CD4+ and CD8+ T cells in tumor protection was assessed in a T-cell depletion experiment on CB6F1 mice. After the tumor challenge with PRAME-expressing CT26 tumor cells, and CD4+ T-cell depletion with the monoclonal anti-CD4 antibody, the mean tumor surface at 1 month after the challenge was markedly higher than that of mice subjected to CD8+ T-cell depletion, suggesting the crucial role of CD4+ T cells in tumor protection (Supplementary Fig. S2, Supplemental Digital Content 2, http://links.lww.com/JIT/A396).
The Specificity of Tumor Protection in CB6F1 Mice Induced by recPRAME+AS15
To assess whether the observed tumor protection was PRAME specific, mice immunized with recPRAME+AS15 were challenged either with CT26-PRAME cells or with CT26 cells expressing an irrelevant antigen, MAGE-A3 (CT26-MAGE-A3). After 2 injections of recPRAME+AS15, CB6F1 mice were specifically protected against a challenge with PRAME-expressing tumor cells, but not against a challenge with tumor cells expressing MAGE-A3 (Fig. 5). The mean tumor size of mice immunized with recPRAME+AS15 and challenged with CT26-PRAME was significantly lower compared with mice challenged with tumor cells expressing MAGE-A3 or mice receiving PBS and challenged with CT26-PRAME or CT26-MAGE-A3.
FIGURE 5.
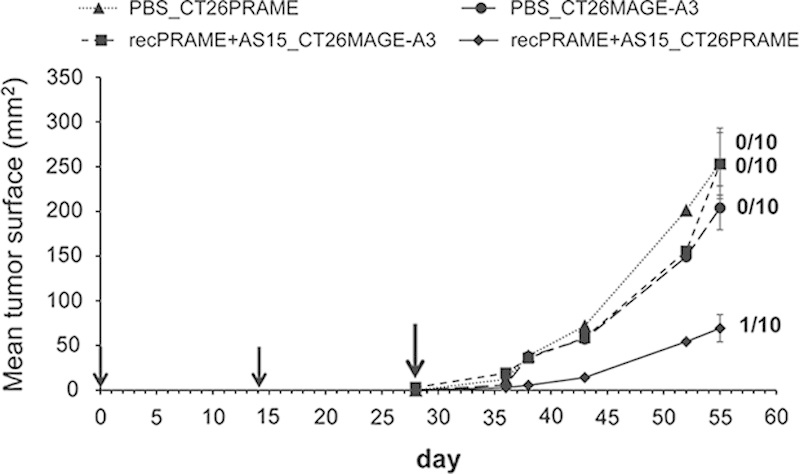
The specificity of tumor protection in CB6F1 mice induced by 2 injections of recPRAME+AS15. Mean tumor sizes at the last time point in each group: (A) recPRAME+AS15_CT26PRAME, 69.3 mm2; recPRAME+AS15_CT26MAGE-A3, 253.0 mm2; phosphate-buffered saline (PBS)_CT26PRAME, 253.7 mm2; PBS_CT26MAGE-A3, 203.9 mm2. Statistically significant differences in mean tumor sizes were observed between groups: recPRAME+AS15_CT26PRAME versus recPRAME+AS15_CT26MAGE-A3, P<0.0001; recPRAME+AS15_CT26PRAME versus PBS_CT26PRAME, P=0.0010; recPRAME+AS15_CT26PRAME versus PBS_CT26MAGE-A3, P=0.0003. Data are expressed as a mean tumor size of 10 mice per group. Error bars represent SEs. Black arrows represent the injection time points. The last black arrow indicates the tumor challenge time point. Numbers of tumor-free mice among the 10 mice included in the analysis per group at the last time point of assessment are indicated. PBS_CT26PRAME indicates mice injected with PBS, challenged (at day 28) with PRAME-expressing tumor cells; PBS_CT26MAGE-A3, mice injected with PBS, challenged (at day 28) with MAGE-A3-expressing tumor cells; recPRAME+AS15_CT26MAGE-A3, mice injected with recPRAME+AS15, challenged (at day 28) with MAGE-A3-expressing tumor cells; recPRAME+AS15_CT26PRAME, mice injected with recPRAME+AS15, challenged (at day 28) with PRAME-expressing tumor cells.
