

Background: The role of phosphorylation for regulating the Rac1 GEF PREX2 is not understood.
Results: PAK phosphorylation of PREX2 downstream of PIP3 and Gβγ reduces PREX2 GEF activity.
Conclusion: Second messengers can initiate negative feedback to decrease Rac1 activation through PAK phosphorylation of PREX2.
Significance: PAK negative regulation of GEFs could represent a broadly utilized mechanism to attenuate Rac1 activation and its outputs.
Keywords: G-protein-coupled receptor (GPCR); guanine nucleotide exchange factor (GEF); insulin; phosphatidylinositide 3-kinase (PI 3-kinase); Rac (Rac GTPase); serine/threonine-protein kinase PAK 1 (PAK1); signal transduction; Phosphatidylinositol-3,4,5-trisphosphate-dependent RAC exchanger 2 (PREX2)
Abstract
Phosphatidylinositol 3,4,5-trisphosphate (PIP3)-dependent Rac exchanger 2 (PREX2) is a guanine nucleotide exchange factor (GEF) for the Ras-related C3 botulinum toxin substrate 1 (Rac1) GTPase, facilitating the exchange of GDP for GTP on Rac1. GTP-bound Rac1 then activates its downstream effectors, including p21-activated kinases (PAKs). PREX2 and Rac1 are frequently mutated in cancer and have key roles within the insulin-signaling pathway. Rac1 can be inactivated by multiple mechanisms; however, negative regulation by insulin is not well understood. Here, we show that in response to being activated after insulin stimulation, Rac1 initiates its own inactivation by decreasing PREX2 GEF activity. Following PREX2-mediated activation of Rac1 by the second messengers PIP3 or Gβγ, we found that PREX2 was phosphorylated through a PAK-dependent mechanism. PAK-mediated phosphorylation of PREX2 reduced GEF activity toward Rac1 by inhibiting PREX2 binding to PIP3 and Gβγ. Cell fractionation experiments also revealed that phosphorylation prevented PREX2 from localizing to the cellular membrane. Furthermore, the onset of insulin-induced phosphorylation of PREX2 was delayed compared with AKT. Altogether, we propose that second messengers activate the Rac1 signal, which sets in motion a cascade whereby PAKs phosphorylate and negatively regulate PREX2 to decrease Rac1 activation. This type of regulation would allow for transient activation of the PREX2-Rac1 signal and may be relevant in multiple physiological processes, including diseases such as diabetes and cancer when insulin signaling is chronically activated.
Introduction
Phosphatidylinositol 3,4,5-trisphosphate (PIP3)2-dependent Rac exchanger 2 (PREX2) is a guanine nucleotide exchange factor (GEF) for the GTPase Ras-related C3 botulinum toxin substrate 1 (Rac1) (1, 2). GEFs such as PREX2 bind to Rac1 and facilitate the exchange of GDP for GTP, and while in the GTP-bound state, Rac1 is able to bind many effectors to carry out various cellular functions (3). For example, GTP-bound Rac1 activates the WASP family verprolin-homologous protein (WAVE) regulatory complex, p21-activated kinases (PAKs), the NADPH oxidase complex, and the p110β catalytic subunit of phosphatidylinositide 3-kinase (PI3K) (4–8). The mechanisms of Rac1 inactivation are generally not as well characterized as the mechanisms of Rac1 activation, but inactivation in some contexts is known to occur through GTPase-activating proteins (GAPs), which increase the rate of hydrolysis of GTP, promoting the inactive GDP-bound state of Rac1, and by guanine nucleotide dissociation inhibitors (GDIs), which sequester inactive Rac1 in the cytosol (3, 9).
PREX2, along with its homolog PREX1, is activated by PIP3 and by β and γ subunits of G-proteins (Gβγ). PIP3 is generated by PI3K activity downstream of receptor tyrosine kinase and G-protein-coupled receptor (GPCR) activation (10–12), and Gβγ is released upon activation of GPCRs (13). PREX2 can be directly activated by both PIP3 and Gβγ in vitro, and expression of both PI3K and Gβγ synergistically activates PREX2 in vivo (1, 2). PIP3 and Gβγ levels at the membrane are regulated by numerous ligand-activated receptors, and PREX proteins have been studied in many of these contexts. PREX2 mediates signaling downstream of the insulin receptor (14), a receptor tyrosine kinase that stimulates PI3K and activates Rac1 and AKT, both of which are critical for regulating glucose metabolism in many tissues (15–19). PREX2 inactivating mutation in mice leads to increased glucose in the blood after glucose or insulin injection and a reduction in AKT phosphorylation in insulin-treated liver and adipose tissue (14). These phenotypes are likely the result of both PREX2 GEF activity toward Rac1 and PREX2 inhibition of the phosphatase and tensin homolog (PTEN), a lipid phosphatase that antagonizes PI3K by dephosphorylating PIP3, therefore reducing AKT activation (14–16, 20). Additionally, PREX2 expression increases the level of platelet-derived growth factor (PDGF)-stimulated Rac activity in porcine aortic endothelial cells, and knockdown of the PREX2b isoform in endothelial cells prevents sphingosine 1-phosphate-stimulated cell migration (1, 21). PREX1 has reported roles in Rac1 activation and cell migration downstream of many ligands, including PDGF, neuregulin, epidermal growth factor (EGF), and N-formyl-methionine-leucyl-phenylalanine (fMLP) (1, 22–27). Furthermore, in differentiated 3T3-L1 mouse adipocytes, the PREX1-Rac1 signal regulates insulin-induced glucose uptake (28).
