

Background: The HVR is important in K-Ras4B signaling.
Results: GTP binding and oncogenic mutations may weaken the HVR-catalytic core interactions.
Conclusion: GTP and some oncogenic mutations (e.g. G12C/G12V/Q61H/E37K) could attenuate HVR-catalytic domain interactions at the switch I/effector binding site by direct or longer-range interactions.
Significance: GTP and specific mutations could prompt exposure of switch I/effector binding site, thereby up-regulating signaling.
Keywords: biophysics, computational biology, nuclear magnetic resonance (NMR), protein conformation, protein dynamic, Ras protein
Abstract
K-Ras4B, a frequently mutated oncogene in cancer, plays an essential role in cell growth, differentiation, and survival. Its C-terminal membrane-associated hypervariable region (HVR) is required for full biological activity. In the active GTP-bound state, the HVR interacts with acidic plasma membrane (PM) headgroups, whereas the farnesyl anchors in the membrane; in the inactive GDP-bound state, the HVR may interact with both the PM and the catalytic domain at the effector binding region, obstructing signaling and nucleotide exchange. Here, using molecular dynamics simulations and NMR, we aim to figure out the effects of nucleotides (GTP and GDP) and frequent (G12C, G12D, G12V, G13D, and Q61H) and infrequent (E37K and R164Q) oncogenic mutations on full-length K-Ras4B. The mutations are away from or directly at the HVR switch I/effector binding site. Our results suggest that full-length wild-type GDP-bound K-Ras4B (K-Ras4BWT-GDP) is in an intrinsically autoinhibited state via tight HVR-catalytic domain interactions. The looser association in K-Ras4BWT-GTP may release the HVR. Some of the oncogenic mutations weaken the HVR-catalytic domain association in the K-Ras4B-GDP/-GTP bound states, which may facilitate the HVR disassociation in a nucleotide-independent manner, thereby up-regulating oncogenic Ras signaling. Thus, our results suggest that mutations can exert their effects in more than one way, abolishing GTP hydrolysis and facilitating effector binding.
Introduction
The Ras family of small GTPases plays an essential role in signal transduction pathways that promote cellular proliferation, survival, growth, and differentiation (1, 2). Ras behaves as a molecular switch by cycling between inactive GDP-bound and active GTP-bound states. Ras activity and subsequent signal propagation are initiated by guanine nucleotide exchange factors that catalyze the exchange of GDP by GTP and are terminated by GTPase activating proteins. GTPase activating proteins accelerate by 5 orders of magnitude GTP hydrolysis compared with the intrinsic GTPase activity, which does not contribute significantly to steady-state levels of Ras-GTP (3, 4). Active, GTP-loaded Ras interacts preferentially with downstream effectors such as Raf kinase (5), phosphatidylinositol 3-kinase (PI3K) (6), and Ral guanine nucleotide dissociation stimulator (7), thereby initiating signaling cascades. Recent structural models of GTP-bound K-Ras4B dimers showed how Ras dimerization may play a role in Raf activation and signaling (8) as validated by cellular assays (9). Aberrant activation of Ras is associated with ∼30% of human cancers. Oncogenic mutations, most commonly observed at amino acid residues Gly-12, Gly-13, and Gln-61, render Ras in a constitutively active GTP-bound state (10), resulting in prolonged activation of downstream signaling, including MAPK (mitogen-activated protein kinase) and PI3K pathways (11, 12).
There are three major Ras isoforms, H-, N-, and K-Ras, with the latter including two splice variants (K-Ras4A and K-Ras4B). Accumulating evidence indicates that K-Ras (86%) is the most frequently mutated isoform in Ras-driven cancers followed by N-Ras (11%) and H-Ras (3%) (13). The sequences of Ras isoforms in their catalytic domains are extremely conserved (sequence identity >89%) (14), whereas their C-terminal membrane-associated hypervariable region (HVR)5 exhibits significant divergence (sequence homology <15%) (Fig. 1A). The diversity of HVR sequences may provide some clues for isoform-specific differences in network signaling and oncogenic potential. K-Ras4B is modified by farnesylation at Cys-185 within the conserved C-terminal CAAX box (C, cysteine; A, aliphatic amino acid; X, any amino acid) (15). This post-translational modification together with the polybasic stretch of the HVR that forms electrostatic interactions with the negatively charged phospholipids in the inner plasma membrane (PM) leaflet allows K-Ras4B to insert the prenyl group in the HVR into a disordered PM (16). Combining computational and experimental studies, we recently showed that the HVR post-translational modifications in K-Ras4B play a pivotal role in targeting microdomains of the PM, suggesting an additional function for HVR in regulation of Ras signaling (17).
FIGURE 1.

Multiple sequence alignment of the amino acids in the H-Ras, N-Ras, K-Ras4A, and K-Ras4B and the architectures of GTP-/GDP-bound K-Ras4B catalytic domain. A, this alignment was produced by the ClustalX2 program. The non-identity of residues in the alignment is indicated by the pink circle. The listed residue numbers refer to H-Ras. A distinguishing feature of the HVR of K-Ras4B is bearing a polybasic stretch. B, a schematic representation of GppNHp-/GDP-bound K-Ras4B catalytic domain. The functional P-loop (residues 10–17), switch I (residues 32–38), and switch II (residues 59–68) of the catalytic domain are colored by lime, pink, and blue, respectively. The positions of residues Gly-12, Gly-13, Gln-61, Glu-37, and Arg-164 are colored by red. The effector binding region is depicted by a pink arrow. C, backbone superimposition of GppNHp (PDB ID 3GFT, cyan)- and GDP-bound (PDB ID 4EPT, light blue) K-Ras4B catalytic domain to the GppNHp-bound H-Ras catalytic domain (PDB ID 4G0N, green) in complex with its effector, RBD of Raf (RafRBD). Residues Gly-12, Gly-13, Gln-61, and Arg-164 are far away from the effector binding region, whereas Glu-37 is within the effector binding region. D, detailed electrostatic interactions between H-Ras and RafRBD. Residues Glu-37 and Asp-38 of H-Ras form electrostatic interactions with residues Arg-59, Arg-67, and Arg-89 of RafRBD. Residues Glu-31 and Asp-33 of H-Ras form electrostatic interactions with residue Lys-84 of RafRBD.
Despite the fundamental importance of HVR in Ras signaling (18–24), no structures of full-length Ras are currently available; Ras structures are determined exclusively with a truncated catalytic domain (25–29). When full-length was attempted, no HVR electron density was obtained. Previously, using nuclear magnetic resonance (NMR) spectroscopy, Thapar et al. (30) revealed that the HVR of H-Ras transiently interacts with the catalytic domain. Furthermore, based on NMR and isothermal titration calorimetry (ITC) data, we hypothesized that the HVR is modulated by the K-Ras4B catalytic domain in a nucleotide-dependent manner (31). However, the details of the interactions of the HVR with the catalytic domain in the GDP-/GTP-bound forms of Ras remained unresolved. In particular, little is understood about how the oncogenic mutations affect the HVR of full-length Ras.
Here, we mainly focus on the association of the HVR with the catalytic domain in solution. Based on earlier NMR and molecular dynamics (MD) simulations data (32), two different full-length K-Ras4B1–185 (hereafter simply referred as K-Ras4B) conformations for the GTP- and the GDP-bound state were selected as initial models. 200 ns of explicit solvent MD simulations were performed for each of the 32 systems (Table 1, in total 6.4 μs), including wild-type K-Ras4B-GTP/GDP (K-Ras4BWT-GTP/GDP) and their mutants (K-Ras4BMut-GTP/GDP) in frequent (G12C, G12D, G12V, G13D, and Q61H) and infrequent (E37K and R164Q) oncogenic states. The simulations show that in solution, K-Ras4BWT-GDP exists in an autoinhibited form characterized by tight interactions between the HVR and the catalytic domain. The HVR partially overlaps the Ras effector binding region and in one conformational species forms an antiparallel β sheet with the β2 strand of the catalytic domain. Upon GTP loading, the HVR associates loosely with the catalytic domain, indicating a lower autoinhibition threshold in K-Ras4BWT-GTP. Some of the oncogenic mutations attenuate the association of the HVR with the catalytic domain, which may facilitate the release of the HVR from the catalytic domain in a nucleotide-independent manner. The attenuation of the HVR-catalytic domain interactions is much stronger for the K-Ras4B-GTP mutants than for the K-Ras4B-GDP mutants. To date, oncogenic mutations were assumed to exert their effects only directly on the catalytic domain. Here, for the first time, our data elucidate how specific oncogenic mutations affect the structural and dynamic behavior of the HVR of K-Ras4B by direct or longer range interactions. We show an additional, previously overlooked, mechanism that contributes to Ras signaling by exposing the switch I/effector binding region, which is occluded by the HVR under physiological conditions. These observations underscore the double-thronged nature of the mutations. They can exert their effects in more than one way, abolishing GTP hydrolysis and facilitating effector binding. Although the bulk of the results are derived from extensive simulations, our NMR data support the two types of HVR-mediated exposure by oncogenic mutations: direct (E37K) and longer range interactions (the most oncogenic mutation, G12D).