Study on Monkeys
The safety and the tolerability of repeated injections of recPRAME+AS15 were assessed in a GLP toxicology study on Cynomolgus monkeys. Extensive toxicology and antibody response data are presented in a separate publication.46 In brief, injections of recPRAME+AS15 were well tolerated and did not induce any local or systemic toxicity. High PRAME-specific antibody responses were induced in all monkeys immunized with recPRAME+AS15. Repeated injections of recPRAME+AS15 also induced PRAME-specific CD4+ and CD69+ (producing at least 1 cytokine) T-cell responses; after the seventh dose, the mean percentage of T cells producing cytokines was statistically significantly higher in monkeys from the recPRAME+AS15 group, compared with the saline control (Fig. 6). These data show that although PRAME mRNA expression is observed in a few normal tissues in monkeys, repeated injections of recPRAME+AS15 induced humoral and T-cell immune responses with no local or systemic toxicities.
FIGURE 6.
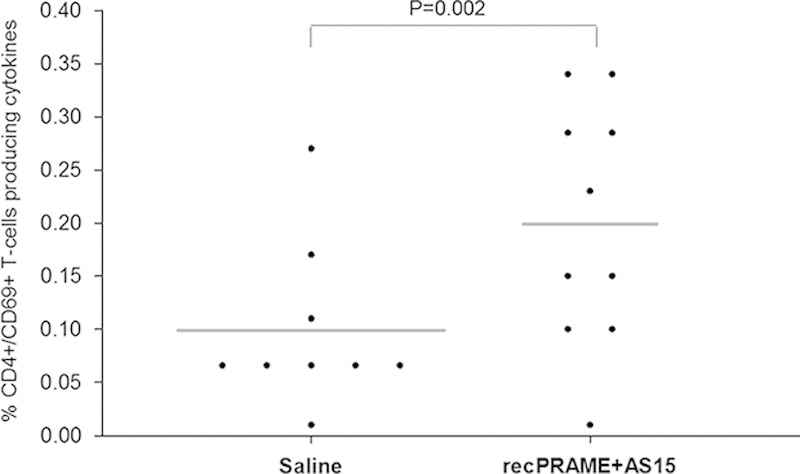
The percentage of T cells (CD4+ and CD69+) producing cytokines in Cynomolgus monkeys injected with saline or recPRAME+AS15 at postdose 7. Dots represent individual percentages of T cells producing cytokines. The horizontal bars represent mean values. recPRAME+AS15 indicates monkeys injected with recPRAME+AS15; Saline, monkeys injected with saline.
DISCUSSION
To support the clinical development of the PRAME cancer immunotherapeutic, we evaluated the immunogenicity of recPRAME+AS15 injections in CB6F1, HLA-A2 HLA-DR1 transgenic mice, and in nonhuman primates (Cynomolgus monkeys). The capacity of repeated injections of recPRAME+AS15 to induce antitumor activity was also evaluated in CB6F1 mice.
The evaluation of the immune responses in CB6F1 mice revealed that injections of recPRAME+AS15 triggered both humoral and cellular PRAME-specific immune responses, with significantly higher antibody titers and percentages of T cells producing cytokines (mostly CD4+) compared with mice injected with PBS, recPRAME alone, or the AS15 immunostimulant alone. These results suggest that the combination of recPRAME with a strong immunostimulant is necessary to induce such comprehensive immune responses. This is consistent with findings from several clinical and nonclinical studies showing that a combination of tumor antigens delivered as peptides or proteins in formulations comprising 1 or more immunostimulants among MPL, QS-21, and CpG enhances humoral and cellular immune responses.49–59
Our data also show that this immune response is persistent. The level of PRAME-specific antibodies and T cells induced by recPRAME+AS15 measured 2 months after the last immunization was still relatively high. In contrast to the antibody levels, the T-cell response was slightly lower than 2 weeks after the last immunization, suggesting that the immune response might decrease with time and that booster injections may be needed.
