

Background: The mechanism of interaction between small molecules and amyloid-β fibrils is unknown.
Results: Molecular modeling on the basis of solid-state NMR reveals that sulindac sulfide intercalates between β-strands of amyloid-β fibrils.
Conclusion: Sulindac sulfide interacts with amyloid-β fibrils in a specific manner and binds to hydrophobic cavities in the core of the fibrils.
Significance: Unraveling how small molecules interfere with amyloidogenic deposits will assist structure-based drug design for neurodegenerative disorders.
Keywords: Alzheimer disease, amyloid, drug design, protein structure, solid-state NMR, magic angle spinning, ligand interactions
Abstract
Alzheimer disease is the most severe neurodegenerative disease worldwide. In the past years, a plethora of small molecules interfering with amyloid-β (Aβ) aggregation has been reported. However, their mode of interaction with amyloid fibers is not understood. Non-steroidal anti-inflammatory drugs (NSAIDs) are known γ-secretase modulators; they influence Aβ populations. It has been suggested that NSAIDs are pleiotrophic and can interact with more than one pathomechanism. Here we present a magic angle spinning solid-state NMR study demonstrating that the NSAID sulindac sulfide interacts specifically with Alzheimer disease Aβ fibrils. We find that sulindac sulfide does not induce drastic architectural changes in the fibrillar structure but intercalates between the two β-strands of the amyloid fibril and binds to hydrophobic cavities, which are found consistently in all analyzed structures. The characteristic Asp23-Lys28 salt bridge is not affected upon interacting with sulindac sulfide. The primary binding site is located in the vicinity of residue Gly33, a residue involved in Met35 oxidation. The results presented here will assist the search for pharmacologically active molecules that can potentially be employed as lead structures to guide the design of small molecules for the treatment of Alzheimer disease.
Introduction
The self-assembly of amyloidogenic proteins into fibrils and oligomers plays a pivotal role in various diseases (1). The deposition of fibrils formed by the amyloid-β peptide (Aβ)3 into plaques in brain tissue is a major pathological hallmark in the progression of neurodegeneration in Alzheimer disease. Aβ peptides are generated through sequential proteolytic cleavages of the amyloid precursor protein (APP) by the β- and γ-secretases (2, 3). This results in the production of Aβ peptides of differing lengths (4), mainly Aβ1–40 and Aβ1–42 (5), and shorter variants, such as Aβ1–39 (6). Soluble oligomers formed by Aβ1–42 represent the toxic species responsible for the decline in cognitive function associated with neurodegeneration (7). Several studies have demonstrated that small molecules can interfere with the solubility of amyloid proteins and are, therefore, potential drug candidates (8, 9). Chronic inflammation significantly enhances Alzheimer disease pathogenesis (10). Fibrillar β-amyloid deposits co-localize with numerous chronic inflammatory mediators and activated microglia in the brain (11). The relation to inflammation suggests that non-steroidal anti-inflammatory drugs (NSAIDs) might be beneficial for the treatment of Alzheimer disease. In fact, epidemiological studies demonstrate a link between the use of anti-inflammatory drugs and the prevalence of Alzheimer disease (12). This work aims to investigate the interaction mechanism of the NSAID sulindac sulfide (Fig. 1a) with Alzheimer peptide Aβ fibrils. In addition to sulindac sulfide (13), NSAIDs, including ibuprofen, indomethacin (13), and flurbiprofen (14), have been identified as γ-secretase modulators. γ-Secretase modulators interfere with APP processing and modify the relative Aβ1–42 population. In particular, sulindac sulfide decreases the relative amount of the amyloid-prone Aβ1–42, whereas the production of shorter, less amyloidogenic Aβ peptides is increased (13–16).
FIGURE 1.

The influence of sulindac sulfide on Aβ. a, chemical structure of the NSAID sulindac sulfide. b, TEM images of 50 μm Aβ fibrils in the absence (bottom panel) and presence (top panel) of a 5-fold molar excess of sulindac sulfide. The fibrillar character is maintained, and deposits of sulindac sulfide can be observed. Scale bar = 200 nm. c, two-dimensional 13C-15N TEDOR spectra of Aβ amyloid fibrils incubated in the presence of a 5-fold molar excess of sulindac sulfide and 1% DMSO (red) and Aβ fibrils incubated with 1% DMSO (black) as a control. Corresponding 13C-13C correlation spectra are shown in supplemental Fig. S1. The obtained 13C line widths are in the order of 120–200 Hz (data not shown). Sequential assignments are obtained from three-dimensional NCACX and NCOCX experiments. Arrows indicate residues that experience large chemical shift changes. d, CSPs induced by sulindac sulfide on the NMR chemical shifts of Aβ fibrils. Differences in chemical shifts (Δδ (ppm)) were calculated for 13C and 15N resonances according to ΔδC = [(δCsul − δCref)2]1/2 and ΔδN = [(2 / 5 × (δNsul − δNref))2]1/2, respectively.
Solution-state NMR structures of the APP-TM (transmembrane) dimer have been solved for the wild-type (17) and a familial mutant (18). NMR and EPR experiments have revealed a potential cholesterol-binding site within the C-terminal Cys99 sequence and highlight the significance of the GXXXG segments for binding (19). It has been suggested that NSAIDs can interact with lipids to form phospholipid complexes (20–22). This may provide a general mechanism for the interaction of APP with small molecules. Reports on the interaction between sulindac sulfide and the APP transmembrane sequence are, however, contradictory. Sulindac sulfide, among other γ-secretase modulators, binds to the APP transmembrane sequence of the Cys99 motif (23) and to Cys100 dimers in the presence of SDS micelles (24). Bacterial reporter assays show that γ-secretase modulators, including sulindac sulfide, bind to the GXXXG dimerization motif and, thereby, attenuate the dimerization of the APP transmembrane sequence (25), a process necessary for proteolytic cleavage (26). However, colloidal aggregation of sulindac sulfide in aqueous solutions can potentially induce nonspecific binding (27, 28). Contradicting data have been reported for the influence of sulindac sulfide on the Aβ peptide itself (29, 30).
So far, it is not understood how sulindac sulfide interacts with amyloids. NMR is a suitable technique to study Aβ-ligand interactions for various Aβ aggregation states (31). Solution-state NMR can be employed to study interactions of Aβ monomers and small molecule (32) or peptide inhibitors (33), nanoparticles (34), and various others. Besides monomers and fibrils, oligomeric intermediates formed by Aβ in solution constitute potential drug targets (35, 36). However, oligomeric intermediates and insoluble fibrils are not detectable by solution-state NMR because their lines are broadened beyond detection. Solid-state NMR spectroscopy is a powerful tool that allows the study of Aβ-small molecule interactions at atomic resolution. In the past, this technique has been applied successfully for the characterization of the interaction between Aβ and the polyphenol epigallocatechin gallate (37), curcumin (38, 39), and catechol-type flavonoids (40) and to study the interface of Congo red and amyloids formed by the prion domain of the HET-s protein (41). In this work, we investigate the interaction between sulindac sulfide and Aβ fibrils using solid-state NMR spectroscopy. On the basis of the gathered NMR data, we employ docking to derive a model for the intercalation of sulindac sulfide with Aβ fibrils.