TABLE 1.
Summary of MD simulation systems (each system ran for 200 ns)
| System name | Ions | Water numbers | Total atoms | r.m.s.d. |
||
|---|---|---|---|---|---|---|
| Full-length | Catalytic domain | HVR | ||||
| Å | ||||||
| Simulation of full-length K-Ras4B-GTP based on Model 1 | ||||||
| WT K-Ras4B-GTP | 0 | 6,614 | 22,853 | 3.28 ± 0.35 | 1.73 ± 0.24 | 8.32 ± 1.04 |
| G12C K-Ras4B-GTP | 0 | 7,130 | 24,405 | 7.34 ± 0.53 | 1.26 ± 0.10 | 20.99 ± 1.21 |
| G12D K-Ras4B-GTP | 1 | 7,134 | 24,419 | 6.05 ± 0.20 | 1.59 ± 0.09 | 16.99 ± 0.51 |
| G12V K-Ras4B-GTP | 0 | 7,132 | 24,416 | 3.48 ± 0.19 | 1.53 ± 0.18 | 9.36 ± 0.51 |
| G13D K-Ras4B-GTP | 1 | 6,626 | 22,895 | 3.83 ± 0.70 | 1.52 ± 0.16 | 11.13 ± 1.10 |
| E37K K-Ras4B-GTP | 2 | 6,686 | 23,078 | 4.60 ± 0.71 | 1.88 ± 0.25 | 7.68 ± 0.69 |
| Q61H K-Ras4B-GTP | 0 | 6,617 | 22,862 | 6.71 ± 0.27 | 1.85 ± 0.18 | 19.17 ± 0.67 |
| R164Q K-Ras4B-GTP | 1 | 6,581 | 22,748 | 4.84 ± 0.20 | 1.55 ± 0.13 | 13.27 ± 0.29 |
| Simulation of full-length K-Ras4B-GTP based on Model 2 | ||||||
| WT K-Ras4B-GTP | 0 | 5,959 | 20,888 | 3.06 ± 0.17 | 1.58 ± 0.12 | 5.24 ± 0.36 |
| G12C K-Ras4B-GTP | 0 | 6,497 | 22,506 | 4.10 ± 0.28 | 1.31 ± 0.15 | 8.79 ± 0.91 |
| G12D K-Ras4B-GTP | 1 | 6,491 | 22,490 | 3.31 ± 0.41 | 1.45 ± 0.33 | 5.20 ± 0.63 |
| G12V K-Ras4B-GTP | 0 | 6,485 | 22,475 | 11.00 ± 0.70 | 1.74 ± 0.24 | 31.42 ± 1.96 |
| G13D K-Ras4B-GTP | 1 | 5,934 | 20,819 | 2.88 ± 0.23 | 1.66 ± 0.25 | 3.89 ± 0.40 |
| E37K K-Ras4B-GTP | 2 | 5,892 | 20,696 | 7.28 ± 0.23 | 1.73 ± 0.10 | 21.77 ± 0.68 |
| Q61H K-Ras4B-GTP | 0 | 5,937 | 20,822 | 2.78 ± 0.56 | 1.64 ± 0.10 | 8.87 ± 1.81 |
| R164Q K-Ras4B-GTP | 1 | 6,021 | 21,068 | 3.02 ± 0.32 | 1.69 ± 0.10 | 8.23 ± 0.94 |
| Simulation of full-length K-Ras4B-GDP based on Model 1 | ||||||
| WT K-Ras4B-GDP | 1 | 5,407 | 19,229 | 3.53 ± 0.51 | 1.55 ± 0.13 | 8.80 ± 1.65 |
| G12C K-Ras4B-GDP | 1 | 5,418 | 19,266 | 3.14 ± 0.30 | 2.00 ± 0.25 | 6.13 ± 1.12 |
| G12D K-Ras4B-GDP | 0 | 5,422 | 19,278 | 3.79 ± 0.22 | 1.50 ± 0.08 | 9.33 ± 0.67 |
| G12V K-Ras4B-GDP | 1 | 5,442 | 19,343 | 4.20 ± 0.59 | 2.30 ± 0.46 | 9.08 ± 1.32 |
| G13D K-Ras4B-GDP | 0 | 5,417 | 19,263 | 2.50 ± 0.25 | 1.58 ± 0.13 | 5.03 ± 0.85 |
| E37K K-Ras4B-GDP | 3 | 5,411 | 19,250 | 8.10 ± 1.80 | 1.53 ± 0.15 | 25.09 ± 3.19 |
| Q61H K-Ras4B-GDP | 1 | 5,416 | 19,256 | 3.51 ± 0.51 | 1.84 ± 0.13 | 8.43 ± 1.31 |
| R164Q K-Ras4B-GDP | 0 | 5,277 | 18,831 | 3.36 ± 0.27 | 1.31 ± 0.10 | 9.26 ± 0.97 |
| Simulation of full-length K-Ras4B-GDP based on Model 2 | ||||||
| WT K-Ras4B-GDP | 1 | 5,002 | 18,014 | 3.85 ± 0.46 | 2.20 ± 0.13 | 7.30 ± 1.50 |
| G12C K-Ras4B-GDP | 1 | 5,012 | 18,048 | 4.19 ± 1.08 | 2.24 ± 0.14 | 10.48 ± 3.07 |
| G12D K-Ras4B-GDP | 0 | 5,019 | 18,069 | 3.48 ± 0.48 | 2.03 ± 0.12 | 6.47 ± 1.47 |
| G12V K-Ras4B-GDP | 1 | 5,043 | 18,146 | 3.44 ± 0.32 | 2.47 ± 0.61 | 5.67 ± 1.58 |
| G13D K-Ras4B-GDP | 0 | 4,981 | 17,955 | 4.99 ± 0.59 | 1.77 ± 0.12 | 11.90 ± 1.98 |
| E37K K-Ras4B-GDP | 3 | 5,093 | 18,296 | 5.50 ± 0.88 | 2.87 ± 0.20 | 13.51 ± 1.02 |
| Q61H K-Ras4B-GDP | 1 | 5,004 | 18,020 | 5.30 ± 0.20 | 2.12 ± 0.14 | 13.13 ± 0.61 |
| R164Q K-Ras4B-GDP | 0 | 4,985 | 17,955 | 4.18 ± 0.52 | 2.77 ± 0.26 | 8.20 ± 0.79 |
Experimental Procedures
Construction of Full-length K-Ras4B-GTP/GDP Models
The catalytic domains of K-Ras4B in complex with a nonhydrolyzable GTP analogue (GppNHp) (PDB ID 3GFT) and GDP (PDB ID 4EPT) (33), respectively, were selected to model the full-length K-Ras4B. In 4EPT, residue Ser-118 was mutated back to native Cys-118, and in 3GFT, residue His-61 was mutated back to native Gln-61. GppNHp was modified to the physiological GTP. The differences of NMR chemical shift perturbations (CSPs) between the full-length (residues 1–185) and the truncated catalytic domain (residues 1–166) in its GDP/GTPγS-bound forms (data deposited in Biological Magnetic Resonance Bank, ID 26635) showed that in the GDP-bound K-Ras4B, most of the changes are observed in the switch I and effector binding regions, β2, and in the C-terminal helix, β5 (32). This indicates that the C-terminal HVR associates extensively with the catalytic domain in the GDP-bound state. However, in the GTPγS-bound form, only very few changes occur in the switch I and effector binding regions (Ile-36) and in proximity to the N-terminal portion (Ser-17 and Ala-18) (32). Based on these observations, we generated four different initial configurations of each K-Ras4B-GTP/GDP with a covalently connected HVR to His-166 for the MD simulations. The analysis of the interaction energy between the HVR and the catalytic domain for each initial configuration revealed that the two models of each K-Ras4B-GDP/GTP are the best conformations of the HVR interacting with the catalytic domain. Therefore, we selected the two models, referred to as models 1 and 2, of each K-Ras4B-GDP/GTP as the templates to investigate the effects of oncogenic mutations on the conformational dynamics of the HVR. Models 1 and 2 of K-Ras4BWT-GDP correspond to the previous configurations 2 and 3 of K-Ras4BWT-GDP, whereas models 1 and 2 of K-Ras4BWT-GTP correspond to the previous configurations 3 and 4 of K-Ras4BWT-GTP (32). Currently, the crystal structures of the catalytic domains of G12D K-Ras4B in both GppNHp-bound and GDP-bound states are available. The root mean square deviation (r.m.s.d.) between the whole catalytic domains of K-Ras4BWT-GppNHp and K-Ras4BG12D-GppNHp (PDB ID 4DSN) (34) and that of K-Ras4BWT-GDP and K-Ras4BG12D-GDP (PDB ID 4EPR) (26) is 0.82 Å and 1.17 Å for all the Cα atoms, respectively, suggesting subtle conformational differences between the catalytic domains of K-Ras4BWT-GppNHp and K-Ras4BG12D-GppNHp as well as those of K-Ras4BWT-GDP and K-Ras4BG12D-GDP. Based on these data, coupled with the unavailability of crystal structures of other mutant K-Ras4B structures, the G12C, G12D, G12V, G13D, E37K, Q61H, and R164Q of each K-Ras4B-GTP/GDP were constructed using the wild-type models. The AMBER ff03 (35) force field was assigned for the proteins. The parameters for GTP and GDP were taken from the AMBER parameter database. The proteins were solvated in a truncated octahedral box with TIP3P (36) water molecules; the box size was set to ensure a distance of at least 10 Å between the protein and the box boundaries. The systems were neutralized using counterions. Apart from neutralizing ions and one cofactor, Mg2+, no other ions were used in the simulations, which excludes a screening effect and likely increases the electrostatic interactions compared with the physiological setting.