PREX2 also has important roles in cancer. PREX2 expression is increased in many different tumor types, and it is one of the most frequently mutated GEFs across all cancer types with especially high rates of mutation in melanoma (20, 29–32). In MCF10A cells, expression of PREX2 and a constitutively active PI3K mutant cooperate to form colonies in soft agar (20). Furthermore, in a melanocyte mouse xenograft model, the expression of certain PREX2 tumor mutations cooperates with neuroblastoma RAS viral (v-Ras) oncogene homolog to accelerate tumorigenesis (29). PREX2 tumor mutations can also escape PTEN-mediated inhibition of breast cancer cell invasion (30). Additionally, PREX2 is the target of a microRNA that is down-regulated in metastatic neuroblastomas and gastric cancer (33, 34).
Despite the known roles of PREX2 in critical cellular signaling pathways and potentially in cancer progression, very little is known about how PREX2 is regulated downstream of PI3K and GPCR activation. Although there are no reports of PREX2 phosphorylation, we hypothesized that phosphorylation is likely to be important for PREX2 regulation and function, especially given that phosphorylation can regulate PREX1 and other Rac GEFS, such as VAV and T-lymphoma invasion and metastasis-inducing protein 1 (Tiam1) (26, 35–41). In this study, we show that following PREX2 activation of Rac1 by PIP3 or Gβγ, PAK phosphorylation of PREX2 accumulates to decrease PREX2 GEF activity by reducing its ability to bind to second messengers at the cell membrane. We also identified the insulin pathway as one signaling context in which this regulation takes place. Taken together, our study shows that PAKs mediate negative regulation of PREX2, allowing for tight control of the length and amplitude of Rac1 activation.
Experimental Procedures
Plasmids and Constructs
Full-length PREX2 was cloned into the pcDNA3.1 V5/His vector (Life Technologies, Inc.), and GST PTEN was cloned into the pGEX4T-1 vector (Amersham Biosciences) as described previously (20). PTEN G129E was made by mutating pCEP4-PTEN, and PREX2 point mutations were made by mutating the full-length V5/His PREX2 pcDNA3.1 backbone using the QuikChange II XL site-directed mutagenesis kit (Agilent). PP1α was amplified from cDNA of HEK293 cells and cloned into the pcDNA3.1 V5/His vector using the TOPO-TA cloning kit (Life Technologies, Inc.). E545K and H1047R mutants of p110α were cloned into the pcDNA3.1 vector. MYC-pCMV6M-PAK1 WT and PAK1 K299R (Addgene plasmids 12209 and 12210) and His6-pET28-PAK2 WT were gifts from Jonathan Chernoff (42). The catalytic subunit of PP2A (pcDNA-PP2A/C-HA) was a gift from Gen Sheng Wu (43). pGEX encoding GST-tagged Rac1 was a gift from Katrin Rittinger. The FLAG-Gβ1/HA-Gγ2 vector in the pVITRO2-mcs(Hygro) backbone was a gift from Jonathan Backer. MYC-Rac1-WT and MYC-Rac1-T17N were gifts from Gary Bokoch (Addgene plasmids 12985 and 12984). The plasmid encoding the V5-CRAF fragment (residues 306–648) was a gift from Poulikos Poulikakos.