Experimental Procedures
Aβ Expression and Purification and Sample Preparation
The uniformly 15N or 15N-13C-labeled Aβ1–40 peptide was recombinantly expressed in Escherichia coli inclusion bodies and purified via reverse-phase chromatography as described previously (42). The construct contains an N-terminal methionine but shows the same biochemical properties as the wild-type peptide (43). To obtain monomeric Aβ in solution, the lyophilized peptide was initially dissolved in 10 mm NaOH, sonicated for 10 min in an ultrasonic bath, and centrifuged for 10 min at 14,800 rpm to remove potential nucleation seeds. The solution was then diluted in 2× buffer (100 mm sodium phosphate and 100 mm NaCl buffer (pH 7.3)) to yield the respective Aβ concentration.
Preparation of NSAIDs
Stock solutions of sulindac sulfide were prepared in dimethyl-sulfoxide (DMSO), and the respective amount of NSAID stock was added to Aβ. The final concentration of DMSO in aqueous solutions was 1%.
Solid-state NMR Sample Preparation
Aβ fibrils were obtained according to a protocol described previously (44). Briefly, monomeric Aβ at a concentration of 50 μm was seeded with sonicated fibrils (10% w/w) and incubated under agitation until the completion of fibrillation. This step was repeated for 11 generations. For solid-state NMR measurements, the last generation was allowed to fibrilize for 2 days before sulindac sulfide was added to the sample. Approximately 10 mg of Aβ fibrils was incubated with a 5-fold molar excess of sulindac sulfide (250 μm, 1% DMSO). After mixing, the sample was kept quiescent for 1 h at room temperature. As a reference, Aβ fibrils were incubated under the same conditions with 1% DMSO. The sulindac sulfide-incubated fibrils were sedimented into a 3.2-mm rotor. The reference fibrils were packed into a 4.0-mm rotor.
Solid-state NMR Measurements
13C-detected assignment experiments were carried out using a Bruker Avance wide-bore spectrometer operating at a 1H Larmor frequency of 700 MHz (16.5 tesla). The spectrometer was equipped with a triple-resonance MAS probe (1H, 13C, 15N). All measurements were recorded at a MAS rotation frequency of 17 kHz for 3.2-mm rotors or 13 kHz for 4.0-mm rotors and a temperature of 12 °C. In all experiments, 1H-13C magnetization transfer was achieved through cross-polarization. Two-dimensional spectra were recorded using proton-driven spin diffusion (45, 46) for 13C-13C magnetization transfer with a mixing time of 200 ms or transferred echo double-resonance (TEDOR) (47, 48) for 13C-15N magnetization transfer. The three-dimensional NCACX and NCOCX (49) experiments were recorded employing TEDOR for 13C-15N magnetization transfer and dipolar assisted rotational resonance (50) for 13C-13C mixing.
To identify 13C atoms in Aβ that are located in the vicinity of the 19F atom of sulindac sulfide, we recorded 13C-19F rotational echo double resonance (REDOR) experiments (supplemental Fig. S8 a) (51). These were recorded on a Bruker Avance III wide-bore spectrometer operating at a 1H Larmor frequency of 600 MHz (14 tesla). The spectrometer was equipped with a triple-resonance cross-polarization MAS probe (1H, 13C, 19F). All measurements were recorded at a MAS rotation frequency of 14.6 kHz and a temperature of 12 °C. In the REDOR experiments, 13C-19F dipolar dephasing was preceded by a 13C-13C proton-driven spin diffusion mixing step. REDOR dephasing times were set to 1.1, 2.2, and 4.4 ms. REDOR spectra recorded with and without dephasing pulses were subtracted to identify fluorine-coupled carbons. In Fig. 2a, REDOR spectra are represented in one-dimensional mode. No 13C-13C cross peaks could be observed in the three-dimensional REDOR experiments because the sensitivity was too low. To detect the characteristic salt bridge between the carboxyl of Asp23 and the amine of Lys28 of mature Aβ fibrils, TEDOR experiments (supplemental Fig. S8 b) were carried out on the basis of the pulse sequence described by Jaroniec et al. (52). All spectra were recorded on a Bruker Avance III narrow-bore spectrometer operating at a 1H Larmor frequency of 750 MHz (17.63 tesla) equipped with a triple-resonance MAS probe (1H, 13C, 15N) at a MAS rotation frequency of 11 kHz and 4 °C. In the experiment, the TEDOR mixing time was set to 7.27 and 15.72 ms.
FIGURE 2.

13C-19F REDOR and 13C-15N TEDOR NMR experiments. a, 13C-19F REDOR NMR experiments recorded to observe dephasing of Aβ 13C nuclei in close proximity to the 19F atom of sulindac sulfide. Experiments were run with mixing times of 1.1 (data not shown), 2.2, and 4.4 ms. A list of all 13C-19F contacts observed can be found in supplemental Table S2. Recoupled and reference spectra were subtracted to identify affected 13C resonances (red). For reference, a one-dimensional 13C experiment without 19F recoupling, including all aliphatic resonances, is shown (gray). b, analysis of the salt bridge involving residues Asp23 and Lys28. 13C-15N TEDOR spectra show cross-peaks between Lys28-Nζ and Asp23-Cγ in the presence (red) and absence of sulindac sulfide (black). A TEDOR mixing time of 15.72 ms is employed. One-dimensional traces extracted at the Nζ chemical shifts are superimposed with the one-dimensional 13C reference spectra of the respective sample (gray).
Transmission Electron Microscopy (TEM)
Images were recorded on an EM 900 from Carl Zeiss SMT. Samples were stained with 4% uranyl acetate solution on formvar/carbon-coated grids.
Molecular Modeling
The 2-fold symmetric (PDB code 2LMN) (53) and the 3-fold symmetric (PDB code 2LMP) (54) Aβ1–40 fibril NMR structural models were employed in molecular modeling studies. To identify internal cavities, each model of the Aβ fibril was analyzed separately. For the calculation of internal cavities, van der Waals volumes were derived using the Voronoi cell method (55, 56). Atomic volumes are calculated on the basis of the allocation of space among atoms using hyperbolic surfaces (applying a cubic lattice of 0.1-Å grid width) and applying the ProtOr (57) atom radius set for protein atoms that were determined analytically from reference structures. Internal cavities are determined analytically from each model structure by a Delaunay triangulation (58). An internal cavity is defined as the buried space within a structure that is big enough to accommodate at least a 1.4-Å radius water-sized probe. The size of a cavity is estimated from the average distance of the cavity center to the neighboring atoms and depicted by the radius of a sphere. The polarity of the cavities is assessed with the program DOWSER (59), which calculates potential positions for water not resolved by the original structure determination approach. This program assesses the hydrophilicity of protein cavities by determining the interaction energy between a water molecule and its surrounding atoms. Water molecules with interaction energies of less than −10 kcal/mol are considered “low-energy water molecules” and selected for output. Internal cavities harboring such an internal water molecule are denoted as polar (blue) and the remaining cavities as hydrophobic (gray) (Fig. 3 and supplemental Figs. S4 and S5).