Molecular Dynamics Simulations
MD simulations were performed using the AMBER 11 package (37). To remove bad contacts in the solvated systems, all systems were subjected to 2000 steps of the steepest descent energy minimization followed by 3000 steps of the conjugate gradient energy minimization with a positional restraint of 500 kcal mol−1 Å−2 imposed on the heavy atoms of the proteins (38). Subsequently, the entire system was minimized without any restraints. After minimization, each system was heated gradually from 0 K to 300 K within 300 ps. This was followed by constant temperature equilibration at 300 K for 700 ps, with a positional restraint of 10 kcal mol−1 Å−2 in the complex in a canonical NVT ensemble.
A total of 6.4-μs MD simulations was performed with periodic boundary conditions using the NPT ensemble; each system was simulated for 200 ns (Table 1). Langevin dynamics (39) was used to maintain the temperature at 300 K with a collision frequency of 1 ps−1, and a Langevin piston was assigned to maintain the pressure at 1 atm. An integration step of 2 fs was set for the MD simulations. Long range electrostatic interactions were incorporated by using the particle mesh Ewald method (40). A cut-off equal to 10 Å was used for short range electrostatics and van der Waals interactions. The SHAKE method (41) was applied to constrain all covalent bonds that involve hydrogen atoms.
Site-directed Mutagenesis, Expression, and Purification
To produce the G12D and E37K mutants, site-directed mutagenesis was performed using the QuikChange® site-directed mutagenesis kit (Stratagene) following the manufacturer's instructions. Full-length recombinant G12D and E37K K-Ras4B mutants were expressed in BL 21 AI Escherichia coli cells and purified according to the published protocol (42). The cells were grown overnight at 18 °C, and K-Ras4B expression was induced with a final isopropyl 1-thio-β-d-galactopyranoside concentration of 0.2 mm along with 0.2% arabinose in M9 media containing 15NH4Cl (CIL Inc.). The protein was purified on Ni2+-Sepharose beads (Pierce). SDS-PAGE electrophoresis was used to determine the purity of the protein.
NMR Experiments
All experiments were carried out at 25 °C using Bruker Avance 900 MHz spectrometer fitted with a cryogenic probe. The NMR buffer contained 10% D2O, 50 mm Tris citrate (pH 6.5), 5 mm MgCl2, 50 mm NaCl, and 10 mm β-mercaptoethanol. Sparky (University of California San Francisco) was used for all K-Ras4B assignments based on CBCACONH and NHCACB NMR spectra. Data processing was done with the NMRPipe software (43). 1H,15N heteronuclear single quantum coherence (HSQC) spectra for G12D and E37K mutants and wild-type K-Ras4B were compared. The mean chemical shift differences were calculated as
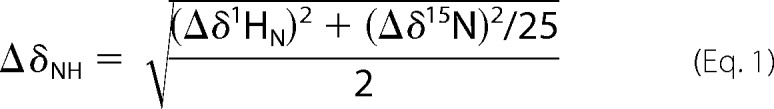 |
Only those chemical shift differences (Δδ) that were greater than the sum of the average of all chemical shift differences and 1 S.D. were considered to be statistically significant.
Results
Overview of Full-length K-Ras4B Structures and Simulations
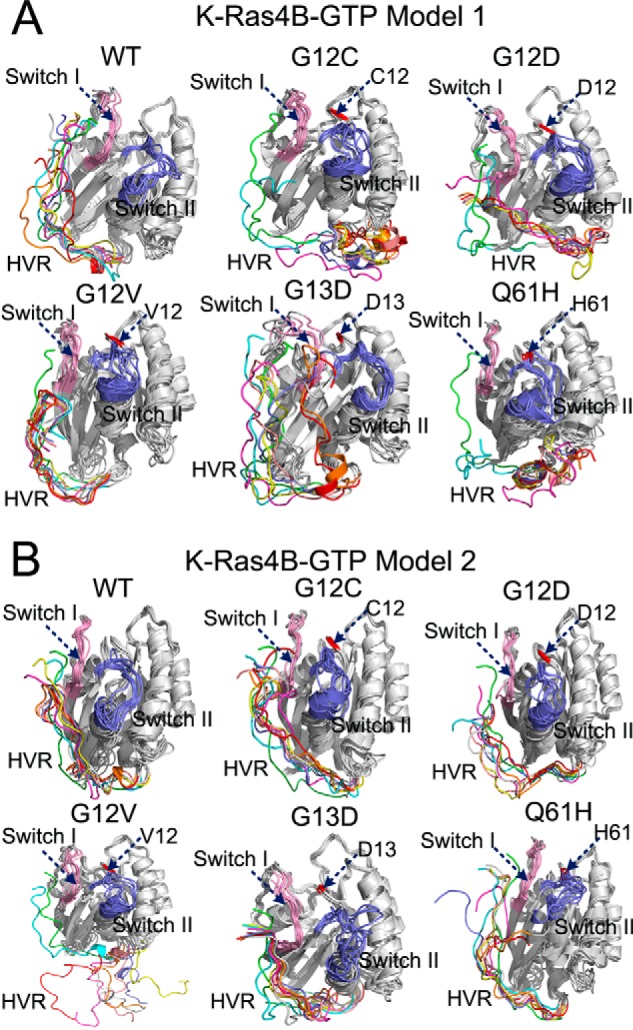
The structure of full-length K-Ras4B is composed of two components, the catalytic domain (residues 1–166) and the HVR (residues 167–188). The functional P-loop (residues 10–17), switch I (residues 32–38), and switch II (residues 59–67) regions constitute the active site for GTP hydrolysis and interaction sites for effector proteins, including Raf and PI3K (Fig. 1B). Residues Gly-12, Gly-13, Gln-61, and Arg-164 are far away from the effector binding region. In contrast, residue Glu-37 in the switch I region is within the effector binding region, which directly engages in electrostatic interactions with the positively charged residues Arg-59 and Arg-67 of the Ras binding domain (RBD) of Raf (RafRBD) (Fig. 1C). The HVR of K-Ras4B is highly positively charged. Therefore, we hypothesized that it might engage in potential electrostatic interactions with negatively charged residues in the catalytic domain that are located in spatial vicinity of the HVR. We performed two independent MD simulations with different starting points (models 1 and 2) for each wild-type and mutant K-Ras4B in the GTP- and GDP-bound states. In the starting points of both K-Ras4B-GTP models, the multiple C-terminal lysine residues on the HVR interacted with the negatively charged residues on the switch I region of the catalytic domain (Fig. 2A). The patterns of HVR-catalytic domain interactions are similar to those of H-Ras-RafRBD interactions (5) (Fig. 1D). However, in the starting points of both K-Ras4B-GDP models (Fig. 2B), the HVR has additional electrostatic interactions with the switch II region of the catalytic domain. These additional HVR contacts in the K-Ras4B-GDP are available because of the conformational rearrangements occurring in the GDP-bound state. The nucleotide-dependent orientation of the HVR with respect to the catalytic domain implies that interactions of the HVR with the catalytic domain are stronger for K-Ras4B-GDP than for K-Ras4B-GTP.