However, CD8+ T-cell responses were weak or time inconsistent, probably due to the genetic background of the mice used in these experiments. This is likely because CD8+ T-cell responses can be detected in some CD1 or OF1 outbred mice injected with recPRAME+AS15 (data not shown). Furthermore, on the basis of the results obtained using adoptive T-cell transfer, it is apparent that CD8+ T cells are important to eradicate an existing tumor; however, other effector cells, such as NK cells or CD4+ T cells, may also play a role. In the current manuscript, we clearly showed that the CD4+ T cells are required for tumor protection.
A similar experiment was conducted in H2-knockout mice transgenic for human HLAs (HLA-A02.01/HLA-DR1 transgenic mice).60 These mice express human HLA Class I (HLA-A02.01) and Class II (HLA-DR1) molecules, the most common HLA alleles in the white population61 and represent a unique in vivo experimental model to study the human immune response without any interference with the mouse MHC response. Although the variability of response is generally higher in transgenic mice compared with regular inbred mice, recPRAME+AS15 induced PRAME-specific CD4+ and CD8+ T cell responses, suggesting that it can potentially induce cellular immune responses in individuals expressing HLA-A02.01 and HLA-DR1.
Because nonhuman primates are considered to be closer to humans than mice in terms of homology to the antigen targeted, it was interesting to characterize the humoral and the cellular immune responses induced by recPRAME+AS15 in these animals. PRAME-specific antibody and T-cell responses were induced in monkeys immunized with recPRAME+AS15. The kinetics of the antibody response in monkeys injected with recPRAME+AS15 revealed that the PRAME-specific antibody levels increased substantially after the fourth injection. The antibody levels did not increase further with any additional immunization, but remained high 3 and 28 days after the seventh injection, indicating that there is no exhaustion of the immune response in these animals after multiple injections.46 Although CD8+ responses were detected in HLA-A02.01/HLA-DR1 transgenic mice, only CD4+CD69+ T cells were measurable in an ex vivo assay in monkeys after a short in vitro stimulation with a pool of overlapping 15-mer peptides covering the whole PRAME sequence. No CD8+ T-cell responses could be detected under the conditions used. Altogether, immunogenicity results of the studies presented in this article suggest that a vaccine combining a recombinant protein with AS15 is not capable of inducing CD8+ T-cell responses. This is further supported by low or undetectable CD8+ responses observed in phase II clinical trials that tested recMAGE-A3+AS15 in patients with NSCLC or melanoma.54,62
After surgery, cancer patients often experience recurrences and development of distant metastases. The capacity to induce a long-term immune memory is a crucial feature of an effective cancer immunotherapy. The immune response induced by 4 injections of recPRAME+AS15 provided a PRAME-specific long-term immune memory able to protect mice against a challenge with PRAME-expressing tumor cells up to 2 months after the last immunization. In mice immunized with recPRAME+AS15, the mean tumor growth remained significantly lower compared with mice injected with PBS, recPRAME alone, or AS15 alone, or was not detectable. These data indicate that both recPRAME and AS15 are required to prevent tumor growth, and that this protection is specific for the PRAME antigen that should be expressed by the tumor. Indeed, after a challenge with a similar tumor expressing another antigen (MAGE-A3), the tumor was not recognized in mice immunized with recPRAME+AS15 and increased in size. In our experiments, we chose a tumor challenge approach, in which tumor-free mice were immunized with recPRAME+AS15 and then challenged with PRAME-expressing tumor cells, as such an approach simulates the adjuvant setting in clinical trials more accurately than would a therapeutic setting approach (ie, first injecting a tumor and then immunizing with recPRAME+AS15). However, unlike mice, patients who underwent surgery and are considered disease free at the time they receive immunotherapy have already been exposed to the tumor. This preexposure is likely to prime the immune system against the targeted antigen or initiate certain immune-suppressive mechanisms. This could possibly impair the efficacy of future immunotherapeutic treatment, although it was not taken into account in the mice experiments. Nevertheless, long-term immunity against the tumor was obtained, which is of particular importance in the context of treating potentially disease-free cancer patients or patients with minimal residual disease undergoing adjuvant treatment and who remain at a high risk of relapse.
