

Background: Current assays for monitoring DNA excision repair in vivo lack sensitivity.
Results: An in vivo excision assay can be used to examine repair of DNA damage induced by many carcinogens and anti-cancer agents.
Conclusion: This methodology provides a sensitive assay for studying nucleotide excision repair in vivo.
Significance: Repair of a broad range of DNA lesions can be readily observed in vivo.
Keywords: apoptosis, carcinogenesis, cell signaling, checkpoint control, DNA damage, DNA damage response, DNA repair, nucleotide excision repair, DNA damage checkpoint, chemotherapy
Abstract
DNA damage by UV and UV-mimetic agents elicits a set of inter-related responses in mammalian cells, including DNA repair, DNA damage checkpoints, and apoptosis. Conventionally, these responses are analyzed separately using different methodologies. Here we describe a unified approach that is capable of quantifying all three responses in parallel using lysates from the same population of cells. We show that a highly sensitive in vivo excision repair assay is capable of detecting nucleotide excision repair of a wide spectrum of DNA lesions (UV damage, chemical carcinogens, and chemotherapeutic drugs) within minutes of damage induction. This method therefore allows for a real-time measure of nucleotide excision repair activity that can be monitored in conjunction with other components of the DNA damage response, including DNA damage checkpoint and apoptotic signaling. This approach therefore provides a convenient and reliable platform for simultaneously examining multiple aspects of the DNA damage response in a single population of cells that can be applied for a diverse array of carcinogenic and chemotherapeutic agents.
Introduction
DNA damage is repaired by many enzyme systems that promote survival and activate cellular signaling pathways in mammalian cells including DNA damage checkpoints, which arrest cell cycle progression and apoptosis that dismantles cellular architecture (1). Commonly, these three phenomena (repair, checkpoint, and apoptosis) are analyzed separately, though it is known that the three responses interface and share some mechanistic features (1–3). Therefore, there is a need to analyze the three responses simultaneously for a deeper understanding of cellular response to DNA damage within the same spatio-temporal framework.
Ultraviolet (UV) light-induced DNA photoproducts are removed from the human genome by the nucleotide excision repair system (4–6). Recently, we developed a method for quantifying the repair of UV-induced DNA damage in vivo with high sensitivity, which can detect repair within minutes of UV irradiation (7, 8). The method consists of observing the nucleotide excision repair-specific ∼30-nt-long oligomers (“nominal 30-mers,” which are actually in the range of 24–32 nucleotides) that had previously only been visualized in vitro (9–12). The method involves cell lysis followed by immunoprecipitation of the nominal 30-mer with antibodies against the repair factor TFIIH which is bound to the oligomer or with antibodies against cyclobutane pyrimidine dimers (CPDs)4 or (6–4) photoproducts [(6–4)PPs] and labeling of the excised oligonucleotide with either radioisotope or non-radioisotopic methods (7, 8, 13). The approach in its original form has been quite useful for characterizing DNA repair in mammalian cells in vivo (7, 8, 13) and also for mapping excision repair events genome-wide (14) but has so far only been used for studying repair of UV-induced DNA damage. Because nucleotide excision repair operates on essentially all types of DNA lesions, in particular on the so-called “bulky DNA adducts” (4, 5), we suggested that with appropriate modifications the method could also be used for investigating other DNA damage repaired by nucleotide excision repair as well.
In this report we describe an improvement of the in vivo excision assay for nucleotide excision repair that does not depend on immunoprecipitation with antibodies against either repair factors or DNA lesions. This modified assay has enabled us to monitor the repair of, in addition to UV photoproducts, the damage induced by carcinogenic chemicals benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE), N-acetoxy-2-acetylaminofluorene (AAF), and formaldehyde as well as the anticancer agent cisplatin. Moreover, in this approach the sensitivity and simplicity of the system has also enabled us to analyze checkpoint and apoptosis simultaneously with repair and using the same cell extract made from only a small number of cells. This integrated approach should be useful for mechanistic studies on DNA repair, checkpoint, and apoptosis in a concerted manner and in a type of damage-independent fashion.
Materials and Methods
Cell Lines and Cell Culture
HeLa cells were obtained from the Korean Cell Line Bank of Seoul National University (Seoul, Korea). The Chinese hamster ovarian (CHO) cell lines AA8 (wild-type) and its derivative UV135 (XPG-deficient), the human melanoma cell line A375, and the human fibroblast cell line GM00200 were purchased from the American Type Culture Collection (Rockville, MD). All cell lines were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum at 37 °C in a 5% CO2 humidified incubator. Cisplatin was obtained from Sigma. BPDE and AAF were obtained from the NCI Chemical Carcinogen Reference Standard Repository (Midwest Research Institute, MO). Bleomycin and mitomycin C (MMC) were purchased from Santa Cruz Biotechnology and Abcam (Cambridge, MA), respectively. All other chemicals were purchased from Sigma.
Genotoxin Exposure
For UV exposures, cells that were grown to near confluency in 100 mm or 150 mm diameter plates were washed with phosphate-buffered saline (PBS, Gibco) and then exposed to the indicated influence of UV radiation using a germicidal lamp that emits primarily 254 nm light. For formaldehyde or MMC treatments, the culture medium was replaced with serum-free medium prior to treatment. For treatments with the other DNA-damaging agents (BPDE, AAF, cisplatin, and bleomycin), the indicated doses of each chemical compound were added directly to the culture medium.