FIGURE 2.

The architectures of full-length GTP-/GDP-bound K-Ras4B. The catalytic domain and the HVR are colored by gray and orange, respectively. The functional P-loop, switch I, and switch II of the catalytic domain are colored lime, pink, and blue, respectively. A, schematic representation of models 1 and 2 of full-length K-Ras4B-GTP. In model 1, the residues Lys-178, Lys-180, Lys-182, and Lys-184 of the HVR form electrostatic interactions with the residues Glu-37, Asp-38, Asp-33, and Glu-31 of the switch I. In model 2, the residues Lys-178, Lys-182, and Lys-184 of the HVR form electrostatic interactions with the residues Glu-37, Asp-38, and Asp-33 of the switch I. B, schematic representation of models 1 and 2 of full-length K-Ras4B-GDP. In model 1 the residues Lys-176, Lys-177, Lys-178, and Lys-180 of the HVR form electrostatic interactions with the residues Glu-37, Asp-38, and Asp-33 of the switch I and Glu62 of the switch II. In model 2 the residues Lys-176, Lys-178, Lys-180, and Lys-182 of the HVR form electrostatic interactions with the residues Glu-37 and Asp-38 of the switch I and Glu62 of the switch II. C, the backbone superimposition of models 1 (HVR colored by red) and 2 (HVR colored by cyan) of full-length K-Ras4B-GTP. The orientation of the HVR with respect to the catalytic domain in the models 1 and 2 is similar. D, the backbone superimposition of models 1 (HVR colored by red) and 2 (HVR colored by cyan) of full-length K-Ras4B-GDP. The orientation of the HVR with respect to the catalytic domain in the models 1 and 2 is markedly different. In model 1, the HVR runs between the β1 strand and the helix α5, whereas in model 2 it runs between the L2 loop and β2 strand.
For each of the 32 systems of full-length K-Ras4B in the different states we performed 200 ns MD simulations (Table 1). Analysis of Cα r.m.s.d. between the wild-type and mutant K-Ras4B revealed that the mutations had subtle effects on the catalytic domains compared with the wild type (Table 1), in agreement with crystallographic studies that demonstrate no significant conformational differences between the wild-type and mutant Ras. Significantly, in all cases, compared with the catalytic domain the HVR had larger r.m.s.d. values during the simulations, which is consistent with its high flexibility. In particular, the r.m.s.d. values of the HVR are markedly larger in the G12C, G12D, G13D, Q61H, and R164Q of model 1 of K-Ras4B-GTP, G12V and E37K of model 2 of K-Ras4B-GTP, E37K of model 1 of K-Ras4B-GDP, and G12C, G13D, E37K, and Q61H of model 2 of K-Ras4B-GDP compared with those in the corresponding wild-type systems. The results indicate that these mutations affected significantly the conformational dynamics of the HVR.
Interactions of the HVR with the Catalytic Domain in K-Ras4BWT-GDP
To reveal the conformational dynamics of the HVR in K-Ras4BWT-GDP, the simulated snapshots sampled at each 25-ns interval over 200 ns of simulation were superimposed on the initial models 1 (Fig. 3A) and 2 (Fig. 3B), respectively. In both models the HVR interacts with the catalytic domain during the entire simulation trajectories. Remarkably, in model 2 of K-Ras4BWT-GDP, the HVR formed an antiparallel β-sheet with the β2 strand of the catalytic domain (Fig. 3B); the latter was in the effector binding region and mimicked its mode of interaction as observed in the co-crystal structure of the RafRBD bound to H-Ras catalytic domain (5) (Fig. 1C). The HVR is disordered. Secondary structure prediction using the DSSP (Dictionary of Secondary Structure of Proteins) method (44) revealed that the transient HVR β-strand conformation stems primarily from six residues (residues 171–176) on the N-terminal HVR (Fig. 4). Analysis of the interaction energy between the HVR and the catalytic domain in the K-Ras4BWT-GDP indicated that the interaction energy in model 2 (−576.43 ± 90.98 kcal/mol, Fig. 5B) were slightly lower than in model 1 (−557.47 ± 87.65 kcal/mol; Fig. 5A). Significantly, in both models the HVR-catalytic domain interactions are dominated by electrostatics. Taken together, these data suggest that in solution, the tight association of the HVR with the catalytic domain renders the K-Ras4BWT-GDP in its autoinhibited form.
FIGURE 3.

Dynamic behaviors of the HVR in the models 1 and 2 of full-length K-Ras4B-GDP. Backbone superimposition of simulated snapshots sampled at each 25-ns interval over 200 ns of simulation to the initial models 1 (A) and 2 (B) of full-length K-Ras4B-GDP in the wild-type (WT), G12C, G12D, G12V, G13D, and Q61H systems. The different colors of HVR represent the different simulation times of snapshots, initial model (green), 25 ns (cyan), 50 ns (magenta), 75 ns (yellow), 100 ns (pink), 125 ns (gray), 150 ns (blue), 175 ns (orange), and 200 ns (red). The same color settings of the HVR are used for Figs. 7 and 8. The locations of mutation sites are colored in red.
FIGURE 4.

Oncogenic mutations disrupt the β sheet of HVR. The percent (%) of HVR β sheet was predicted using the DSSP (Dictionary of Secondary Structure of Proteins) method on the model 2 of wild-type, G12C, G12D, G12V, G13D, and Q61H K-Ras4B-GDP. The β sheet of HVR of K-Ras4BWT-GDP stems from six residues (residues 171–176) on the N-terminal HVR, which is impaired by the oncogenic mutations.
FIGURE 5.

The interaction energy between the HVR and the catalytic domain in the models 1 (A) and 2 (B) of K-Ras4B-GDP and models 1 (C) and 2 (D) of K-Ras4B-GTP in their WT and mutant states. In all cases the electrostatic interactions contribute most to the HVR-catalytic domain interactions. vdW, van der Waals.
Specific Oncogenic Mutations May Weaken the HVR-Catalytic Domain Interactions in the K-Ras4B-GDP
Given the association of the HVR with the catalytic domain in K-Ras4BWT-GDP, we next investigated the impact of frequent oncogenic mutations, including G12C, G12D, G12V, G13D, and Q61H mutations, on the conformational ensemble of K-Ras4B-GDP. For each oncogenic mutant, the simulated snapshots were superimposed to the initial models 1 (Fig. 3A) and 2 (Fig. 3B). As in K-Ras4BWT-GDP, the HVR is retained on the catalytic domain in both models of K-Ras4BMut-GDP. However, the most remarkable difference found in the HVR between the wild-type and mutant K-Ras4B was an impairment of the HVR β-sheet in model 2 of K-Ras4BMut-GDP (Fig. 4). To quantitatively measure the influence of oncogenic mutations on the HVR, the interaction energy between the HVR and the catalytic domain was calculated. As shown in Fig. 5A, in model 1 of K-Ras4B-GDP, analysis of the average interaction energy reveals that the G12D, G12V, and Q61H mutations attenuate the HVR-catalytic domain interactions compared with the wild type. In contrast, the G12C and G13D mutations strengthened the HVR-catalytic domain interactions. However, in model 2 of K-Ras4B-GDP (Fig. 5B), all oncogenic mutations weakened the HVR-catalytic domain interactions with the exception of the G12V mutant; the average interaction energy in the latter is comparable in magnitude with that of the wild type. However, as shown in Fig. 5, A and B, considering the large error bars in the calculated interaction energy, the frequent oncogenic mutations may not significantly affect the HVR-catalytic domain interactions in the K-Ras4B-GDP.