In humans, PRAME mRNA expression was detected in a few normal tissues.20,28,63 In nonhuman primates (Cynomolgus monkeys), the PRAME mRNA expression pattern shares some similarities with humans: namely, the antigen can be detected in the adrenal gland, the testis, and the ovaries of both species (unpublished data available to GSK). Furthermore, the monkey PRAME homologous protein presents >90% identity with the human PRAME protein. Thus, before the clinical evaluation of the PRAME cancer immunotherapeutic was launched, the safety of repeated injections of recPRAME+AS15 was evaluated in a GLP toxicity study in nonhuman primates (Cynomolgus monkeys).46 In this previous study, no signs of inflammation or systemic toxicity were observed after injections of full human doses of recPRAME+AS15 in monkeys. recPRAME+AS15 also induced PRAME-specific antibodies; however, these results should be interpreted with caution due to the higher dose/body mass ratio used compared with the intended administration in humans.
To date, only a few reports on the immunogenicity of PRAME in patients with solid tumors have been published.64,65 Our results support these very scarce reports on the PRAME tumor antigen as a potential target for immunotherapy of solid tumors.
In conclusion, the results presented in this manuscript indicate that recPRAME+AS15 induced a comprehensive immune response in CB6F1 and HLA-A02.01/HLA-DR1 transgenic mice and provided PRAME-specific long-term protection of CB6F1 mice against a tumor challenge. recPRAME+AS15 was well tolerated and did not induce any signs of systemic toxicity in nonhuman primates.
ACKNOWLEDGMENTS
The authors thank Stéphane Veenstra (study monitor for the study on monkeys); Marie-Pierre Malice for statistical analyses; Yves Renaux and André De Groote (Laboratory Animal Science) for the management of laboratory animals (injections of the animals, collection of samples); Olivier Gruselle for providing the data on PRAME expression in monkeys; Romain Piccininno, Carole François, and Aurélie Delplanque (the R&D preclinical cancer immunology team) for technical assistance; and Maria Morales Aira, Delphine Guillaume, and Véronique Gobin from the GSK formulation team who were in charge of the PRAME formulations. The authors also thank Urszula Miecielica, PhD and Mihai Surducan, PhD (XPE Pharma & Science), for providing medical writing services, and Sophie Timmery, PhD (XPE Pharma & Science, on behalf of GSK), for editorial support in preparing this manuscript.
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURES
GlaxoSmithKline Biologicals SA was the funding source and was involved in all stages of the study conduct and analysis. GlaxoSmithKline Biologicals SA also funded all costs associated with the development and the publishing of the present manuscript.
All authors are employed by the GSK group of companies. C.G., L.S., and J.L. declare stock ownership in the GSK group of companies. The remaining authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website, www.immunotherapy-journal.com.