Cell Lysis and in Vivo Excision Assay
At the indicated time points, cells were washed with ice-cold PBS and harvested with a cell scraper. Cells were pelleted by centrifugation, resuspended in a 10× packed cell volume of a Triton X-100 lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, and 1% Triton X-100), and incubated on a rotating mixer for 15 min at 4 °C. Soluble lysates were then obtained by centrifugation at 20,000 × g for 1 h and transferred to new tubes. Small portions (10–20%) of the lysates were saved for immunoblot analysis. The insoluble pellets were subjected to isolation of genomic DNA for immunoslot blot assays (described below). The soluble cell lysates were incubated with RNase A (5–10 μg/ml) and RNase T1 (5–10 units/ml) for 20 min at 37 °C and then treated with 0.15 mg/ml of proteinase K for 30 min at 55 °C. The reactions were subsequently extracted with phenol/chloroform precipitated in ethanol. After centrifugation, the pellets were washed with 70% ethanol, resuspended in TE buffer, and then subjected to labeling with terminal deoxynucleotidyl transferase and the nucleotide analog biotin-11-dUTP as described previously with a few modifications (7). Briefly, the extracted DNA was incubated with 20 units of terminal deoxynucleotidyl transferase (New England Biolabs), 10 μm of biotin-11-dUTP (Biotium, Inc.), and 0.25 mm CoCl2 in 50 μl of 1× Terminal Transferase Reaction Buffer (New England Biolabs) for 30 min at 37 °C. The reactions were stopped by addition of 10 mm EDTA, incubated with RNase A (10–20 μg/ml)/RNase T1 (30–50 units/ml) for 20 min at 37 °C, and then treated with 0.4 mg/ml of proteinase K in the presence of 0.4% SDS. The reactions were subsequently subjected to phenol/chloroform extraction followed by ethanol precipitation. The labeled DNAs were loaded onto 12% TBE-urea gels and run in 1× TBE at 150 V. Following gel electrophoresis, the DNAs were transferred onto a modified nylon membrane (Zeta-Probe membranes by Bio-Rad or Biodyne membrane by Pierce) in 0.5× TBE buffer at 300 mA for 1 h and then fixed to the membrane by UV cross-linking. Detection of biotin-labeled DNA was achieved with the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific) using an ImageQuant LAS 4000 Mini apparatus (GE Healthcare Life Sciences). The relative intensities of the bands were analyzed by using ImageQuant TL Software (GE Healthcare Life Sciences).
Immunoslot Blot Assay
The insoluble pellets obtained during the preparation of cell lysates (described above) were resuspended in a 10× packed cell volume of PBS and then incubated with 0.5 μg/ml of RNase A for 2 min at room temperature. Proteinase K was added to a final concentration of 0.25 μg/ml. Genomic DNA was then isolated using the QIAamp DNA Mini kit (Qiagen) and then subjected to immuno-slot blot analysis as described previously (15, 16). Briefly, purified genomic DNA was denatured and then loaded onto a nitrocellulose membrane. The membrane was baked and then probed with anti-CPD (Kamiya Biomedical) or anti-(6–4)PP (Cosmo Bio) antibodies. Following incubation with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody, chemiluminescent signals were detected with ECL reagent (GE Healthcare Life Sciences) using an ImageQuant LAS 4000 Mini apparatus (GE Healthcare Life Sciences). The relative intensities of the signals were quantified with ImageQuant TL Software (GE Healthcare Life Sciences). To confirm equal loading of genomic DNA, membranes were stained with SYBR Gold (Invitrogen).
Protein Immunoblotting
Soluble cell lysates were separated by SDS-PAGE and transferred to Hybond ECL membranes (GE Healthcare Life Sciences), probed with the indicated antibodies, and detected with ECL reagent (GE Healthcare Life Sciences) using an ImageQuant LAS 4000 Mini apparatus (GE Healthcare Life Sciences). The antibodies were obtained from commercial sources. Anti-p53 antibody was purchased from Santa Cruz. All of the other antibodies were obtained from Cell Signaling Technology.
Immunoprecipitation of Excised Oligomers
Detection of excision products containing specific UV photoproducts was done as previously described (7, 8). Briefly, 5 μl of protein G Dynabeads slurry (Invitrogen) and anti-rabbit Dynabeads slurry (Invitrogen) were mixed, washed with Wash Buffer I (20 mm Tris-Cl, pH 8.0, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100, and 0.1% SDS), resuspended in 20 μl of IP buffer (20 mm Tris-Cl, pH 8.0, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100, and 0.5% sodium deoxycholate) containing 1 μl of rabbit anti-mouse IgG (Abcam) and anti-CPD or anti-(6–4)PP antibody, and incubated for 3 h at 4 °C. The isolated beads were incubated with the purified DNA in 100 μl of IP buffer overnight at 4 °C and then subjected to sequential washes with Wash Buffer I, Wash Buffer II (20 mm Tris-Cl, pH 8.0, 2 mm EDTA, 500 mm NaCl, 1% Triton X-100, and 0.1% SDS), Wash Buffer III (10 mm Tris-Cl pH 8.0, 1 mm EDTA, 150 mm LiCl, 1% Nonidet P-40, and 1% sodium deoxycholate), Wash Buffer IV (100 mm Tris-Cl pH 8.0, 1 mm EDTA, 500 mm LiCl, 1% Nonidet P-40, and 1% sodium deoxycholate), and twice with TE buffer (10 mm Tris-Cl pH 8.0, 1 mm EDTA). The oligonucleotides containing UV photoproducts were eluted with 100 μl of Elution Buffer (50 mm NaHCO3, 1% SDS, and 20 μg/ml of glycogen) at 65 °C for 15 min, followed by phenol/chloroform extraction and ethanol precipitation. The isolated oligonucleotides were then 3′-end labeled with biotin and analyzed as described above.