FIGURE 3.

Packing analysis of Aβ structures. a and b, distribution of internal cavities in 3-fold symmetric Aβ1–40 fibrils (PDB code 2LMP, model 1) (54) (a) and 2-fold symmetric Aβ1–40 fibrils (PDB code 2LMN, model 1) (53) (b). Shown is a cross-section perpendicular to the fibril axis. Each cavity is depicted as a sphere. The radius of the sphere corresponds to the average distance from the cavity center to the Aβ van der Waals surface. Water-containing polar cavities are colored in blue and hydrophobic cavities in gray. The approximate position of cavity clusters #1 to #5 is indicated. The protein backbones are depicted as schematics, with side chains drawn as lines. Residues that are as close as ≤6 Å to the 19F atom of sulindac sulfide are highlighted using sticks. Residues Ile32 and Val36, which define cavity clusters, are represented in purple. Ala30, Leu34, and Val39 are shown in green. Phe19 is colored red. Termini are marked with N or C.
For induced fit docking (60, 61), the region between the two most hydrophobic cavity clusters (termed #1-#5) near Ile32 (#2) and Val36 (#3) from the first NMR model was targeted. Docking was performed using the Schrödinger Maestro suite (62) following a standard protocol. Subsequently, a grid with auto-assigned box size was generated. The resulting box is centered near Leu34 for the 3-fold symmetric structure and Ile32 and Val36 for the 2-fold symmetric structure. Induced fit docking was done with the program Glide with single precision and with no constraints applied (63). To simulate receptor flexibility, side chain orientations were optimized to take into account ligand binding. At last, a Glide redocking was applied for the refined structure. Docking poses are ranked and filtered by means of the Glide score. This score assesses the binding probability and accounts for steric clashes, van der Waals and coulomb energy, lipophilicity, H-bonding, and bond rotation ability, among others. Only the energetically most favorable docking poses (with theoretical energies <30 kcal/mol higher than the best pose) are considered for further analysis.
Results
To probe the interaction between Aβ fibrils and sulindac sulfide (Fig. 1a), we titrated sulindac sulfide to preformed fibrils. Fibrillar Aβ is observed by TEM in the presence of the NSAID (Fig. 1b). The resulting 13C-15N (Fig. 1c) and 13C-13C (supplemental Fig. S1) correlation spectra show well dispersed peaks. The three-dimensional NCACX and NCOCX experiments allowed sequential assignment of resonances for residues Gln15-Val40 for both samples (representative strip plots are shown in supplemental Fig. S2; all assigned resonances are listed in supplemental Table S1). We detected only one set of resonances for both samples, indicating that the fibrils exist in one conformation.
Sulindac sulfide has no significant effect on the fibrillar structure of Aβ because the spectra of both samples are relatively similar (Fig. 1c and supplemental Fig. S1). However, small but defined chemical shift perturbations (CSPs) are observed in the presence of sulindac sulfide, indicating specific interactions of the NSAID with the fibrils. This is remarkable because a non-quantitative and nonspecific binding of sulindac sulfide to Aβ fibrils would result in peak-splitting and line-broadening. Fig. 1d shows CSPs, Δδ (parts per million), upon addition of sulindac sulfide. We observe changes in chemical shift in particular for side chain resonances of Lys16, Val18-Cβ, Phe19-Cβ, Phe20-Cβ, Asn27-Cγ, and Met35-Cβ as well as for the backbone resonances of Phe19, Phe20, Ala21 and Gly33. CSPs reflect ligand binding but could as well be a consequence of local or global structural rearrangements. To unambiguously probe ligand binding, we recorded 13C-19F REDOR experiments employing the NMR-active properties of the 19F atom of sulindac sulfide. In general, only aliphatic resonances of Aβ could be detected. For comparison, a one-dimensional 13C spectrum containing assignments for all aliphatic resonances is represented (Fig. 2a). The strongest dephasing effects are observed for the Cγ resonances of Val18 or Val39. However, because of spectral overlap, these two peaks cannot be discriminated. Smaller signal attenuations are observed for Cγ of Val24 and Val36, Cβs of Ala21 or Ala30 (overlap) as well as for Cγ2 and Cδ1 of Ile32 at longer mixing times. These resonances exhibit the most severe dephasing effects. The respective residues must therefore be located close to the fluorine atom of sulindac sulfide.
To gain further information on the potential effects of sulindac sulfide on the Aβ fibril structure, we investigated the effect on the salt bridge, which is typically formed between the side chains of residues Asp23 and Lys28 (53, 54). From sequential assignments, the chemical shift of the carboxylic group of Asp23 in the presence of sulindac sulfide was assigned to 177.8 ppm and to 178.0 ppm for the reference fibrils. On the basis of one-dimensional 15N spectra, the chemical shift of Lys28-Nζ was found to be 33.9 ppm and 34.3 ppm for the two preparations, respectively. In both samples, a cross-peak between Lys28-Nζ and Asp23-Cγ was detected, implying the presence of a salt bridge in both cases (Fig. 2b).
In the following, the CSPs in the two-dimensional 13C-13C proton-driven spin diffusion and 13C-15N TEDOR as well as the 13C-19F REDOR contacts were used as restraints to derive a model for sulindac sulfide in complex with Aβ1–40 fibrils. We used both a 2-fold (53) and 3-fold symmetric (54) Aβ1–40 fibril NMR structure as reference structures for modeling and docking experiments because they show the highest correlation with our chemical shifts (supplemental Fig. S3) compared with all structures and models analyzed (44, 53, 54, 64–68). The NMR structures contained 10 models each. The architecture of the Aβ1–40 fibrils obviously differs comparing the 2-fold and 3-fold-symmetric structures. In each structure, Aβ adopts a β-strand—turn—β-strand fold. Fibrils are stabilized by hydrogen bonds connecting individual β-strands along the fibril axis. The 3-fold symmetric fibril structure contains three stacks of Aβ molecules in a triangular form. The 2-fold symmetric fibril structure is built from two antiparallel stacks of Aβ molecules. To detect potential binding sites for the hydrophobic sulindac sulfide, we performed a packing density analysis for each Aβ structure. In fact, all models revealed large packing defects. On average, we found eight cavities per stack of four Aβ molecules in both structures (supplemental Figs. S4 and S5), clustering in five different regions within one stack (clusters #1-#5) (Fig. 3). To dissect polar from unpolar cavities, we applied the tool DOWSER (59) to calculate positions of internal water molecules that are not resolved in the NMR structures. We found that the most hydrophobic cavities in each structure clustered in the rigid core region, with calculated water occupancies of <10% in cluster #2 and <30% in cluster #3 that are located on both sides of Phe-19 (supplemental Table S3). The 3-fold symmetric structure seemed to be more tightly packed around clusters #2 and #3, featuring fewer cavities in comparison with the 2-fold symmetric structure. At the same time, other clusters in the 3-fold symmetric structure seemed to contain a larger numbers of cavities. Clusters of polar cavities were found in the flexible turn region between residues Glu22 and Ile31 (cluster #1) and toward the termini of the peptide sequence (cluster #4). The two structures differed in their interface architecture between the termini to the adjacent loop region. Although, in the 3-fold symmetric structure, cavities of cluster #4 were stabilized through interactions with the loop, in the 2-fold symmetric structure, fewer polar cavities were found because the structure opened and cavities could become exposed. Cluster #5 (around M35) with polar cavities was located at the intersheet/contact region between termini and the loop region (3-fold symmetric structure) or at the interface of the antiparallel β-sheets (2-fold symmetric structure). In the intersheet region, where the contact interface between 3-fold and 2-fold symmetric structure was apparently different, the amount of cavities per stack (of four Aβ molecules) and their polarity did not differ. The amount of cavities and the respective water contents per cluster #1-#5 are shown in supplemental Table S3.