REFERENCES
- 1.Aldrich JF, Lowe DB, Shearer MH, et al. Vaccines and immunotherapeutics for the treatment of malignant disease. Clin Dev Immunol. 2010;2010:697158.doi:10.1155/2010/697158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bilusic M, Gulley JL. Endpoints, patient selection, and biomarkers in the design of clinical trials for cancer vaccines. Cancer Immunol Immunother. 2012;61:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vergati M, Intrivici C, Huen NY, et al. Strategies for cancer vaccine development. J Biomed Biotechnol. 2010;2010:596432.doi:10.1155/2010/596432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palumbo MO, Kavan P, Miller WH, Jr, et al. Systemic cancer therapy: achievements and challenges that lie ahead. Front Pharmacol. 2013;4:57.doi:10.3389/fphar.2013.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charoentong P, Angelova M, Efremova M, et al. Bioinformatics for cancer immunology and immunotherapy. Cancer Immunol Immunother. 2012;61:1885–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finn OJ. Cancer immunology. N Engl J Med. 2008;358:2704–2715. [DOI] [PubMed] [Google Scholar]
- 7.Laheru DA, Pardoll DM, Jaffee EM. Genes to vaccines for immunotherapy: how the molecular biology revolution has influenced cancer immunology. Mol Cancer Ther. 2005;4:1645–1652. [DOI] [PubMed] [Google Scholar]
- 8.Hammer GE, Kanaseki T, Shastri N. The final touches make perfect the peptide-MHC class I repertoire. Immunity. 2007;26:397–406. [DOI] [PubMed] [Google Scholar]
- 9.Buonaguro L, Petrizzo A, Tornesello ML, et al. Translating tumor antigens into cancer vaccines. Clin Vaccine Immunol. 2011;18:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palena C, Schlom J. Vaccines against human carcinomas: strategies to improve antitumor immune responses. J Biomed Biotechnol. 2010;2010:380697.doi:10.1155/2010/380697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–839. [DOI] [PubMed] [Google Scholar]
- 12.Andersen MH, Junker N, Ellebaek E, et al. Therapeutic cancer vaccines in combination with conventional therapy. J Biomed Biotechnol. 2010;2010:237623.doi:10.1155/2010/237623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimberu PM, Leonhardt RM. Cancer immunotherapy takes a multi-faceted approach to kick the immune system into gear. Yale J Biol Med. 2011;84:371–380. [PMC free article] [PubMed] [Google Scholar]
- 14.Postow M, Callahan MK, Wolchok JD. Beyond cancer vaccines: a reason for future optimism with immunomodulatory therapy. Cancer J. 2011;17:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weir GM, Liwski RS, Mansour M. Immune modulation by chemotherapy or immunotherapy to enhance cancer vaccines. Cancers. 2011;3:3114–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang CL, Benencia F, Coukos G. Whole tumor antigen vaccines. Semin Immunol. 2010;22:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubensky TW, Jr, Reed SG. Adjuvants for cancer vaccines. Semin Immunol. 2010;22:155–161. [DOI] [PubMed] [Google Scholar]
- 18.Kirkwood JM, Butterfield LH, Tarhini AA, et al. Immunotherapy of cancer in 2012. CA Cancer J Clin. 2012;62:309–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tefit JN, Serra V. Outlining novel cellular adjuvant products for therapeutic vaccines against cancer. Expert Rev Vaccines. 2011;10:1207–1220. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda H, Lethe B, Lehmann F, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6:199–208. [DOI] [PubMed] [Google Scholar]
- 21.Tajeddine N, Gala JL, Louis M, et al. Tumor-associated antigen preferentially expressed antigen of melanoma (PRAME) induces caspase-independent cell death in vitro and reduces tumorigenicity in vivo. Cancer Res. 2005;65:7348–7355. [DOI] [PubMed] [Google Scholar]
- 22.Atanackovic D, Luetkens T, Kloth B, et al. Cancer-testis antigen expression and its epigenetic modulation in acute myeloid leukemia. Am J Hematol. 2011;86:918–922. [DOI] [PubMed] [Google Scholar]
- 23.Ding K, Wang XM, Fu R, et al. PRAME gene expression in acute leukemia and its clinical significance. Cancer Biol Med. 2012;9:73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doolan P, Clynes M, Kennedy S, et al. Prevalence and prognostic and predictive relevance of PRAME in breast cancer. Breast Cancer Res Treat. 2008;109:359–365. [DOI] [PubMed] [Google Scholar]