To ascertain the influence of oncogenic mutations on the HVR, we compared the 1H,15N HSQC spectra of the wild-type and K-Ras4BG12D-GDP; the latter is the most frequent oncogenic mutant. The CSPs for backbone amides between the wild-type and G12D mutant occur for residues in the P-loop (Gly-10, Gly-15), L2 (Val-29), switch I (Glu-33, Ile-36), switch II (Gly-60, Met-67), α2 (Asp-69), β6 (Ser-145), α5 (His-166), and HVR (Lys-178, Lys-179) (Fig. 6). Most of the residues that are perturbed significantly are in the vicinity of the P-loop (residues 10–17) where the G12D mutation is located, suggesting that the G12D mutation alters the conformation of the functional P-loop, switch I and switch II regions. Remarkably, G12D can also perturb the HVR, which is relatively far away from the P-loop, indicating that the G12D mutation affects the HVR-catalytic domain interactions by longer range interactions (45–47).
FIGURE 6.
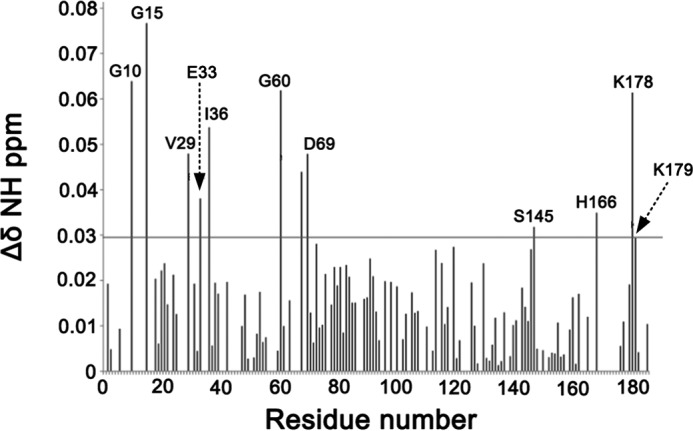
The experimental NMR CSPs for K-Ras4BG12D-GDP. Residual chemical shifts obtained after the overlay of 1H,15N HSQC NMR spectra of K-Ras4BWT-GDP and that of K-Ras4BG12D-GDP. The horizontal line in the graph shows the sum of mean CSP and S.D. The CSPs above the horizontal line are considered significance.
Interactions of the HVR with the Catalytic Domain in K-Ras4BWT-GTP
To reveal the conformational dynamics of the HVR in K-Ras4BWT-GTP, the simulated snapshots sampled at each 25-ns interval over the 200-ns trajectory were superimposed on the initial models 1 (Fig. 7A) and 2 (Fig. 7B). Similar to K-Ras4BWT-GDP, the HVR can interact with the catalytic domain along the simulations in both K-Ras4BWT-GTP models. Because the contacts between the HVR and the catalytic domain in the K-Ras4BWT-GDP outweigh those of K-Ras4BWT-GTP, we hypothesized that the HVR interacts loosely with the catalytic domain in K-Ras4BWT-GTP. To test this hypothesis, the interaction energies between the HVR and the catalytic domain in both models of K-Ras4BWT-GTP were calculated. The interaction energies were −530.13 ± 103.16 and −470.34 ± 56.81 kcal/mol for models 1 (Fig. 5C) and 2 (Fig. 5D) of K-Ras4BWT-GTP, respectively, which were higher than those of models 1 (−557.47 ± 87.65 kcal/mol; Fig. 5A) and 2 (−576.43 ± 90.98 kcal/mol, Fig. 5B) of K-Ras4BWT-GDP. Taken together, these data suggest that the loose interaction of the HVR with the catalytic domain in the GTP-bound form may facilitate the disassociation of the HVR from the catalytic domain.
FIGURE 7.
Dynamic behaviors of the HVR in the models 1 and 2 of full-length K-Ras4B-GTP. Backbone superimposition of simulated snapshots sampled at each 25-ns interval over 200 ns simulation to the initial models 1 (A) and 2 (B) of full-length K-Ras4B-GTP in the WT, G12C, G12D, G12V, G13D, and Q61H systems. The locations of mutation sites are colored in red.
Specific Oncogenic Mutations Significantly Affect the HVR-Catalytic Domain Interactions in K-Ras4B-GTP
Because of the loose engagement of the HVR with the catalytic domain in K-Ras4BWT-GTP, we forecasted that the oncogenic mutations would have pronounced effects on the conformational dynamics of the HVR. To test this likelihood, the simulated snapshots in each oncogenic mutant were superimposed onto the initial models 1 (Fig. 7A) and 2 (Fig. 7B). The orientation of the HVR with respect to the catalytic domain in K-Ras4B-GTP underwent extensive changes in the oncogenic mutants. Remarkably, in the G12C, G12D, and Q61H mutants of model 1 and the G12D and G12V mutants of model 2, we observed that the HVR is released from the catalytic domain during the simulations. We calculated the interaction energy between the HVR and the catalytic domain for each oncogenic mutant. As shown in Fig. 5, C and D, analysis of average interaction energy revealed that in both models all oncogenic mutations reduced the interaction compared with the wild type. For example, in model 1, the interaction energies for G12C, G12D, G12V, G13D, and Q61H mutant were −348.31 ± 96.15, −436.98 ± 109.32, −473.72 ± 85.47, -447.22 ± 118.07 kcal/mol, and −378.18 ± 106.65 kcal/mol, respectively, which were larger than the wild type (−530.13 ± 103.16 kcal/mol). In model 2, the interaction energies for G12C, G12D, G12V, G13D, and Q61H mutants were −401.74 ± 98.41, −398.70 ± 80.26, −222.24 ± 119.59, −452.43 ± 78.74, and −386.78 ± 105.31 kcal/mol, respectively, which were also larger than the wild type (−470.34 ± 56.81 kcal/mol). However, as shown in Fig. 5, C and D, considering the large error bars in the calculated interaction energy, only the frequent oncogenic G12V mutation in the model 2 significantly affected the HVR-catalytic domain interactions in the K-Ras4B-GTP. Cumulatively, these results suggest that specific oncogenic mutations markedly affect the HVR-catalytic domain interactions in the K-Ras4B-GTP, which may accelerate the release of the HVR from the catalytic domain and further up-regulate signaling.
The Effect of the E37K Mutation on Full-length K-Ras4B
Residue Glu-37 is situated on the switch I region. In both K-Ras4B-GTP/GDP (Fig. 2), Glu-37 engaged in electrostatic interactions with the lysine residues on the C-terminal HVR. Recently, an infrequent K-Ras oncogenic mutation, E37K, has been detected in patients of chronic myelomonocytic leukemia (48). We hypothesized that the inverse charge of residue 37 could have a large effect on the overall structure. To test this hypothesis, the simulated snapshots sampled at each 25-ns interval over 200 ns of simulation were superimposed to the initial models 1 and 2 of K-Ras4B-GTP/GDP, respectively. As shown in Fig. 8A, the HVR can interact with the catalytic domain in model 1 of K-Ras4BE37K-GTP but was released in model 2. As for the simulations of K-Ras4BE37K-GDP (Fig. 8B), the HVR was released from the catalytic domain in model 1. Although the HVR was capable of interacting with the catalytic domain in model 2, it exhibited large fluctuations during the simulations. We calculated the interaction energy between the HVR and the catalytic domain. The E37K mutation resulted in a significant loss of interaction, i.e. increasing the average interaction energy in both GDP/GTP-bound forms compared with models 1 and 2 of K-Ras4BWT-GTP/GDP (Fig. 5). However, considering the error bars in the calculated interaction energy, only the E37K mutation in the model 1 of K-Ras4B-GDP significantly affected the HVR-catalytic domain interactions.
FIGURE 8.

Effects of E37K and R164Q mutations on the dynamic behaviors of HVR. Backbone superimposition of simulated snapshots sampled at each 25-ns interval over 200 ns of simulation to the initial model in the models 1 and 2 of K-Ras4BE37K-GTP (A), the models 1 and 2 of K-Ras4BE37K-GDP (B), the models 1 and 2 of K-Ras4BR164Q-GTP (C), and the models 1 and 2 of K-Ras4BR164Q-GDP (D). The locations of mutation sites are colored in red.