Clusters #2 and #3 contain large hydrophobic cavities, which could, in principle, bury a sulindac sulfide molecule. The exact size of the individual cavities depends on the rotameric state of Phe19. The 3-fold symmetric and the 2-fold symmetric structures differ slightly in the position of Phe19 with respect to the registry of the second β-sheet containing residues Ile32 to Val36 (supplemental Fig. S6). In the 3-fold symmetric structure, Phe19 is oriented more toward Ile32, whereas, in the 2-fold symmetric structure, Phe19 is pointing toward Leu34 and is, therefore, located more central in the core region. Surprisingly, the size, polarity, and distribution of the detected cavities is very similar in all analyzed structural models even though there are slight differences between the two models.
To test whether the hydrophobic cavities in clusters #2 and #3 are indeed suitable binding sites for sulindac sulfide, we applied an induced fit docking approach where the sulindac sulfide and side chains of Aβ were kept flexible. The size and accessibility of these cavities depends on the rotameric state of Phe19. In fact, docking into cluster #3 of the 3-fold symmetric structure allows identification of two scenarios that fit to the distance restraints obtained from the NMR analysis. In particular, we find that the distance of the sulindac sulfide 19F atom to the methyl groups of Ile32, Leu34, or Val36 is smaller than 6 Å. In pose 1, sulindac sulfide lies in the Gly33 groove and is orientated along the fibril axes, being close to Ile32 and Leu34. The aromatic side chains of Phe19 are directed toward Ala30 (Fig. 4a, top panel). In pose 2, sulindac sulfide has rotated into a position with its conjugated ring system parallel to the β-sheets and orthogonal to Phe19 so that the 19F atom approaches Leu34 and Val36. (Fig. 4a, bottom panel). Both poses suggest an aromatic π stacking interaction between the conjugated ring system of sulindac sulfide and the Phe19 side chain. Similar interactions are obtained from docking to cluster #2 of the 2-fold symmetric structure (Fig. 4b). In pose 1, sulindac sulfide is orientated along the fibril axes, with its conjugated ring system parallel to Phe19, analogous to pose 1 in the 3-fold symmetric structure. Again, the 19F atom is positioned in the Gly33 groove close to Ile32 and Leu34. In contrast to the 3-fold symmetric structure, Phe19 is positioned here on the other side of sulindac sulfide, close to Leu34, thereby shielding Val36 so that sulindac sulfide cannot change into a position to contact Val36 (Fig. 4b, top panel). An additional docking to cluster #3 yields poses 2 and 3, where the normal of the conjugated ring system of sulindac sulfide is orientated perpendicularly to the fibril axes. Thereby, sulindac sulfide extends into the terminal region (#4). The conjugated ring system is positioned parallel to the β-sheets and orthogonal to Phe19. Sulindac sulfide is either orientated in a way that the 19F atom faces Phe19, Leu34, and Val36 (Fig. 4b, center panel) or that the 19F atom is oriented toward the fibril exterior, contacting Val36 and Val39 (Fig. 4b, bottom panel).
FIGURE 4.
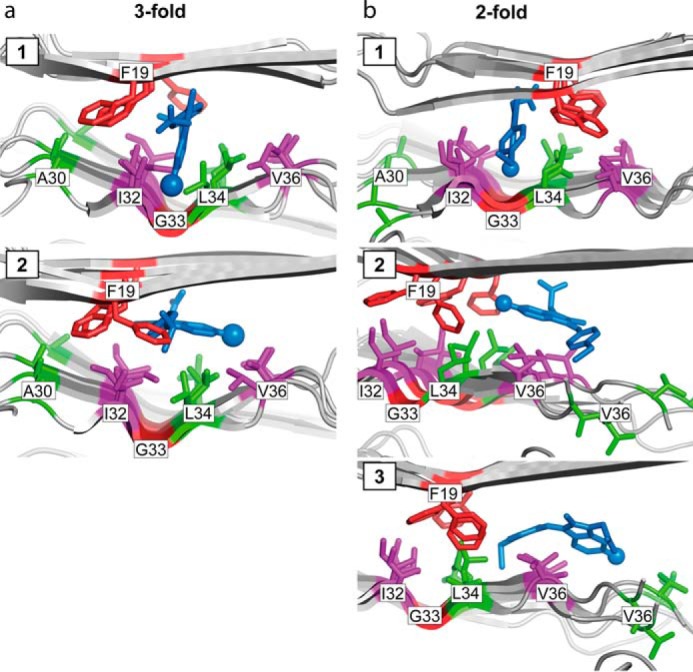
Induced fit docking of sulindac sulfide to Aβ. a and b, induced fit docking of sulindac sulfide to the hydrophobic cavity cluster #3 of the 3-fold symmetric Aβ1–40 fibril structure (PDB code 2LMP, model 1, poses 1 and 2) (54) (a) and to the hydrophobic cavity clusters #2 and #3 of the 2-fold symmetric Aβ1–40 structure (PDB code 2LMN, model 1; for cluster #2, pose 1 and cluster #3, poses 2 and 3) (53) (b). Sulindac sulfide is depicted as blue sticks and the protein backbones as a schematic. Residues within 6 Å of the 19F atom of sulindac sulfide are depicted as sticks in purple for the cavity cluster defining residues Ile32 and Val36 and in green for Ala30, Leu34, and Val39. Phe19 and Gly33 are colored in red.
Discussion
Sulindac sulfide-incubated Aβ fibrils are highly similar to control fibrils, implying that sulindac sulfide has no significant effect on fibril structure. Both the NMR chemical shift patterns (Fig. 1c and supplemental Fig. S1) and the morphology in TEM images (Fig. 1b) are maintained. Analysis of 13C chemical shifts predicts β-strands as the main secondary structural element in both fibril preparations (supplemental Fig. S7). In addition, chemical shift analysis by torsion angle likeliness obtained from shift and sequence similarities+ (TALOS+) (69) yields secondary structural propensities that predict the presence of two β-strands typically observed in specific regions within Aβ1–40 fibrils (44, 53, 54, 67, 70) and oligomers (71, 72). Furthermore, we find, by TEDOR experiments, that the Asp23-Lys28 salt bridge remains intact in the presence of sulindac sulfide (Fig. 2b). Even though the overall fibrillar character is maintained, we observe defined chemical shift changes, potentially indicating local conformational changes (Fig. 1d). Previous reports have stated that small molecules such as curcumin (39) are able to disrupt the characteristic salt bridge in Aβ1–42 fibrils. Judging from EM data, curcumin has a more drastic effect on the general fibril architecture compared with sulindac sulfide because it destroys Aβ1–42 fibrils (39). We note that only one set of resonances is observed for the sulindac sulfide-incubated Aβ fibrils. No resonance splitting is observed for cross-peaks. Rather, resonances move to new positions, indicating that each peptide in a fibril interacts specifically with one or more NSAID molecules.