Results and Discussion
Simple Method for Monitoring DNA Repair, Checkpoint, and Apoptosis from a Single Cell Culture
We have previously described a method for isolation and quantification of excised oligonucleotides (nominal 30-mers) generated in vivo by nucleotide excision repair using a variety of cell lysis methods (7, 8, 13). We have expanded the utility of this assay by using a milder cell lysis method and by using both the soluble and insoluble fractions to monitor repair, the checkpoint response, and apoptosis.
A schematic of our approach is shown in Fig. 1. Cells are exposed to UV (or another DNA-damaging agent) and following incubation periods of various lengths cells are lysed in an isotonic buffer containing a non-ionic detergent and then centrifuged to separate the soluble nominal 30-mer excision products and soluble cellular proteins from the insoluble fraction which contains chromatin and cellular organelles. Genomic DNA is purified from this insoluble fraction and then probed by immuno-slot blotting with DNA lesion-specific antibodies to monitor the removal of damage from the genome.
FIGURE 1.
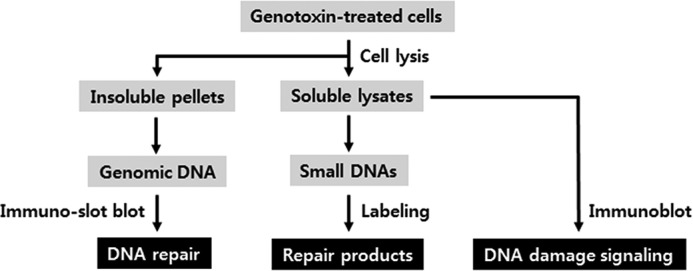
An integrated approach for monitoring the repair of genomic DNA damage, the generation of excised oligonucleotide repair products, and the activation of DNA damage signaling pathways in a single culture of cells. Following the exposure of cells to DNA-damaging agents, cells are lysed with a non-ionic detergent and soluble and insoluble fractions separated by centrifugation. The insoluble pellet is subjected to extraction of genomic DNA to measure the presence of lesions via immuno-slot blot assay. The soluble cell lysate is processed for extraction and visualization of the oligonucleotide products of excision repair or used in immunoblot assays for monitoring DNA damage response signaling (checkpoint and apoptosis).
The soluble fraction is used to monitor DNA repair and various DNA damaging signaling pathways, including DNA damage checkpoints that delay cell cycle progression and apoptosis, which induces cell death in damaged cells. DNA repair activity is examined by detecting the excised, damage-containing oligonucleotide products of nucleotide excision repair that are released from genomic DNA during repair. The remaining soluble lysate is probed for DNA damage checkpoint activation by immunoblotting for phosphorylation of the DNA damage checkpoint kinases Chk1 and Chk2 and for apoptosis by immunoblotting for proteolytic cleavage of caspase 3 and poly-ADP-ribose polymerase (PARP).
An application of this method using UV-irradiated HeLa cells is shown in Fig. 2. Cells were harvested and lysed at the indicated time points following irradiation. Soluble and insoluble fractions were then prepared and used to probe for nucleotide excision repair. Fig. 2A shows repair as measured by the chemiluminescence signal of biotin-labeled excision products that were either probed directly or immunoprecipitated with either (6–4) photoproduct or CPD antibodies. Fig. 2B shows the repair of the same photoproducts as measured by the remaining damage in chromosomal DNA by immuno-slot blot (15). Several points of interest are evident in this figure. First, the post-labeling/chemiluminescence method is quite sensitive, capable of detecting repair at the earliest time points analyzed (15 min) when repair as probed by slot-blot of the genomic DNA fraction is inconclusive because while the excised oligonucleotide assay measures repair with virtually zero background the immuno-slot blot assay measures repair as the difference between two large numbers (15) and it is not capable of unambiguously detecting repair when only a small fraction of the damage is repaired. This is in particular evident in the case of CPD repair, which is virtually undetectable for up to 4 h by slot blot assay (Fig. 2B). In contrast, the repair of CPD is detectable at the earliest time point (15 min) by the excision assay and follows an essentially linear kinetics afterward (Fig. 2A). Secondly, the (6–4)PP is excised at a much faster rate than CPDs, in agreement with previous in vivo (17) and in vitro (18) observations. During the first hour following UV irradiation, the (6–4)PP repair increases linearly as measured by both post-excision labeling/chemiluminescence assay and the slot blot assay. After the 1-h time point the excised oligomer signal gradually decreases because as we have previously reported the excised oligomer is degraded to small oligonucleotides containing the photoproducts that are no longer detectable by the excision assay, which measures mostly the primary excision products of 24- to 32-nt in length and the primary degradation products of ∼20-nt but not smaller fragments (7, 8, 13). Finally, we note that the improvement we have made in the excision assay has enabled us to detect the nominal 30-mer without immunoprecipitation and the mild lysis procedure has reduced the background at higher molecular weight region of the gel, which arises from genomic contamination in the soluble fraction.