To reveal the impact of the E37K mutation on the K-Ras4B, we compared the 1H,15N HSQC spectra of the wild-type and K-Ras4BE37K-GDP. The marked CSPs for backbone amides between the wild-type and E37K mutant occurred for residues in β1 (Tyr-4), P-loop (Gly-10, Ser-17), L2 (Leu-23–Gln-25, Phe-28, Val-29), β2 (Ser-39, Arg-41, Lys-42), β3 (Thr-50, Thr-51, Asp-54, Thr-58), switch II (Gly-60, Glu-62, Met-67), β4 (Cys-80), and α3 (Arg-97, Lys-101) (Fig. 9A). The most remarkable difference between models 1 and 2 was the orientation of the HVR with respect to the catalytic domain. In model 1 (Fig. 9B), the HVR ran between β1 and α2 regions, whereas in model 2 (Fig. 9C) it ran between L2 and β2 regions. L2 and β2 are involved in binding Raf CRD and RafRBD (5), respectively. Thus, in model 2 of K-Ras4B-GDP, the HVR partially overlaps the Ras effector binding region. The experimental NMR CSPs showed that the E37K mutation was capable of affecting the L2 and β2 regions of the catalytic domain, which suggests that model 2 prevails over model 1 in characterizing the HVR-catalytic domain interactions.
FIGURE 9.

The experimental NMR CSPs for K-Ras4BE37K-GDP. A, residual chemical shifts obtained after the overlay of 1H,15N HSQC NMR spectra of K-Ras4BWT-GDP and that of K-Ras4BE37K-GDP. The horizontal line in the graph shows the sum of mean CSP and S.D. The CSPs above the horizontal line are considered significant. A schematic representation of the regions undergoing significant CSPs (in magenta) are mapped on the models 1 (B) and 2 (C) of full-length K-Ras4B-GDP.
The Effect of the R164Q Mutation on Full-length K-Ras4B
Residue Arg-164 is located on the C-terminal helix α5. In full-length K-Ras4B-GTP/GDP, Arg-164 formed electrostatic interactions with both residues Asp-47 and Glu-49 on the L3 loop that connects β2 to β3. Recently, using microarray analysis, Smith et al. (49) identified a new infrequent mutation of K-Ras, R164Q, in sporadic colorectal tumors. We anticipate that substitution of arginine by glutamine disrupts electrostatic interactions between Arg-164 and residues Asp-47 and Glu-49, which in turn may resonate in the entire structure. To test this idea, simulated snapshots sampled at each 25-ns interval over 200-ns simulation were superimposed onto the initial models 1 and 2 of K-Ras4BR164Q-GTP/GDP, respectively. As shown in Fig. 8C, the HVR changed the orientation with respect to the catalytic domain in model 1 of K-Ras4BR164Q-GTP, but it retained the interactions with the catalytic domain in model 2. In the simulations of K-Ras4BR164Q-GDP (Fig. 8D), the HVR interacted with the catalytic domain in both models, although the C-terminal HVR illustrates large fluctuations during the simulations. In addition, significant conformational changes also occur in the switch I and switch II of model 2. The average interaction energy between the HVR and the catalytic domain showed that the R164Q mutation reduces the HVR-catalytic domain interactions in both GDP-/GTP-bound forms compared with the models 1 and 2 of K-Ras4BWT-GTP/GDP (Fig. 5). However, considering the large error bars in the calculated interaction energy, the R164Q mutation may not significantly affect the HVR-catalytic domain interactions in both GDP-/GTP-bound K-Ras4B.
Discussion
Currently structures of full-length Ras remain undetermined. Although there have been considerable efforts in elucidating the interactions of post-translational modifications (farnesylation, geranylgeranylation, palmitoylation, and phosphorylation) of the HVRs of Ras isoforms with different PMs (50–53), not much is known about how the HVR interacts with the catalytic domain of Ras. Here, combining MD simulations and NMR, we propose that K-Ras4B-GDP is in its autoinhibited form via the interactions of the HVR with the catalytic domain, thereby locking K-Ras4B in an inactive state. The exchange of GDP by GTP produces an active K-Ras4B-GTP featured by the release of HVR from the catalytic domain.
The phenomenon of autoinhibition has been commonly observed in enzymes consisting of several domains. Evolution has exploited this mechanism to reduce protein activity in resting cells. Examples include the AMP-activated protein kinase (AMPK), where the engagement of an autoinhibitory domain with the catalytic domain shifts the kinase to an autoinhibited state with much lower activity than that of the kinase domain alone (54). In Akt, the pleckstrin homology domain couples with the kinase domain to lock the Akt in an inactive conformation (55, 56). In c-Abl-tyrosine kinase, the N-terminal SH3-SH2 domains, acting as an autoinhibitory “cap,” binds to the kinase domain, and the loss of this autoinhibition turns c-Abl into an oncogenic protein (57). As in the case of Ran, a Ras-like small GTPase, the determination of Ran structures in complex with different nucleotides unraveled a C-terminal α-helix interacting with the Ran binding domain in the GDP-bound form (58). Conversely, the Ran-GppNHp complex indicates that GTP binding induces a conformational switch that may detach the C-terminal helix form the Ran binding domain (59). Consistently, biochemical analysis demonstrates that K-Ras4B forms an assembly that is similar to that of Ran.
Previously, based on NMR and ITC, we revealed that the HVR of K-Ras4B is the primary binding site for calmodulin (CaM), a ubiquitous Ca2+-binding protein, and the affinity of K-Ras4B to CaM depends on nucleotide binding to K-Ras4B (31). We showed that little or no CaM binding to K-Ras4B was observed in the GDP-bound form, suggesting that the HVR may be sequestered by the catalytic domain in the K-Ras4B-GDP to prevent it from binding CaM. In contrast, in the GTPγS-bound form, K-Ras4B can interact with CaM with micromolar affinity, pointing to an HVR disassociated state in K-Ras4B-GTP.
Based on the NMR CSPs data (32), we generated two initial configurations of each full-length K-Ras4B-GTP/GDP for the MD simulations. Superimposition of both GTP-bound models showed that the orientation of the HVR with respect to the catalytic domain was very similar (Fig. 2C). However, we observed a significant difference in the orientation of the HVR with respect to the catalytic domain between the two K-Ras4B-GDP models (Fig. 2D). In model 1, the HVR ran between the β2 strand and the α2 helix, whereas in model 2 it ran between the L2 loop and the β2 strand. In the K-Ras4B-GDP model 2 simulation, the six N-terminal HVR residues (residues 171–176) formed an antiparallel β sheet with the β2 strand of the catalytic domain. This β sheet formation enhances the interaction of the HVR with the catalytic domain. The interaction energy between the HVR and the catalytic domain was calculated on the basis of each simulated trajectory. It is lower in both models of K-Ras4BWT-GDP than K-Ras4BWT-GTP, in agreement with experimental data. Moreover, the average interaction energy of model 2 of K-Ras4B-GDP was lower than that of the model 1 of K-Ras4B-GDP by ∼20 kcal/mol, indicating that the interaction of HVR with the catalytic domain is stronger in model 2 than in model 1. The additional HVR β strand in model 2 may be responsible for augmenting the HVR-catalytic domain interaction.
We carried out simulations of oncogenic mutations, including frequent (G12C, G12D, G12V, G13D, and Q61H) and infrequent (E37K and R164Q) mutations to explore their effects on the full-length K-Ras4B-GDP/-GTP. Because of the loose interaction of the HVR with the catalytic domain in K-Ras4B-GTP, mutations such as G12C, G12V, Q61H, and E37K caused the disassociation of the HVR from the catalytic domain during the simulations. Analysis of the average interaction energy between the HVR and the catalytic domain demonstrated that all oncogenic mutants reduced the interaction energy compared with K-Ras4BWT-GTP. In contrast, in all cases except that of E37K in model 1 of K-Ras4B-GDP, the HVR retained the interactions with the catalytic domain; however, all oncogenic mutants in model 2 disrupted the N-terminal β sheet extension by the HVR, which may facilitate the HVR release.