Upon addition of sulindac sulfide, CSPs are observed in particular for hydrophobic residues such as Val18, Phe19 and Phe20, Gly33, and Met35 but also for the polar side chain of Lys16. The most dramatic CSPs are detected for Lys16 and Gly33 (Fig. 1d). 13C-19F REDOR experiments yield unambiguous distance restraints. The REDOR experiments show that sulindac sulfide binds in the vicinity of methyl groups of Val18 or Val39 as well as Ile31 and Ile32 Cγ2.
To identify trends and avoid bias, we use both the 2-fold symmetric (PDB code 2LMN) (53) and the 3-fold symmetric (PDB code 2LMP) (54) Aβ1–40 fibrils as reference structures for modeling and docking experiments. The polymorphism of these structures may differ from the fibrils investigated in this study. However, they show the highest similarities of all three wild-type Aβ1–40 fibril structures currently available (53, 54, 64). Within the two Aβ fibril structural ensembles, several hydrophobic cavities are detected that are large enough to potentially host a sulindac sulfide molecule. We did not find significant deviations in the distribution, size, and polarity of the cavities between the analyzed structural ensembles and models. Therefore, changes in fibril polymorphism do not significantly affect the distribution of hydrophobic patches in amyloid fibrils. We conclude that the two employed structures provide a suitable basis for docking experiments, which is also supported by experimental NMR data, although high-resolution structures of a better defined polymorph will allow more accurate docking in the future.
An induced fit docking reveals that the NSAID can interact with fibrils in three different ways, and all models fulfill the NMR restraints. The apparently ambiguous REDOR restraints suggest that more than one sulindac sulfide molecule might be involved in binding. In the docking poses that are in best agreement with the NMR restraints, sulindac sulfide intercalates between the two β-strands of the Aβ fibril, with the long axis of the molecule either parallel (Fig. 4, a and b, pose 1) or perpendicular (Fig. 4, a, pose 2, and b, poses 2 and 3) to the fibril axis. Therefore, residue Phe19 seems to play a crucial role in sulindac sulfide binding because its rotameric state has an influence on the size and shape of cavities in clusters #2 and #3. Furthermore, the aromatic side chain is involved in π stacking with the conjugated ring system of the NSAID.
Pose 1 of the docking approach suggests that sulindac sulfide fits into the groove formed by Gly33 (Fig. 4, a and b, top panels). Therefore, the large CSPs may be attributed to Gly33 backbone atoms experiencing a change in chemical environment or even undergoing conformational changes to accommodate the NSAID. In theoretical studies, this glycine residue has been suggested to be involved in oxidation of Aβ1–42 because of its close proximity to the side chain of Met35 (73, 74). Furthermore, Gly33 has been reported as the key amino acid for Aβ toxicity and to be responsible for driving Aβ into neurotoxic conformations (75). Both residues, Gly33 and Met35, have been hypothesized to stabilize reactive oxygen species (76, 77). Sulindac sulfide might, therefore, act by binding to the hydrophobic pocket in the vicinity of Gly33, and preventing oxidation of the Aβ peptide. Oxidation of Aβ1–42 reduces fibril assembly and aggregation because of the increased polarity introduced by the methionine sulfoxide (78). In accordance, we find that the Aβ-sulindac sulfide complex exists in a stable fibrillar state.
CSPs for Lys16 may be explained by a recent docking study involving sulindac sulfide and Aβ fibrils that suggested that sulindac sulfide may bind weakly to a shallow, solvent-exposed pocket in the vicinity of Lys16 and Val18 and does not interfere with fibrillation (30). This binding mode may, in addition, account for the large REDOR signal detected for the overlapping resonances Val18/Val39 Cγ (Fig. 2a). We cannot exclude docking of sulindac sulfide to the fibril surface because our REDOR and CSP data are also in agreement with the blind docking model proposed by Yesuvadian et al. (30).
Sulindac sulfide has been reported to form colloidal aggregates above a critical micelle concentration in solution (28). This phenomenon is commonly observed for hydrophobic compounds (79, 80). The size of small-molecule colloidal particles is typically on the order of 50–600 nm (28). These colloids bind unfolded proteins in a promiscuous manner (79, 80) and have been shown to lead to precipitation and inhibition of protein function (81–83). However, the effect of sulindac sulfide on Aβ fibril chemical shifts reported here indicates not a promiscuous but a specific interaction. Aggregates of the compound are observed in TEM images (Fig. 1b), implying the presence of colloids in solution. This is expected because the concentration used (250 μm) lies above the critical micelle concentration of 50–100 μm (28). To account for the experimental single set of resonances, we must assume that individual NSAID molecules dissociate from the colloidal complexes and interact specifically with Aβ fibrils.
Binding of sulindac sulfide in different cavities for different Aβ peptides would result in splitting of the resonances and line-broadening. We observe, however, narrow lines, indicating that the small molecule must either bind simultaneously to different cavities or exchange between these cavities. Relaxation experiments will be carried out in the future to differentiate between these two scenarios.
In conclusion, we suggest that the NSAID sulindac sulfide is able to interact with Aβ fibrils in a rather specific manner. We find that several cavities can accommodate a sulindac sulfide molecule. This is supported by defined CSPs and 13C-19F REDOR contacts. Sulindac sulfide does not induce drastic architectural changes to the non-toxic fibrillar structure, as indicated by NMR and EM. In addition, the characteristic Asp23-Lys28 salt bridge and the length and positioning of the β-strands are not affected. Molecular modeling suggests that sulindac sulfide intercalates between the two β-strands at, presumably, more than one position. The presented data contribute to the elucidation of the mechanism by which small molecules bind insoluble amyloids. This understanding is crucial for the design of pharmacologically relevant molecules that interfere with Aβ species and that might, in the future, be employed for the treatment of Alzheimer disease.
Author Contributions
E. P. expressed and purified isotopically labelled peptide samples, designed and performed NMR experiments, and carried out the data analysis. R. S. implemented the solid-state NMR experiments. J. M. L. d. A. recorded the initial experiments and assisted with data analysis. G. A. O. carried out the fluorine REDOR experiments. G. M. performed biochemical experiments. H. J. B. and P. W. H. performed and analyzed the molecular modeling and docking. All authors discussed the results. B. R. and E. P. conceived the project and wrote the paper with input from all authors.
Supplementary Material
Acknowledgments
This work was performed in the framework of SFB-1035/Project-B07 and SFB740/Project-B6 (German Research Foundation). The computer time necessary for calculation of docking experiments was provided by Norddeutscher Verbund für Hoch- und Höchstleistungsrechner Project bec00085.