FIGURE 2.

Analysis of genomic DNA, excised oligonucleotide repair products, and DNA damage checkpoint signaling in UV-irradiated cells. A, analysis of excised oligomers released from genomic DNA following UV irradiation. HeLa cells were exposed to 10 J/m2 of UV (254 nm) light and harvested at the indicated time points. Following cell lysis and centrifugation the soluble fractions of the cells were processed for isolation and labeling of excised oligonucleotide repair products. To measure the total excised oligomers, the purified small DNA molecules were directly labeled with biotin, separated by gel electrophoresis, transferred to a membrane, and then detected with HRP-conjugated streptavidin (left). For detection of excision products containing (6–4)PPs or CPDs, the purified small DNA molecules were immunoprecipitated with anti-(6–4)PP (middle) or anti-CPD (right) antibodies prior to biotinylation. Data are presented as the mean values ± S.D. obtained from at least three independent experiments. B, immuno-slot blot assay for measuring the removal of photoproducts from genomic DNA. Genomic DNA was isolated from the insoluble pellets in A and then used in immuno-slot blot assays with antibodies against (6–4)PPs and CPDs. The Sybr-Gold staining shows equal loading of total genomic DNA. A quantitative analysis of immuno-slot blot assays in which the signal at time zero was set to 100 is also shown. The average (and S.D.) level of photoproduct removal from three independent experiments is shown. C, analysis of UV-induced DNA damage checkpoint signaling. A small portion of the soluble fractions prepared in A were analyzed by SDS-PAGE and immunoblotting with the indicated antibodies.
Fig. 2C shows that the soluble fraction that was used to detect the excision product can also be used to probe for checkpoint response as measured by the rapid Chk1 (Checkpoint kinase 1) and p53 phosphorylation followed by the slower Chk2 phosphorylation, which reflects activation of the DNA damage sensor kinases ATR and ATM by stalled replication forks and double-strand breaks, respectively (2, 19, 20). Finally, Fig. 2C also shows that the soluble fraction can be used to probe for activation of apoptotic signaling, which is monitored by immunoblotting with antibodies that recognize cleavage products of the apoptotic protease caspase-3 and one of its major targets, PARP (Poly-ADP-ribose polymerase). To recapitulate, gentle lysis of UV irradiated cells followed by separation of the soluble fraction can be used to analyze the three main cellular responses to DNA damage: repair, checkpoint, and apoptosis.
Analysis of Cellular Response to Chemical Carcinogens
Our assay for detecting the nominal 30-mer generated by nucleotide excision repair has so far been used only for measuring repair of UV-induced photoproducts (7, 8, 13). Because nucleotide excision repair operates on virtually all base lesions and in particular on bulky base adducts generated by numerous chemical carcinogens (4–6), we wished to investigate whether the assay can be used to measure repair of damage caused by those chemical carcinogens as well. Thus, we analyzed the repair of damage caused by two of the most commonly used experimental carcinogens, BPDE and AAF. Both of these carcinogens generate bulky adducts on guanine residues that are repaired by nucleotide excision repair (21–23). Fig. 3, A and B shows that excision of damage induced by these carcinogens can be detected in a dose-dependent manner in HeLa cells. Fig. 3A (middle panel) shows a longer exposure of the BPDE excision gel which reveals that the excised oligonucleotides were detectable with a dose as low as 0.05 μm of this carcinogen. Moreover, immunoblot analysis of the same lysates that were used for detecting the excised BPDE oligonucleotides showed that Chk1 was similarly phosphorylated in a dose-dependent manner.
FIGURE 3.

Detection of excised oligomers containing bulky base adducts in vivo. A, HeLa cells were treated with different concentrations of BPDE (0.01–10 μm), harvested 1 h later, and then processed for analysis of excision products and DNA damage checkpoint signaling. For UV-induced excision products, cells were exposed to 10 J/m2 of UV and harvested after 1 h of incubation. Immunoblotting was performed with antibodies against the indicated proteins (bottom). B, CHO wild-type (CHO-WT) and XPG-deficient (CHO-XPG) cells were treated with different concentrations of BPDE (0.05, 0.1, 0.5, 1, 5 μm) for 1 h and then analyzed for excision and DNA damage checkpoint signaling. C, A375 cells were treated for 1 h with BPDE (0, 1.25, 2.5 μm) and analyzed for excision repair. For detection of UV-induced excision products, cells were exposed to 10 J/m2 of UV and then harvested 1 h later. D, normal human fibroblasts (GM00200 cells) were treated for 1 h with BPDE (0, 0.25, 0.5, 1 μm) and analyzed for excision. E, HeLa cells were treated with AAF (1–10 μm) for 1 h and analyzed for excised oligonucleotides. Lane 6 represents the excised oligonucleotides generated in HeLa cells exposed to 10 J/m2 of UV and then harvested 1 h later.
We investigated the general applicability of the method to mammalian cell lines by additional excision assays. First, we show that a WT-CHO cell line also generates the nominal 30-mer in a BPDE dose-dependent manner while the XPG mutant CHO cell line fails to produce the excision products (Fig. 3C) consistent with the specificity of the assay. Secondly, all commonly used human cell lines tested similarly generate the nominal 30-mer upon treatment with this carcinogen. Fig. 3, D and E show that the excision repair-specific oligomers were readily observed in BPDE-treated human melanoma (Fig. 3D) and fibroblast (Fig. 3E) cell lines.