Here, we propose new insights into K-Ras4B regulation. As shown in Fig. 10A, in resting cells K-Ras4B is predominantly GDP-bound. The farnesylated HVR of K-Ras4BWT-GDP formed an antiparallel β sheet through interaction with the β2 strand of the catalytic domain. A dynamic equilibrium existed between the released and catalytic domain-bound K-Ras4B-GDP HVR, but the equilibrium constant of koff (unreleased) was significantly larger than the kon (released), rendering the full-length K-Ras4B-GDP primarily in the unreleased, autoinhibited form. The released HVR had the ability to insert the farnesyl into the PM. At the PM, guanine nucleotide exchanged GDP for GTP to produce the active K-Ras4B-GTP, which can bind its effectors to initiate downstream signaling. GTPase-activating proteins accelerated GTP→GDP hydrolysis to produce the inactive K-Ras4B-GDP, terminating signaling. When detached from the PM, K-Ras4BWT-GDP was autoinhibited by its β sheet-forming HVR, which interacts with the catalytic domain. Oncogenic mutants (Fig. 10B) disrupted the β sheet, weakening the interactions between the HVR and the catalytic domain, which shifted the equilibrium to the released HVR state. Thus, oncogenic mutations elevated the released K-Ras4B-GDP HVR population compared with the WT, thereby up-regulating downstream signaling and oncogenic cell growth. The absence of crystal structures of full-length K-Ras4B may have obscured this secondary, although critical HVR-mediated oncogenic mechanism that we were able to unravel through a combination of simulations and NMR. We note that our current model of full-length K-Ras4B in both GDP- and GTP-bound states has no farnesyl group attached to residue Cys-185 and no MD simulations of full-length K-Ras4B-menbrane complex are included. Therefore, the geometrical organization and dynamics of the HVR-GTPase interactions with respect to the membrane may alter and are yet to be established. In addition, the relative energetics of the HVR-GTPase and the farnesyl-HVR membrane interactions are currently unknown. These may influence the unreleased-released HVR equilibrium.
FIGURE 10.

The mechanisms of wild-type and oncogenic K-Ras4B regulation. In resting cells, K-Ras4BWT (A) is predominantly GDP-bound, and the farnesylated HVR of K-Ras4BWT-GDP forms an antiparallel β sheet with the β2 sheet of the catalytic domain. In the cytoplasma pool, K-Ras4BWT-GDP is in a dynamic equilibrium between the released (kon) and unreleased (koff) HVR states (kon ≪ koff), which renders the full-length K-Ras4B-GDPWT primary in the unreleased, autoinhibited form. The released HVR state of K-Ras4BWT-GDP can insert the farnesylated HVR into the PM, where the polybasic stretch of the HVR forms electrostatic interactions with the negatively charged phospholipids in the inner leaflet of PM. At the PM, guanine nucleotide exchanges (GEF) exchange GDP for GTP to produce the active K-Ras4BWT-GTP, which can bind its effectors, e.g. Raf, PI3K, and Ral guanine nucleotide dissociation stimulator (RalGDS), to initiate downstream signaling. GTPase activating proteins (GAP) accelerate GTP→GDP hydrolysis to produce the inactive K-Ras4BWT-GDP, thereby terminating signaling. After disassociation from the PM, K-Ras4BWT-GDP is autoinhibited again by the interactions of its HVR β sheet with the catalytic domain to complete catalytic process. B, In the oncogenic states, the mutations impair the β sheet secondary structure of HVR and weaken the interactions between the HVR and the catalytic domain. This effect shifts the equilibrium to the released HVR state of K-Ras4BMut-GDP and increases the population of the released HVR state of K-Ras4BMut-GDP compared with the wild type. As a result, more population of K-Ras4BMut-GDP can traffic to the PM to recruit its effectors, thereby leading to the increase of downstream signaling associated with oncogenic cell growth.
Author Contributions
S. L., H. J., and R. N. conceived and designed the study. A. B. and V. G. performed the NMR experiments. S. L., H. J., J. Z., and R. N. performed MD simulations. S. L., H. J., V. G., and R. N. prepared and wrote the manuscript. All authors edited and approved the manuscript.
This work was supported by the National Basic Research Program of China (973 Program) Grant 2015CB910403 (to J. Z.), National Natural Science Foundation of China Grants 81322046, 81302689, and 81473137 (to J. Z.), Shanghai Rising-Star Program Grant 13QA1402300) (to J. Z.), Program for New Century Excellent Talents in University (NCET-12-0355) (to J. Z.), National Institutes of Health Grant R01 CA135341 (to V. G.), American Cancer Society Grant RGS-09-057-01-GMC (to V. G.). This work was also supported in whole or in part with Federal funds from the NCI-Frederick, National Institutes of Health under Contract HHSN261200800001E (to R. N.). This work was supported in part by the Intramural Research Program of NCI-Frederick, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article.
The resonance assignments have been deposited in the Biological Magnetic Resonance Bank (BMRB) NMR chemical shift data bank under BMRB ID 26635.
- HVR
- hypervariable region
- PM
- plasma membrane
- ITC
- isothermal titration calorimetry
- MD
- molecular dynamics
- CSP
- chemical shift perturbation
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- r.m.s.d.
- root mean square deviation
- HSQC
- heteronuclear single quantum coherence
- RBD
- Ras binding domain
- CaM
- calmodulin
- GppNHp
- guanosine 5'-[β,γ-imido] triphosphate.
References
- 1.Cherfils J., and Zeghouf M. (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 2.Hernández-Alcoceba R., del Peso L., and Lacal J. C. (2000) The Ras family of GTPases in cancer cell invasion. Cell. Mol. Life Sci. 57, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bos J. L., Rehmann H., and Wittinghofer A. (2007) Review GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 4.Boriack-Sjodin P. A., Margarit S. M., Bar-Sagi D., and Kuriyan J. (1998) The structural basis of the activation of Ras by Sos. Nature 394, 337–343 [DOI] [PubMed] [Google Scholar]
- 5.Fetics S. K., Guterres H., Kearney B. M., Buhrman G., Ma B., Nussinov R., and Mattos C. (2015) Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pacold M. E., Suire S., Perisic O., Lara-Gonzalez S., Davis C. T., Walker E. H., Hawkins P. T., Stephens L., Eccleston J. F., and Williams R. L. (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 103, 931–943 [DOI] [PubMed] [Google Scholar]
- 7.Vetter I. R., Linnemann T., Wohlgemuth S., Geyer M., Kalbitzer H. R., Herrmann C., and Wittinghofer A. (1999) Structural and biochemical analysis of Ras-effector signaling via RalGDS. FEBS Lett. 451, 175–180 [DOI] [PubMed] [Google Scholar]
- 8.Muratcioglu S., Chavan T. S., Freed B. C., Jang H., Khavrutskii L., Freed R. N., Dyba M. A., Stefanisko K., Tarasov S. G., Gursoy A., Keskin O., Tarasova N. I., Gaponenko V., and Nussinov R. (2015) GTP-dependent K-Ras dimerization. Structure 23, 1325–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nan X., Tamgüney T. M., Collisson E. A, Lin L.-J., Pitt C., Galeas J., Lewis S., Gray J. W., McCormick F., and Chu S. (2015) Ras-GTP dimers activate the mitogen-activated protein kinase (MAPK) pathway. Proc. Natl. Acad. Sci. U.S.A. 112, 7996–8001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., and Der C. J. (2014) Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nussinov R., Tsai C.-J., and Mattos C. (2013) “Pathway drug cocktail”: targeting Ras signaling based on structural pathways. Trends Mol. Med. 19, 695–704 [DOI] [PubMed] [Google Scholar]
- 12.Samatar A. A., and Poulikakos P. I. (2014) Targeting RAS–ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov. 13, 928–942 [DOI] [PubMed] [Google Scholar]
- 13.Prior I. A., Lewis P. D., and Mattos C. (2012) A comprehensive survey of ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castellano E., and Santos E. (2011) Functional specificity of Ras isoforms: so similar but so different. Genes Cancer 2, 216–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long S. B., Casey P. J., and Beese L. S. (2000) The basis for K-Ras4B binding specificity to protein farnesyltransferase revealed by 2 Å resolution ternary complex structures. Structure 8, 209–222 [DOI] [PubMed] [Google Scholar]
- 16.Brunsveld L., Waldmann H., and Huster D. (2009) Membrane binding of lipidated Ras peptides and proteins: the structural point of view. Biochim. Biophys. Acta 1788, 273–288 [DOI] [PubMed] [Google Scholar]