This work was supported by the Center for Integrated Protein Science Munich, the Helmholtz-Gemeinschaft, and the Deutsche Forschungsgemeinschaft (Grant Re1435). This work was also supported by Canadian Institute of Health Research Grant MOP-133411 and infrastructure (to G. M.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Results, Figures S1–S8, Tables S1–S3, and References.
- Aβ
- amyloid-β
- APP
- amyloid precursor protein
- NSAID
- non-steroidal anti-inflammatory drug
- DMSO
- dimethyl sulfoxide
- MAS
- magic angle spinning
- TEDOR
- transferred echo double resonance
- REDOR
- rotational echo double resonance
- TEM
- transmission electron microscopy.
References
- 1.Bucciantini M., Giannoni E., Chiti F., Baroni F., Formigli L., Zurdo J., Taddei N., Ramponi G., Dobson C. M., and Stefani M. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511 [DOI] [PubMed] [Google Scholar]
- 2.Haass C. (2004) Take five: BACE and the γ-secretase quartet conduct Alzheimer's amyloid β-peptide generation. EMBO J. 23, 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang J., Lemaire H. G., Unterbeck A., Salbaum J. M., Masters C. L., Grzeschik K. H., Multhaup G., Beyreuther K., and Müller-Hill B. (1987) The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736 [DOI] [PubMed] [Google Scholar]
- 4.Olsson F., Schmidt S., Althoff V., Munter L. M., Jin S., Rosqvist S., Lendahl U., Multhaup G., and Lundkvist J. (2014) Characterization of intermediate steps in amyloid β (Aβ) production under near-native conditions. J. Biol. Chem. 289, 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roher A. E., Lowenson J. D., Clarke S., Woods A. S., Cotter R. J., Gowing E., and Ball M. J. (1993) β-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 10836–10840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prelli F., Castaño E., Glenner G. G., and Frangione B. (1988) Differences between vascular and plaque core amyloid in Alzheimer's disease. J. Neurochem. 51, 648–651 [DOI] [PubMed] [Google Scholar]
- 7.Selkoe D. J. (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bieschke J., Russ J., Friedrich R. P., Ehrnhoefer D. E., Wobst H., Neugebauer K., and Wanker E. E. (2010) EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bieschke J., Herbst M., Wiglenda T., Friedrich R. P., Boeddrich A., Schiele F., Kleckers D., Lopez del Amo J. M., Grüning B. A., Wang Q., Schmidt M. R., Lurz R., Anwyl R., Schnoegl S., Fändrich M., Frank R. F., Reif B., Günther S., Walsh D. M., and Wanker E. E. (2012) Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101 [DOI] [PubMed] [Google Scholar]
- 10.Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G. M., Cooper N. R., Eikelenboom P., Emmerling M., Fiebich B. L., Finch C. E., Frautschy S., Griffin W. S., Hampel H., Hull M., Landreth G., Lue L., Mrak R., Mackenzie I. R., McGeer P. L., O'Banion M. K., Pachter J., Pasinetti G., Plata-Salaman C., Rogers J., Rydel R., Shen Y., Streit W., Strohmeyer R., Tooyoma I., Van Muiswinkel F. L., Veerhuis R., Walker D., Webster S., Wegrzyniak B., Wenk G., and Wyss-Coray T. (2000) Inflammation and Alzheimer's disease. Neurobiol. Aging 21, 383–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGeer P. L., and McGeer E. G. (1995) The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Brain Res. Rev. 21, 195–218 [DOI] [PubMed] [Google Scholar]
- 12.in t' Veld B. A., Ruitenberg A., Hofman A., Launer L. J., van Duijn C. M., Stijnen T., Breteler M. M., and Stricker B. H. (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med. 345, 1515–1521 [DOI] [PubMed] [Google Scholar]
- 13.Weggen S., Eriksen J. L., Sagi S. A., Pietrzik C. U., Ozols V., Fauq A., Golde T. E., and Koo E. H. (2003) Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid β 42 production by direct modulation of γ-secretase activity. J. Biol. Chem. 278, 31831–31837 [DOI] [PubMed] [Google Scholar]
- 14.Eriksen J. L., Sagi S. A., Smith T. E., Weggen S., Das P., McLendon D. C., Ozols V. V., Jessing K. W., Zavitz K. H., Koo E. H., and Golde T. E. (2003) NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ 42 in vivo. J. Clin. Invest. 112, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi Y., Hayashi I., Tominari Y., Rikimaru K., Morohashi Y., Kan T., Natsugari H., Fukuyama T., Tomita T., and Iwatsubo T. (2003) Sulindac sulfide is a noncompetitive γ-secretase inhibitor that preferentially reduces Aβ 42 generation. J. Biol. Chem. 278, 18664–18670 [DOI] [PubMed] [Google Scholar]
- 16.Wanngren J., Ottervald J., Parpal S., Portelius E., Strömberg K., Borgegård T., Klintenberg R., Juréus A., Blomqvist J., Blennow K., Zetterberg H., Lundkvist J., Rosqvist S., and Karlström H. (2012) Second generation γ-secretase modulators exhibit different modulation of Notch β and Aβ production. J. Biol. Chem. 287, 32640–32650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nadezhdin K. D., Bocharova O. V., Bocharov E. V., and Arseniev A. S. (2012) Dimeric structure of transmembrane domain of amyloid precursor protein in micellar environment. FEBS Lett. 586, 1687–1692 [DOI] [PubMed] [Google Scholar]
- 18.Chen W., Gamache E., Rosenman D. J., Xie J., Lopez M. M., Li Y. M., and Wang C. (2014) Familial Alzheimer's mutations within APPTM increase Aβ42 production by enhancing accessibility of ϵ-cleavage site. Nat. Commun. 5, 3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett P. J., Song Y., Van Horn W. D., Hustedt E. J., Schafer J. M., Hadziselimovic A., Beel A. J., and Sanders C. R. (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manrique-Moreno M., Moreno M. M., Garidel P., Suwalsky M., Howe J., and Brandenburg K. (2009) The membrane-activity of Ibuprofen, Diclofenac, and Naproxen: a physico-chemical study with lecithin phospholipids. Biochim. Biophys. Acta 1788, 1296–1303 [DOI] [PubMed] [Google Scholar]
- 21.Hüsch J., Dutagaci B., Glaubitz C., Geppert T., Schneider G., Harms M., Müller-Goymann C. C., Fink L., Schmidt M. U., Setzer C., Zirkel J., Rebmann H., Schubert-Zsilavecz M., and Abdel-Tawab M. (2011) Structural properties of so-called NSAID-phospholipid-complexes. Eur. J. Pharm. Sci. 44, 103–116 [DOI] [PubMed] [Google Scholar]