- 25.Heighway J, Knapp T, Boyce L, et al. Expression profiling of primary non-small cell lung cancer for target identification. Oncogene. 2002;21:7749–7763. [DOI] [PubMed] [Google Scholar]
- 26.Szczepanski MJ, DeLeo AB, Luczak M, et al. PRAME expression in head and neck cancer correlates with markers of poor prognosis and might help in selecting candidates for retinoid chemoprevention in pre-malignant lesions. Oral Oncol. 2013;49:144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Baren N, Chambost H, Ferrant A, et al. PRAME, a gene encoding an antigen recognized on a human melanoma by cytolytic T cells, is expressed in acute leukaemia cells. Br J Haematol. 1998;102:1376–1379. [DOI] [PubMed] [Google Scholar]
- 28.Kessler JH, Beekman NJ, Bres-Vloemans SA, et al. Efficient identification of novel HLA-A(*)0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J Exp Med. 2001;193:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costessi A, Mahrour N, Sharma V, et al. The human EKC/KEOPS complex is recruited to Cullin2 ubiquitin ligases by the human tumour antigen PRAME. PLoS One. 2012;7:e42822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costessi A, Mahrour N, Tijchon E, et al. The tumour antigen PRAME is a subunit of a Cul2 ubiquitin ligase and associates with active NFY promoters. EMBO J. 2011;30:3786–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quintarelli C, Dotti G, Hasan ST, et al. High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood. 2011;117:3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffioen M, Kessler JH, Borghi M, et al. Detection and functional analysis of CD8+ T cells specific for PRAME: a target for T-cell therapy. Clin Cancer Res. 2006;12:3130–3136. [DOI] [PubMed] [Google Scholar]
- 33.Li L, Giannopoulos K, Reinhardt P, et al. Immunotherapy for patients with acute myeloid leukemia using autologous dendritic cells generated from leukemic blasts. Int J Oncol. 2006;28:855–861. [PubMed] [Google Scholar]
- 34.Rezvani K, Yong AS, Tawab A, et al. Ex vivo characterization of polyclonal memory CD8+ T-cell responses to PRAME-specific peptides in patients with acute lymphoblastic leukemia and acute and chronic myeloid leukemia. Blood. 2009;113:2245–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amir AL, van der Steen DM, van Loenen MM, et al. PRAME-specific allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clin Cancer Res. 2011;17:5615–5625. [DOI] [PubMed] [Google Scholar]
- 36.The European Parliament and the Council of the European Union. Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. Official Journal of the European Union. 2010;L 276:33-79. Available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2010:276:0033:0079:en:PDF. Accessed February 21, 2014.
- 37.Organisation for Economic Co-operation and Development (OECD). OECD Principles on Good Laboratory Practice (as revised in 1997). 1998. Available at: http://search.oecd.org/officialdocuments/displaydocumentpdf/?doclanguage=en&cote=env/mc/chem(98)17. Accessed February 21, 2014. [Google Scholar]
- 38.The European Parliament and the Council of the European Union. Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances. Official Journal of the European Union. 2004;L 50:44-50. Available at: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32004L0010&from=EN. Accessed February 21, 2014.
- 39.Ministère de l’Emploi et de la Solidarité. Arrêté du 14 mars 2000 relatif aux bonnes pratiques de laboratoire. 2000. Available at: http://ansm.sante.fr/var/ansm_site/storage/original/application/dd4641e3edd1885e60f0337d4c385b5e.pdf. Accessed February 21, 2014.
- 40.The Council of the European Communities. Council Directive 86/609/EEC of 24 November 1986 on the approximation of laws, regulations and administrative provisions of the Member States regarding the protection of animals used for experimental and other scientific purposes 1986. Available at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:31986L0609:en:HTML. Accessed February 21, 2014.