Kinetics of Formation and Degradation of the Excised Oligonucleotide
Previously, we found that the primary excision products (nominal 30-mer) of UV-induced photoproducts were degraded to smaller oligonucleotides of <20 nt with a half-life of about 1 h (8). We wished to determine if this also occurred with DNA damage induced by chemical carcinogens. Hence, we followed the time course of the appearance and disappearance of BPDE excision products. Fig. 4A shows that the level of BPDE nominal 30-mers reaches a maximum intensity at 1 h following treatment and then goes down with a half-life of ∼1 h. For comparison, in Fig. 1B we show the kinetics of formation and decay of (6–4) photoproduct nominal 30-mer that was determined in a previous study (7, 8). As is apparent, the kinetics of formation and degradation of nominal 30-mers carrying BPDE or (6–4) photoproducts are very similar, indicating that the BPDE-guanine adduct is repaired as efficiently as the (6–4) photoproduct. Interestingly, analyses of checkpoint/apoptosis response of BPDE treated cells (Fig. 4A, bottom panel) reveal Chk1 phosphorylation is an early response to DNA damage, consistent with the notion that the ATR-Chk1 axis is activated by ssDNA signal generated by stalled replication forks or by nucleotide excision repair gaps (19, 24) whereas Chk2 phosphorylation represents activation of the ATM-Chk2 axis by double-strand breaks which appear at slower kinetics as a consequence of replication fork collapse or apoptosis-initiated chromatin fragmentation (2, 25). Indeed, in contrast to the rapid Chk1 phosphorylation, Chk2 phosphorylation appears to exhibit kinetics similar to that of PARP cleavage (Fig. 4A, bottom).
FIGURE 4.

Time-course analysis of excision generation following BPDE treatment. A, HeLa cells were treated with 1 μm of BPDE, harvested at the indicated time points, and then processed for analysis of excised oligonucleotides. Immunoblotting was performed with antibodies against the indicated proteins (bottom). B, quantitative analysis (average and standard deviation) of three independent experiments performed as described in A. The highest BPDE-excision product signal was arbitrarily set to a value of 100 for normalization of the signals from other time points.
Simultaneous Analysis of Repair, Checkpoint, and Apoptosis in Response to Anti-cancer Drugs
The majority of chemotherapeutic drugs kill cancer cells by damaging DNA and hence it is of importance to be able to analyze cellular responses to DNA damage by simultaneously analyzing for repair, checkpoint, and apoptosis. We carried out such analyses with the three most commonly used anti-cancer drugs, cisplatin, mitomycin C, and bleomycin.
Cisplatin introduces DNA damage in the form of Pt-GpG, Pt-ApG, and Pt-GpXpG intrastrand and G-Pt-G interstrand crosslinks (26). In vitro studies have demonstrated that intrastrand crosslinks induced by cisplatin are removed from DNA in the form of short oligomers by the nucleotide excision repair system (26–30). We therefore applied our combined DNA damage response assay to cisplatin treated cells. Fig. 5, A and B shows that the nominal 30-mer of nucleotide excision repair is generated by cisplatin in a dose-dependent manner and in parallel with excision repair, checkpoint activation, and apoptosis can be detected by immunoblotting for Chk1 and Chk2 phosphorylation and PARP cleavage, respectively (Fig. 5A, bottom panel). As in the case of BPDE damage, cisplatin damage excision is also detectable in other mammalian cell lines including human fibroblasts (Fig. 5C) and WT-CHO but not XPG mutant-CHO cells (Fig. 5D), as would be expected from the specificity of the in vivo excision assay. Interestingly, when the formation and degradation kinetics of cisplatin oligomer excision products were analyzed (Fig. 5, E and F) they exhibited CPD excision product-like behavior (7, 8) in contrast to BPDE oligomers, which demonstrated (6–4) photoproduct-like behavior (see Fig. 4). The cisplatin adducts are removed at a slow rate and as a consequence the primary excision products are detectable for up to 6 h after which degradation dominates and the total detectable excised oligomers decay, as was observed with CPDs in previous studies (7, 8, 13). Finally, we note that even though cisplatin makes G-Pt-G interstrand crosslinks (26), our post-excision labeling assay does not detect products that could be assigned to interstrand crosslink repair.
FIGURE 5.

Examination of excision repair products induced by cisplatin. A, HeLa cells were treated for 4 h with cisplatin (0.1, 0.3, 0.6, 0.9 mm) and then analyzed for excision repair. Lane 1 shows the excised oligonucleotides that were generated in HeLa cells following exposure to 5 J/m2 of UV and then harvested after 30 min incubation. Immunoblotting was performed with antibodies against the indicated proteins (bottom). B, quantitative analysis (average and standard deviation) of three independent experiments as shown in A. C, human melanoma cells (A375) were treated with different concentrations of cisplatin, harvested 4 h later, and then analyzed for excision products. D, CHO wild-type (CHO-WT) and XPG-deficient (CHO-XPG) cells were treated with different concentrations of cisplatin (0.1, 0.3, 0.6, 0.9 mm), harvested 4 h later, and then processed for analysis of excision repair. Small portions of cell lysates were analyzed by immunoblotting with antibodies against the indicated proteins (bottom). E, HeLa cells were treated with cisplatin (0.6 mm), harvested at the indicated time points, and then analyzed for excision products. Small portions of cell lysates were analyzed by immunoblotting with antibodies against the indicated proteins (bottom). F, quantitative analysis (average and standard deviation) of three independent assays as shown in E.