- 22.Lichtenberger L. M., Zhou Y., Jayaraman V., Doyen J. R., O'Neil R. G., Dial E. J., Volk D. E., Gorenstein D. G., Boggara M. B., and Krishnamoorti R. (2012) Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: characterization of interaction of NSAIDs with phosphatidylcholine. Biochim. Biophys. Acta 1821, 994–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kukar T. L., Ladd T. B., Bann M. A., Fraering P. C., Narlawar R., Maharvi G. M., Healy B., Chapman R., Welzel A. T., Price R. W., Moore B., Rangachari V., Cusack B., Eriksen J., Jansen-West K., Verbeeck C., Yager D., Eckman C., Ye W., Sagi S., Cottrell B. A., Torpey J., Rosenberry T. L., Fauq A., Wolfe M. S., Schmidt B., Walsh D. M., Koo E. H., and Golde T. E. (2008) Substrate-targeting γ-secretase modulators. Nature 453, 925–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botev A., Munter L. M., Wenzel R., Richter L., Althoff V., Ismer J., Gerling U., Weise C., Koksch B., Hildebrand P. W., Bittl R., and Multhaup G. (2011) The amyloid precursor protein C-terminal fragment C100 occurs in monomeric and dimeric stable conformations and binds γ-secretase modulators. Biochemistry 50, 828–835 [DOI] [PubMed] [Google Scholar]
- 25.Richter L., Munter L. M., Ness J., Hildebrand P. W., Dasari M., Unterreitmeier S., Bulic B., Beyermann M., Gust R., Reif B., Weggen S., Langosch D., and Multhaup G. (2010) Amyloid β 42 peptide (Aβ42)-lowering compounds directly bind to Aβ and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc. Natl. Acad. Sci. U.S.A. 107, 14597–14602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khalifa N. B., Van Hees J., Tasiaux B., Huysseune S., Smith S. O., Constantinescu S. N., Octave J. N., and Kienlen-Campard P. (2010) What is the role of amyloid precursor protein dimerization? Cell Adh. Migr. 4, 268–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beel A. J., Barrett P., Schnier P. D., Hitchcock S. A., Bagal D., Sanders C. R., and Jordan J. B. (2009) Nonspecificity of binding of γ-secretase modulators to the amyloid precursor protein. Biochemistry 48, 11837–11839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett P. J., Sanders C. R., Kaufman S. A., Michelsen K., and Jordan J. B. (2011) NSAID-based γ-secretase modulators do not bind to the amyloid-β polypeptide. Biochemistry 50, 10328–10342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirohata M., Ono K., Naiki H., and Yamada M. (2005) Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer's β-amyloid fibrils in vitro. Neuropharmacology 49, 1088–1099 [DOI] [PubMed] [Google Scholar]
- 30.Yesuvadian R., Krishnamoorthy J., Ramamoorthy A., and Bhunia A. (2014) Potent γ-secretase inhibitors/modulators interact with amyloid-β fibrils but do not inhibit fibrillation: a high-resolution NMR study. Biochem. Biophys. Res. Commun. 447, 590–595 [DOI] [PubMed] [Google Scholar]
- 31.Prade E., Lopez del Amo J.-M., and Reif B. (2014) Advances in Biological Solid-State NMR: Proteins and Membrane-Active Peptides (Separovic F., and Naito A., eds), pp. 533–555, The Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 32.Choi J. S., Braymer J. J., Nanga R. P., Ramamoorthy A., and Lim M. H. (2010) Design of small molecules that target metal-Aβ species and regulate metal-induced Aβ aggregation and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 21990–21995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rezaei-Ghaleh N., Andreetto E., Yan L. M., Kapurniotu A., and Zweckstetter M. (2011) Interaction between amyloid β peptide and an aggregation blocker peptide mimicking islet amyloid polypeptide. PLoS ONE 6, e20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoo S. I., Yang M., Brender J. R., Subramanian V., Sun K., Joo N. E., Jeong S. H., Ramamoorthy A., and Kotov N. A. (2011) Inhibition of amyloid peptide fibrillation by inorganic nanoparticles: functional similarities with proteins. Angew. Chem. Int. Ed. Engl. 50, 5110–5115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vivekanandan S., Brender J. R., Lee S. Y., and Ramamoorthy A. (2011) A partially folded structure of amyloid-β(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramamoorthy A., and Lim M. H. (2013) Structural characterization and inhibition of toxic amyloid-β oligomeric intermediates. Biophys. J. 105, 287–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez del Amo J. M., Fink U., Dasari M., Grelle G., Wanker E. E., Bieschke J., and Reif B. (2012) Structural properties of EGCG-induced, nontoxic Alzheimer's disease Aβ oligomers. J. Mol. Biol. 421, 517–524 [DOI] [PubMed] [Google Scholar]
- 38.Masuda Y., Fukuchi M., Yatagawa T., Tada M., Takeda K., Irie K., Akagi K., Monobe Y., Imazawa T., and Takegoshi K. (2011) Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid β-protein fibrils. Bioorg. Med. Chem. 19, 5967–5974 [DOI] [PubMed] [Google Scholar]
- 39.Mithu V. S., Sarkar B., Bhowmik D., Das A. K., Chandrakesan M., Maiti S., and Madhu P. K. (2014) Curcumin alters the salt bridge-containing turn region in amyloid β(1–42) aggregates. J. Biol. Chem. 289, 11122–11131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sato M., Murakami K., Uno M., Nakagawa Y., Katayama S., Akagi K., Masuda Y., Takegoshi K., and Irie K. (2013) Site-specific inhibitory mechanism for amyloid β42 aggregation by catechol-type flavonoids targeting the Lys residues. J. Biol. Chem. 288, 23212–23224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schütz A. K., Soragni A., Hornemann S., Aguzzi A., Ernst M., Böckmann A., and Meier B. H. (2011) The amyloid-Congo red interface at atomic resolution. Angew. Chem. Int. Ed. Engl. 50, 5956–5960 [DOI] [PubMed] [Google Scholar]
- 42.Dasari M., Espargaro A., Sabate R., Lopez del Amo J. M., Fink U., Grelle G., Bieschke J., Ventura S., and Reif B. (2011) Bacterial inclusion bodies of Alzheimer's disease β-amyloid peptides can be employed to study native-like aggregation intermediate states. ChemBioChem 12, 407–423 [DOI] [PubMed] [Google Scholar]
- 43.Walsh D. M., Thulin E., Minogue A. M., Gustavsson N., Pang E., Teplow D. B., and Linse S. (2009) A facile method for expression and purification of the Alzheimer's disease-associated amyloid β-peptide. FEBS J. 276, 1266–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez del Amo J. M., Schmidt M., Fink U., Dasari M., Fändrich M., and Reif B. (2012) An asymmetric dimer as the basic subunit in Alzheimer's disease amyloid β fibrils. Angew. Chem. Int. Ed. Engl. 51, 6136–6139 [DOI] [PubMed] [Google Scholar]
- 45.Bloembergen N. (1949) On the interaction of nuclear spins in a crystalline lattice. Physica 15, 386–426 [Google Scholar]
- 46.Szeverenyi N. M., Sullivan M. J., and Maciel G. E. (1982) Observation of spin exchange by two-dimensional Fourier transform 13C cross polarization-magic-angle spinning. J. Magn. Reson. 47, 462–475 [Google Scholar]
- 47.Hing A. W., Vega S., and Schaefer J. (1992) Transferred-echo double-resonance NMR. J. Magn. Reson. 96, 205–209 [Google Scholar]