- 17.Jang H., Abraham S. J., Chavan T. S., Hitchinson B., Khavrutskii L., Tarasova N. I., Nussinov R., and Gaponenko V. (2015) Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J. Biol. Chem. 290, 9465–9477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorfe A. A., Babakhani A., and McCammon J. A. (2007) Free energy profile of H-ras membrane anchor upon membrane insertion. Angew. Chem. Int. Ed. Engl. 46, 8234–8237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorfe A. A., Babakhani A., and McCammon J. A. (2007) H-ras protein in a bilayer: interaction and structure perturbation. J. Am. Chem. Soc. 129, 12280–12286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janosi L., Li Z., Hancock J. F., and Gorfe A. A. (2012) Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc. Natl. Acad. Sci. U.S.A. 109, 8097–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guzmán C., Šolman M., Ligabue A., Blaževitš O., Andrade D. M., Reymond L., Eggeling C., and Abankwa D. (2014) The efficacy of Raf kinase recruitment to the GTPase H-ras depends on H-ras membrane conformer-specific nanoclustering. J. Biol. Chem. 289, 9519–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentry L. R., Nishimura A., Cox A. D., Martin T. D., Tsygankov D., Nishida M., Elston T. C., and Der C. J. (2015) Divergent Roles of CAAX motif-signaled posttranslational modifications in the regulation and subcellular localization of Ral GTPases. J. Biol. Chem. 290, 22851–22861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evelyn C. R., Biesiada J., Duan X., Tang H., Shang X., Papoian R., Seibel W. L., Nelson S., Meller J., and Zheng Y. (2015) Combined rational design and a high throughput screening platform for identifying chemical inhibitors of a ras-activating enzyme. J. Biol. Chem. 290, 12879–12898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuld N. J., Vervacke J. S., Lorimer E. L., Simon N. C., Hauser A. D., Barbieri J. T., Distefano M. D., and Williams C. L. (2014) The chaperone protein SmgGDS interacts with small GTPases entering the prenylation pathway by recognizing the last amino acid in the CAAX motif. J. Biol. Chem. 289, 6862–6876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrem J. M., Peters U., Sos M. L., Wells J. A., and Shokat K. M. (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milroy L. G., and Ottmann C. (2014) The renaissance of Ras. ACS Chem. Biol. 9, 2447–2458 [DOI] [PubMed] [Google Scholar]
- 27.Rosnizeck I. C., Spoerner M., Harsch T., Kreitner S., Filchtinski D., Herrmann C., Engel D., König B., and Kalbitzer H. R. (2012) Metal-bis(2-picolyl)amine complexes as state 1(T) inhibitors of activated Ras protein. Angew. Chem. Int. Ed. Engl. 51, 10647–10651 [DOI] [PubMed] [Google Scholar]
- 28.Araki M., Shima F., Yoshikawa Y., Muraoka S., Ijiri Y., Nagahara Y., Shirono T., Kataoka T., and Tamura A. (2011) Solution structure of the state 1 conformer of GTP-bound H-Ras protein and distinct dynamic properties between the state 1 and state 2 conformers. J. Biol. Chem. 286, 39644–39653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shima F., Ijiri Y., Muraoka S., Liao J., Ye M., Araki M., Matsumoto K., Yamamoto N., Sugimoto T., Yoshikawa Y., Kumasaka T., Yamamoto M., Tamura A., and Kataoka T. (2010) Structural basis for conformational dynamics of GTP-bound Ras protein. J. Biol. Chem. 285, 22696–22705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thapar R., Williams J. G., and Campbell S. L. (2004) NMR characterization of full-length farnesylated and non-farnesylated H-Ras and its implications for Raf activation. J. Mol. Biol. 343, 1391–1408 [DOI] [PubMed] [Google Scholar]
- 31.Abraham S. J., Nolet R. P., Calvert R. J., Anderson L. M., and Gaponenko V. (2009) The hypervariable region of K-Ras4B is responsible for its specific interactions with calmodulin. Biochemistry 48, 7575–7583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chavan T. S., Jang H., Khavrutskii L., Abraham S. J., Banerjee A. Freed B. C., Johannessen L., Tarasov S. G., Gaponenko V., Nussinov R., and Tarasova N. I. (2015) High affinity interaction of K-Ras4B hypervariable region with Ras active site. Biophys. J. 10.1158/1538-7445.AM2014-3224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Q., Burke J. P., Phan J., Burns M. C., Olejniczak E. T., Waterson A. G., Lee T., Rossanese O. W., and Fesik S. W. (2012) Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew. Chem. Int. Ed. Engl. 51, 6140–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maurer T., Garrenton L. S., Oh A., Pitts K., Anderson D. J., Skelton N. J., Fauber B. P., Pan B., Malek S., Stokoe D., Ludlam M. J., Bowman K. K., Wu J., Giannetti A. M., Starovasnik M. A, Mellman I., Jackson P. K., Rudolph J., Wang W., and Fang G. (2012) Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. U.S.A. 109, 5299–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., and Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 37.Case D. A., Cheatham T. E. 3rd, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., and Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu S., Huang W., Wang Q., Shen Q., Li S., Nussinov R., and Zhang J. (2014) The structural basis of ATP as an allosteric modulator. PLoS Comput. Biol. 10, e1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X., and Brooks B. R. (2003) Self-guided Langevin dynamics simulation method. Chem. Phys. Lett. 381, 512–518 [Google Scholar]
- 40.Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 41.Ryckaert J.-P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 42.Abraham S. J., Muhamed I., Nolet R., Yeung F., and Gaponenko V. (2010) Expression, purification, and characterization of soluble K-Ras4B for structural analysis. Protein Expr. Purif. 73, 125–131 [DOI] [PubMed] [Google Scholar]
- 43.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 44.Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 45.Lu S., Li S., and Zhang J. (2014) Harnessing allostery: a novel approach to drug discovery. Med. Res. Rev. 34, 1242–1285 [DOI] [PubMed] [Google Scholar]
- 46.Nussinov R., and Tsai C.-J. (2013) Allostery in disease and in drug discovery. Cell 153, 293–305 [DOI] [PubMed] [Google Scholar]
- 47.Tsai C.-J., and Nussinov R. (2014) A unified view of “how allostery works.” PLoS Comput. Biol. 10, e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernard V., Gebauer N., Dinh T., Stegemann J., Feller A. C., and Merz H. (2014) Applicability of next-generation sequencing to decalcified formalin-fixed and paraffin-embedded chronic myelomonocytic leukaemia samples. Int. J. Clin. Exp. Pathol. 7, 1667–1676 [PMC free article] [PubMed] [Google Scholar]
- 49.Smith G., Bounds R., Wolf H., Steele R. J., Carey F. A., and Wolf C. R. (2010) Activating K-Ras mutations outwith “hotspot” codons in sporadic colorectal tumours: implications for personalised cancer medicine. Br. J. Cancer 102, 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arozarena I., Calvo F., and Crespo P. (2011) Ras, an actor on many stages: posttranslational modifications, localization, and site-specified events. Genes Cancer 2, 182–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basso A. D., Kirschmeier P., and Bishop W. R. (2006) Lipid posttranslational modifications: farnesyl transferase inhibitors. J. Lipid Res. 47, 15–31 [DOI] [PubMed] [Google Scholar]
- 52.Rowinsky E. K., Windle J. J., and Von Hoff D. D. (1999) Ras protein farnesyltransferase: a strategic target for anticancer therapeutic development. J. Clin. Oncol. 17, 3631–3652 [DOI] [PubMed] [Google Scholar]
- 53.Hancock J. F. (2003) Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4, 373–384 [DOI] [PubMed] [Google Scholar]
- 54.Chen L., Jiao Z.-H., Zheng L.-S., Zhang Y.-Y., Xie S.-T., Wang Z.-X., and Wu J.-W. (2009) Structural insight into the autoinhibition mechanism of AMP-activated protein kinase. Nature 459, 1146–1149 [DOI] [PubMed] [Google Scholar]
- 55.Wu W.-I., Voegtli W. C., Sturgis H. L., Dizon F. P., Vigers G. P., and Brandhuber B. J. (2010) Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 5, e12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu S., Deng R., Jiang H., Song H., Li S., Shen Q., Huang W., Nussinov R., Yu J., and Zhang J. (2015) The mechanism of ATP-dependent allosteric protection of Akt kinase phosphorylation. Structure 23, 1725–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagar B., Hantschel O., Young M. A., Scheffzek K., Veach D., Bornmann W., Clarkson B., Superti-Furga G., and Kuriyan J. (2003) Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112, 859–871 [DOI] [PubMed] [Google Scholar]
- 58.Partridge J. R., and Schwartz T. U. (2009) Crystallographic and biochemical analysis of the Ran-binding zinc finger domain. J. Mol. Biol. 391, 375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vetter I. R., Nowak C., Nishimoto T., Kuhlmann J., and Wittinghofer A. (1999) Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature 398, 39–46 [DOI] [PubMed] [Google Scholar]