Mitomycin C is another drug that makes both intrastrand and interstrand lesions by attacking the N2 position of guanine (31, 32). Fig. 6A shows that mitomycin adducts are removed from DNA by the canonical dual incision. As in the case of cisplatin interstrand crosslinks there is no evidence of mitomycin C interstrand crosslink repair by our post-excision labeling assay. Treatment of cells with bleomycin, which introduces breaks in DNA but does not form stable base adducts (33, 34), did not generate the nominal 30-mer diagnostic of nucleotide excision repair but caused large scale DNA fragmentation (Fig. 6B). Consistent with the general notion that UV-mimetic agents primarily activate the ATR-Chk1 axis and secondarily the ATM-Chk2 pathway and the reverse order for radiomimetic agents (1, 19, 20, 25), we observe a strong and relatively rapid Chk1 phosphorylation by mitomycin treatment and conversely strong Chk2 phosphorylation at relatively low dose of bleomycin and only a weak signal of Chk1 phosphorylation at higher doses of the drug (Fig. 6, A and B, lower panels).
FIGURE 6.

Detection of excised oligonucleotides following treatment with the anti-cancer compounds mitomycin C (MMC) and bleomycin. A, HeLa cells were treated with the indicated concentrations of mitomycin C (1, 10, 30, 100 μm), harvested after 1 h incubation, and then analyzed for the generation of excised oligonucleotides. Immunoblotting was performed with antibodies against the indicated proteins (bottom). B, HeLa cells were treated with bleomycin (0.7, 3, 17, 83 μm) for 1 h and then processed for detection of excised oligonucleotides. Lane 7 in A and B show excision repair products generated within 1 h after the exposure HeLa cells to 5 J/m2 of UV. Immunoblotting was performed with antibodies against the indicated proteins (bottom).
Analysis of Formaldehyde Induced DNA Damage Response
Many industrial, household, and medicinal products contain formaldehyde-based polymers from which the highly reactive monomeric form is released in sufficient quantities which reacts with cellular constituents to produce a variety of lesions (35). The most prominent among these are DNA base adducts, DNA-protein crosslinks, and protein-protein crosslinks. Though there is genetic evidence supporting a role for the nucleotide excision repair system in promoting resistance to formaldehyde (36, 37), the DNA lesions targeted by nucleotide excision repair are not known and several studies have reported that protein-DNA crosslinks are repaired at similar rates in normal and excision repair-deficient formaldehyde-treated human cells (38–40). We therefore used our assay to investigate the role of nucleotide excision repair of formaldehyde damage as well as the DNA damage response elicited by formaldehyde. Fig. 7 shows the results of our assay for simultaneous detection of repair, checkpoint, and apoptosis induced by formaldehyde. As seen in Fig. 7A, the nominal 30-mer representing excision of DNA adducts is observed over a narrow dose range and at higher doses the signal disappears (Fig. 7, A and B) presumably because of the extensive DNA-protein and protein-protein crosslinking that traps the excision product in the insoluble fraction. In contrast to excision, checkpoint activation as measured by Chk1 and Chk2 phosphorylation and PARP cleavage is observed at higher concentrations of formaldehyde and diminishes as a result of extensive crosslinking at only the highest concentration used. Of significance, the nominal 30-mer was similarly observed with other human cell lines treated with formaldehyde (Fig. 7, C and D) consistent with the general utility of the method. Time course analyses of repair in formaldehyde-treated cells showed only a small time window for damage removal (Fig. 7, E and F). We note however that even though formaldehyde causes extensive DNA-protein crosslinks, our assay is incapable of detecting the repair of these damages. In fact, it has been shown that human excision repair is capable of only excising small peptide-DNA crosslinks but not those of proteins in the range of >10 kDa (37, 41, 42). This limitation notwithstanding, we believe that our assay would be useful in studying the formation and repair of many DNA damaging agents including other carcinogens and chemotherapeutic drugs.
FIGURE 7.

Detection of excision repair products following treatment with formaldehyde. A, HeLa cells were treated with various concentrations of formaldehyde (0.1, 0.2, 0.4, 0.6, 0.8, 1 mm), harvested after 3 h incubation, and then analyzed for the generation of excision products. Immunoblotting was performed with antibodies against the indicated proteins (bottom). B, quantitative analysis (average and standard deviation) of three independent experiments as shown in A. C, detection of excised oligonucleotides in A375 cells treated for 3 h with formaldehyde at different concentrations (0.2, 0.4, 0.6, 0.8 mm). Lane 1 shows the generation of repair products 1 h after exposure of HeLa cells to 5 J/m2 of UV. D, GM00200 cells were treated at different concentrations (0.2, 0.4, 0.6, 0.8 mm), harvested 3 h later, and analyzed for excised oligomers. E, HeLa cells were treated with formaldehyde (0.4 mm), harvested at the indicated time points, and then analyzed for excision repair products. Lane 8 shows the excised oligonucleotides induced 1 h following exposure to 5 J/m2 of UV. F, quantitative analysis (average and standard deviation) of three independent experiments as shown in E.