- 48.Hing A. W., Vega S., and Schaefer J. (1993) Measurement of heteronuclear dipolar coupling by transferred-echo double-resonance NMR. J. Magn. Reson. 103, 151–162 [Google Scholar]
- 49.Castellani F., van Rossum B. J., Diehl A., Rehbein K., and Oschkinat H. (2003) Determination of solid-state NMR structures of proteins by means of three-dimensional 15N-13C-13C dipolar correlation spectroscopy and chemical shift analysis. Biochemistry 42, 11476–11483 [DOI] [PubMed] [Google Scholar]
- 50.Takegoshi K., Nakamura S., and Terao T. (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 [Google Scholar]
- 51.Gullion T., and Schaefer J. (1989) Rotational-echo double-resonance NMR. J. Magn. Reson. 81, 196–200 [DOI] [PubMed] [Google Scholar]
- 52.Jaroniec C. P., Filip C., and Griffin R. G. (2002) 3D TEDOR NMR experiments for the simultaneous measurement of multiple carbon-nitrogen distances in uniformly 13C,15N-labeled solids. J. Am. Chem. Soc. 124, 10728–10742 [DOI] [PubMed] [Google Scholar]
- 53.Petkova A. T., Yau W. M., and Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry 45, 498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paravastu A. K., Leapman R. D., Yau W. M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goede A., Preissner R., and Frömmel C. (1997) Voronoi cell: new method for allocation of space among atoms: elimination of avoidable errors in calculation of atomic volume and density. J. Comput. Chem. 18, 1113–1123 [Google Scholar]
- 56.Rother K., Hildebrand P. W., Goede A., Gruening B., and Preissner R. (2009) Voronoia: analyzing packing in protein structures. Nucleic Acids Res. 37, D393–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsai J., and Gerstein M. (2002) Calculations of protein volumes: sensitivity analysis and parameter database. Bioinformatics 18, 985–995 [DOI] [PubMed] [Google Scholar]
- 58.Delaunay B. N. (1934) Sur la sphère vide. B. Acad. Sci. USSR 7, 793–800 [Google Scholar]
- 59.Zhang L., and Hermans J. (1996) Hydrophilicity of cavities in proteins. Proteins 24, 433–438 [DOI] [PubMed] [Google Scholar]
- 60.Sherman W., Day T., Jacobson M. P., Friesner R. A., and Farid R. (2006) Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 49, 534–553 [DOI] [PubMed] [Google Scholar]
- 61.Sherman W., Beard H. S., and Farid R. (2006) Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 67, 83–84 [DOI] [PubMed] [Google Scholar]
- 62.Schrödinger Release 2014-1: Maestro, version 9.7, Schrödinger, LLC, New York, NY, 2014 [Google Scholar]
- 63.Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., and Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring: 1: method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 64.Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., and Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 66.Niu Z., Zhao W., Zhang Z., Xiao F., Tang X., and Yang J. (2014) The molecular structure of Alzheimer β-amyloid fibrils formed in the presence of phospholipid vesicles. Angew. Chem. Int. Ed. Engl. 53, 9294–9297 [DOI] [PubMed] [Google Scholar]
- 67.Bertini I., Gonnelli L., Luchinat C., Mao J., and Nesi A. (2011) A new structural model of Aβ40 fibrils. J. Am. Chem. Soc. 133, 16013–16022 [DOI] [PubMed] [Google Scholar]
- 68.Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., and Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tycko R. (2006) Molecular structure of amyloid fibrils: insights from solid-state NMR. Q. Rev. Biophys. 39, 1–55 [DOI] [PubMed] [Google Scholar]
- 71.Chimon S., Shaibat M. A., Jones C. R., Calero D. C., Aizezi B., and Ishii Y. (2007) Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid. Nat. Struct. Mol. Biol. 14, 1157–1164 [DOI] [PubMed] [Google Scholar]
- 72.Ahmed M., Davis J., Aucoin D., Sato T., Ahuja S., Aimoto S., Elliott J. I., Van Nostrand W. E., and Smith S. O. (2010) Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rauk A., Armstrong D. A., and Fairlie D. P. (2000) Is oxidative damage by β-amyloid and prion peptides mediated by hydrogen atom transfer from glycine α-carbon to methionine sulfur within β-sheets? J. Am. Chem. Soc. 122, 9761–9767 [Google Scholar]
- 74.Rauk A., and Armstrong D. A. (2000) Influence of β-sheet structure on the susceptibility of proteins to backbone oxidative damage: preference for αc-centered radical formation at glycine residues of antiparallel β-sheets. J. Am. Chem. Soc. 122, 4185–4192 [Google Scholar]
- 75.Harmeier A., Wozny C., Rost B. R., Munter L. M., Hua H., Georgiev O., Beyermann M., Hildebrand P. W., Weise C., Schaffner W., Schmitz D., and Multhaup G. (2009) Role of amyloid-β glycine 33 in oligomerization, toxicity, and neuronal plasticity. J. Neurosci. 29, 7582–7590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanski J., Varadarajan S., Aksenova M., and Butterfield D. A. (2002) Role of glycine-33 and methionine-35 in Alzheimer's amyloid β-peptide 1–42-associated oxidative stress and neurotoxicity. Biochim. Biophys. Acta 1586, 190–198 [DOI] [PubMed] [Google Scholar]
- 77.Brunelle P., and Rauk A. (2002) The radical model of Alzheimer's disease: specific recognition of Gly29 and Gly33 by Met35 in a β-sheet model of Aβ: an ONIOM study. J. Alzheimers Dis. 4, 283–289 [DOI] [PubMed] [Google Scholar]
- 78.Hou L., Kang I., Marchant R. E., and Zagorski M. G. (2002) Methionine 35 oxidation reduces fibril assembly of the amyloid aβ-(1–42) peptide of Alzheimer's disease. J. Biol. Chem. 277, 40173–40176 [DOI] [PubMed] [Google Scholar]
- 79.McGovern S. L., Caselli E., Grigorieff N., and Shoichet B. K. (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 45, 1712–1722 [DOI] [PubMed] [Google Scholar]
- 80.Coan K. E., and Shoichet B. K. (2008) Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 130, 9606–9612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hagerman A. E., and Butler L. G. (1981) The specificity of proanthocyanidin-protein interactions. J. Biol. Chem. 256, 4494–4497 [PubMed] [Google Scholar]
- 82.Feng B. Y., Toyama B. H., Wille H., Colby D. W., Collins S. R., May B. C., Prusiner S. B., Weissman J., and Shoichet B. K. (2008) Small-molecule aggregates inhibit amyloid polymerization. Nat. Chem. Biol. 4, 197–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ono K., Li L., Takamura Y., Yoshiike Y., Zhu L., Han F., Mao X., Ikeda T., Takasaki J., Nishijo H., Takashima A., Teplow D. B., Zagorski M. G., and Yamada M. (2012) Phenolic compounds prevent amyloid β-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem. 287, 14631–14643 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.