- 41.Destexhe E, Grosdidier E, Baudson N, et al. Non-clinical safety evaluation of single and repeated intramuscular administrations of MAGE-A3 cancer immunotherapeutic in rabbits and Cynomolgus monkeys. J Appl Toxicol. 2015;35:717–728. [DOI] [PubMed] [Google Scholar]
- 42.Wang M, Bronte V, Chen PW, et al. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–4692. [PMC free article] [PubMed] [Google Scholar]
- 43.Aebig JA, Mullen GE, Dobrescu G, et al. Formulation of vaccines containing CpG oligonucleotides and alum. J Immunol Methods. 2007;323:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weeratna RD, Makinen SR, McCluskie MJ, et al. TLR agonists as vaccine adjuvants: comparison of CpG ODN and Resiquimod (R-848). Vaccine. 2005;23:5263–5270. [DOI] [PubMed] [Google Scholar]
- 45.Weigel BJ, Rodeberg DA, Krieg AM, et al. CpG oligodeoxynucleotides potentiate the antitumor effects of chemotherapy or tumor resection in an orthotopic murine model of rhabdomyosarcoma. Clin Cancer Res. 2003;9:3105–3114. [PubMed] [Google Scholar]
- 46.Garcon N, Silvano J, Kuper CF, et al. Non-clinical safety evaluation of repeated intramuscular administration of the AS15 immunostimulant combined with various antigens in rabbits and Cynomolgus monkeys. J Appl Toxicol. 2015. DOI 10.1002/jat.3167. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fisher RA. Statistical Methods for Research Workers, 14th ed New York, NY: Hafner Publishing Company; 1973. [Google Scholar]
- 48.Dean AM, Voss D. Design and Analysis of Experiments (Springer Texts in Statistics). New-York: Springer; 2000. [Google Scholar]
- 49.Atanackovic D, Altorki NK, Cao Y, et al. Booster vaccination of cancer patients with MAGE-A3 protein reveals long-term immunological memory or tolerance depending on priming. Proc Natl Acad Sci U S A. 2008;105:1650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atanackovic D, Altorki NK, Stockert E, et al. Vaccine-induced CD4+ T cell responses to MAGE-3 protein in lung cancer patients. J Immunol. 2004;172:3289–3296. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Zhang L, Wen W, et al. Induction of HCA587-specific antitumor immunity with HCA587 protein formulated with CpG and ISCOM in mice. PLoS One. 2012;7:e47219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cluff CW. Monophosphoryl lipid A (MPL) as an adjuvant for anti-cancer vaccines: clinical results. Adv Exp Med Biol. 2010;667:111–123. [DOI] [PubMed] [Google Scholar]
- 53.Daayana S, Elkord E, Winters U, et al. Phase II trial of imiquimod and HPV therapeutic vaccination in patients with vulval intraepithelial neoplasia. Br J Cancer. 2010;102:1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kruit WH, Suciu S, Dreno B, et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol. 2013;31:2413–2420. [DOI] [PubMed] [Google Scholar]
- 55.Speiser DE, Lienard D, Rufer N, et al. Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest. 2005;115:739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valmori D, Souleimanian NE, Tosello V, et al. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc Natl Acad Sci U S A. 2007;104:8947–8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Ojik H, Kruit W, Portielje J, et al. Phase I/II study with CpG 7909 as adjuvant to vaccination with MAGE-3 protein in patients with MAGE-3 positive tumors (abstract). Ann Oncol. 2002;13(suppl 5):157–162.11863097 [Google Scholar]
- 58.Vantomme V, Dantinne C, Amrani N, et al. Immunologic analysis of a phase I/II study of vaccination with MAGE-3 protein combined with the AS02B adjuvant in patients with MAGE-3-positive tumors. J Immunother. 2004;27:124–135. [DOI] [PubMed] [Google Scholar]
- 59.Zhang L, Chen J, Song X, et al. Cancer/testis antigen HCA587-derived long peptide vaccine generates potent immunologic responses and antitumor effects in mouse model. Oncol Res. 2013;21:193–200. [DOI] [PubMed] [Google Scholar]
- 60.Pajot A, Michel ML, Fazilleau N, et al. A mouse model of human adaptive immune functions: HLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol. 2004;34:3060–3069. [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez-Galarza FF, Christmas S, Middleton D, et al. Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 2011;39:D913–D919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vansteenkiste J, Zielinski M, Linder A, et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: phase II randomized study results. J Clin Oncol. 2013;31:2396–2403. [DOI] [PubMed] [Google Scholar]
- 63.Kilpinen S, Autio R, Ojala K, et al. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9,783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008;9:R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pollack SM, Li Y, Blaisdell MJ, et al. NYESO-1/LAGE-1s and PRAME are targets for antigen specific T cells in chondrosarcoma following treatment with 5-Aza-2-deoxycitabine. PLoS One. 2012;7:e32165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weber JS, Vogelzang NJ, Ernstoff MS, et al. A phase 1 study of a vaccine targeting preferentially expressed antigen in melanoma and prostate-specific membrane antigen in patients with advanced solid tumors. J Immunother. 2011;34:556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]