Conclusions
In this study, we describe a simple method for examining the three main cellular responses to DNA damage by carcinogens and chemotherapeutic drugs that form base adducts: nucleotide excision repair, DNA damage checkpoints, and apoptosis. Excision repair is probed by detection of the nucleotide excision repair specific 24–32 nt-long oligomers (“nominal 30-mer”) by post-excision biotin tagging and chemiluminescence. Checkpoint and apoptosis are examined by immunoblotting for Chk1 and Chk2 phosphorylation and for PARP cleavage, respectively. The method has certain advantages for probing for these endpoints over existing methods. First, the in vivo excision repair assay probes for the production of unique size oligonucleotides that are generated by the nucleotide excision repair system in response to DNA damage. The production of these oligomers occurs over a background of virtually zero and is capable of measuring repair of as little as 0.1% of UV damage. In contrast, slot blot (15) or ELISA (43) assays measure the decrease of damage-specific signal intensity in terms of a difference between two large numbers and as a consequence are much less sensitive than the in vivo excision assay. Secondly, both slot blot and ELISA require damage-specific antibodies whereas the excision assay does not and hence can be used for all damages that are processed by nucleotide excision repair with extreme sensitivity. Finally, the simplified method we describe for preparing cell lysates is compatible for detecting repair by chemiluminescence and checkpoint and apoptosis by immunoblotting using the same soluble fraction of the cell lysate from as few as 5 × 105 cells. The assay is thus suitable for screening for these phenomena of genotoxic responses in clinical or epidemiological settings and for assessing the temporal order of these three critical cellular responses to DNA damage using the same experimental platform.
Author Contributions
S-Y. K. and J-H. C. carried out the experiments and analyzed the results. J-H. C., S-K. K., M. K., and A. S. conceived of the idea for the project, analyzed the results, and wrote the paper.
This work was supported by National Institutes of Health Grant GM32833 (to A. S.) and the Korea Research Institute of Standards and Science under the project “Development of Bio-clinical Measurement Standards” (15011024) (to J-H. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- CPD
- cyclobutane pyrimidine dimer
- (6–4)PP
- (6–4)photoproducts
- BPDE
- benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide
- AAF
- N-acetoxy-2-acetylaminofluorene
- CHO
- Chinese Hamster Ovary
- XPG
- Xeroderma pigmentosum Group G
- MMC
- mitomycin C
- PARP
- poly-ADP ribose polymerase
- Chk1/Chk2
- Checkpoint Kinase 1/2
- ATR
- ataxia telangiectasia mutated and Rad3-related
- ATM
- ataxia telangiectasia mutated.
References
- 1.Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., and Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist C. J., and Kastan M. B. (2004) Initiating cellular stress responses. Cell. 118, 9–17 [DOI] [PubMed] [Google Scholar]
- 3.Sirbu B.M., and Cortez D. (2013) DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect. Biol. 5, a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sancar A. (1996) DNA excision repair. Annu. Rev. Biochem. 65, 43–81 [DOI] [PubMed] [Google Scholar]
- 5.Wood R. D. (1997) Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 [DOI] [PubMed] [Google Scholar]
- 6.Reardon J. T., and Sancar A. (2005) Nucleotide excision repair. Prog. Nucleic Acids Res. Mol. Biol. 79, 183–235 [DOI] [PubMed] [Google Scholar]
- 7.Choi J. H., Gaddameedhi S., Kim S. Y., Hu J., Kemp M. G., and Sancar A. (2014) Highly specific and sensitive method for measuring nucleotide excision repair kinetics of ultraviolet photoproducts in human cells. Nucleic Acids Res. 42, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J., Choi J. H., Gaddameedhi S., Kemp M. G., Reardon J. T., and Sancar A. (2013) Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J. C., Svoboda D. L., Reardon J. T., and Sancar A. (1992) Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer. Proc. Natl. Acad. Sci. U.S.A. 89, 3664–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Svoboda D. L., Taylor J. S., Hearst J. E., and Sancar A. (1993) DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J. Biol. Chem. 268, 1931–1936 [PubMed] [Google Scholar]
- 11.Mu D., Park C. H., Matsunaga T., Hsu D. S., Reardon J. T., and Sancar A. (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J. Biol. Chem. 270, 2415–2418 [DOI] [PubMed] [Google Scholar]
- 12.Kemp M. G., Reardon J. T., Lindsey-Boltz L. A., and Sancar A. (2012) Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kemp M. G., Gaddameedhi S., Choi J. H., Hu J., and Sancar A. (2014) DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair. J. Biol. Chem. 289, 26574–26583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu J., Adar S., Selby C. P., Lieb J. D., and Sancar A. (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wani A. A., D'Ambrosio S. M., and Alvi N. K. (1987) Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem. Photobiol. 46, 477–482 [DOI] [PubMed] [Google Scholar]
- 16.Gaddameedhi S., Kemp M. G., Reardon J. T., Shields J. M., Smith-Roe S. L., Kaufmann W. K., and Sancar A. (2010) Similar nucleotide excision repair capacity in melanocytes and melanoma cells. Cancer Res. 70, 4922–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell D. L. (1988) The relative cytotoxicity of (6–4) photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol. 48, 51–57 [DOI] [PubMed] [Google Scholar]
- 18.Reardon J. T., and Sancar A. (2003) Recognition and repair of the cyclobutane thymine dimer, a major cause of skin cancers, by the human excision nuclease. Genes Dev. 17, 2539–2551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cimprich K. A., and Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tibbetts R. S., Brumbaugh K. M., Williams J. M., Sarkaria J. N., Cliby W. A., Shieh S. Y., Taya Y., Prives C., and Abraham R. T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hess M. T., Gunz D., and Naegeli H. (1996) A repair competition assay to assess recognition by human nucleotide excision repair. Nucleic Acids Res. 24, 824–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gunz D., Hess M. T., and Naegeli H. (1996) Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J. Biol. Chem. 271, 25089–25098 [DOI] [PubMed] [Google Scholar]
- 23.Mu D., Bertrand-Burggraf E., Huang J. C., Fuchs R. P. P., Sancar A., and Fuchs B. P. (1994) Human and E.coli excinucleases are affected differently by the sequence context of acetylaminofluorene-guanine adduct. Nucleic Acids Res. 22, 4869–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindsey-Boltz L. A., Kemp M.G., Reardon J. T., DeRocco V., Iyer R. R., Modrich P., and Sancar A. (2014) Coupling of human DNA excision repair and the DNA damage checkpoint in a defined in vitro system. J. Biol. Chem. 289, 5074–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitagawa R., and Kastan M. B. (2005) The ATM-dependent DNA damage signaling pathway. Cold Spring Harb.Symp. Quant. Biol. 70, 99–109 [DOI] [PubMed] [Google Scholar]
- 26.Jamieson E.R., and Lippard S.J. (1999) Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem. Rev. 99, 2467–2498 [DOI] [PubMed] [Google Scholar]
- 27.Huang J.C., Zamble D.B., Reardon J.T., Lippard S.J., and Sancar A. (1994) HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 91, 10394–10398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamble D.B., Mu D., Reardon J.T., Sancar A., and Lippard S.J. (1996) Repair of cisplatin–DNA adducts by the mammalian excision nuclease. Biochemistry. 35, 10004–10013 [DOI] [PubMed] [Google Scholar]
- 29.Wang D., Hara R., Singh G., Sancar A., and Lippard S.J. (2003) Nucleotide excision repair from site-specifically platinum-modified nucleosomes. Biochemistry. 42, 6747–6753 [DOI] [PubMed] [Google Scholar]
- 30.Moggs J.G., Yarema K.J., Essigmann J.M., and Wood R.D. (1996) Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J. Biol. Chem. 271, 7177–7186 [DOI] [PubMed] [Google Scholar]
- 31.Sanderson B.J., and Shield A.J. (1996) Mutagenic damage to mammalian cells by therapeutic alkylating agents. Mutat. Res. 355, 41–57 [DOI] [PubMed] [Google Scholar]
- 32.Tomasz M., and Palom Y. (1997) The mitomycin bioreductive antitumor agents: cross-linking and alkylation of DNA as the molecular basis of their activity. Pharmacol. Ther. 76, 73–87 [DOI] [PubMed] [Google Scholar]
- 33.Chen J., and Stubbe J. (2005) Bleomycins: towards better therapeutics. Nat. Rev. Cancer. 5, 102–112 [DOI] [PubMed] [Google Scholar]
- 34.Povirk L.F. (1996) DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 355, 71–89 [DOI] [PubMed] [Google Scholar]
- 35.Swenberg J.A., Moeller B.C., Lu K., Rager J.E., Fry R.C., and Starr T.B. (2013) Formaldehyde carcinogenicity research: 30 years and counting for mode of action, epidemiology, and cancer risk assessment. Toxicol. Pathol. 41, 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Graaf B., Clore A., and McCullough A.K. (2009) Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair 8, 1207–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakano T., Katafuchi A., Matsubara M., Terato H., Tsuboi T., Masuda T., Tatsumoto T., Pack S.P., Makino K., Croteau D.L., Van Houten B., Iijima K., Tauchi H., and Ide H. (2009) Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 284, 27065–27076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grafstrom R.C., Fornace A. Jr, and Harris C.C. (1984) Repair of DNA damage caused by formaldehyde in human cells. Cancer Res. 44, 4323–4327 [PubMed] [Google Scholar]
- 39.Speit G., Schütz P., and Merk O. (2000) Induction and repair of formaldehyde-induced DNA-protein crosslinks in repair-deficient human cell lines. Mutagenesis. 15, 85–90 [DOI] [PubMed] [Google Scholar]
- 40.Quievryn G., and Zhitkovich A. (2000) Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis. 21, 1573–1580 [PubMed] [Google Scholar]
- 41.Reardon J.T., Cheng Y., and Sancar A. (2006) Repair of DNA-protein cross-links in mammalian cells. Cell. Cycle. 5, 1366–1370 [DOI] [PubMed] [Google Scholar]
- 42.Baker D.J., Wuenschell G., Xia L., Termini J., Bates S.E., Riggs A.D., and O'Connor T.R. (2007) Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem. 282, 22592–22604 [DOI] [PubMed] [Google Scholar]
- 43.Mitchell D.L. (1988) The induction and repair of lesions produced by the photolysis of (6–4) photoproducts in normal and UV-hypersensitive human cells. Mutat. Res. 194, 227–237 [DOI] [PubMed] [Google Scholar]