Antibodies
Mouse V5 primary antibody (R960-25) was purchased from Life Technologies, Inc. Mouse M2 FLAG (F1804), vinculin (V9131), and actin (A5316) antibodies, in addition to mouse anti-V5-agarose affinity (A7345) and anti-M2-FLAG-agarose affinity (A2220) gels were purchased from Sigma. Mouse tubulin (MMS-410P) was purchased from Covance. Rabbit PTEN clone 138G6 (9559), total AKT (9272), p308 AKT (4056), p473 AKT (9271), total ribosomal S6 (2271), p235/236 S6 (2211), ERK1/2 (9102), p202/204 ERK1/2 (4376) and total EGFR (4267) primary antibodies were purchased from Cell Signaling Technologies. The rabbit polyclonal Ser(P)-1107 PREX2 antibody to amino acids 1098–1116 (GHDTISNRDpSYSDCNSNRN) was made in collaboration with Cell Signaling Technologies. Mouse Rac1 (05-389) and p338 RAF1/CRAF (05-538) primary antibodies were purchased from Millipore. Mouse MYC clone 9E10 (sc-40) primary antibody was purchased from Santa Cruz Biotechnology. Anti-His HRP antibody (34460) was purchased from Qiagen. The PREX2 rabbit polyclonal antibody was previously reported (20). Secondary antibodies directed against rabbit and mouse IgG conjugated to HRP were purchased from Pierce.
Cell Lines, Transfection, and Drug Treatments
HEK293 and U87MG cells were cultured in DMEM (Cellgro) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 IU penicillin, and 100 μg/ml streptomycin (Cellgro). DBTRG-05MG cells were cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS, 100 IU penicillin, and 100 μg/ml streptomycin. When indicated, cells were starved for 16 h in the appropriate media without FBS before treatment. Transfections were performed with Lipofectamine 2000 (Life Technologies, Inc.) following the manufacturer's protocol. Unless otherwise indicated, insulin (Sigma) treatments were for 30 min at 5 μg/ml. For treatment with a small molecule inhibitor, the indicated concentrations were incubated with the cells for 15 min prior to addition of insulin or for 30 min prior to harvesting if no insulin was added.
In Vitro λ-Phosphatase Assay
For endogenous PREX2, HEK293 cells were harvested in lysis buffer (20 mm HEPES, pH 7.4, 0.25% Nonidet P-40, 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, 1× eukaryotic protease inhibitor mixture (Sigma)). The lysates were vortexed, sonicated, centrifuged at 4 °C for 30 min, and then combined with 5 μl of PREX2 antibody and 20 μl of protein A/G PLUS-agarose beads (Santa Cruz Biotechnology), rotating at 4 °C for 4 h. The beads were then washed five times in lysis buffer. For analysis of exogenous PREX2, HEK293 cells expressing V5 PREX2 were harvested in lysis buffer (25 mm Tris, pH 7.5, 0.2% Triton X-100, 200 mm KCl, 10% glycerol, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, 1× eukaryotic protease inhibitor mixture). The lysates were vortexed, sonicated, centrifuged at 4 °C for 30 min, and then incubated with V5-agarose beads (Sigma) for 4 h rotating at 4 °C. The beads were then washed five times in lysis buffer. For both V5 PREX2 and endogenous PREX2, the phosphatase reactions were set up in 100 μl using 1 μl of λ-phosphatase from New England Biolabs along with the supplied 10× λ-phosphatase buffer and 10× MnCl2 diluted in water. The reactions were then incubated at 30 °C for 20 min and were terminated with 2× Laemmli sample buffer (125 mm Tris, pH 6.8, 4% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.05% bromphenol blue).
Mass Spectrometry
V5 PREX2 was expressed in HEK293 cells. The cells were starved and then treated with either 500 nm GDC0941 (referred to as the unstimulated state), 5 μg/ml insulin, or insulin and 100 nm calyculin A (both of these conditions are referred to as the stimulated state). The cells were lysed in buffer containing 25 mm Tris, pH 7.5, 0.2% Triton X-100, 200 mm KCl, 10% glycerol, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, and 1× eukaryotic protease inhibitor mixture. The lysate was vortexed, sonicated, centrifuged at 4 °C for 30 min, and then pre-cleared with protein A/G PLUS-agarose beads and mouse IgG (Santa Cruz Biotechnology). The beads were removed, and V5 PREX2 was isolated on V5-agarose beads by incubating them with the lysate for 4 h rotating at 4 °C. The beads were then washed five times in lysis buffer and resuspended in 2× Laemmli sample buffer. For each condition, two samples of ∼3 μg of V5 PREX2 were run by SDS-PAGE, and the bands were excised. The protein bands were digested overnight with sequencing grade trypsin or chymotrypsin (Promega). These digests were analyzed separately, and phosphopeptides were enriched using titanium dioxide spin tips (Protean) followed by microcapillary liquid chromatography/tandem mass spectrometry (LC-MS/MS). Enriched peptides were introduced to the Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) in positive ion collision-induced dissociation mode using a data-dependent acquisition with an EASY-NLC II nanoflow HPLC (Proxeon) (75 mm inner diameter × 15 cm, C18 column) at 300 nl/min. The MS/MS spectra were searched against the reversed and concatenated SwissProt (Version 2012_3, Uniprot) protein database and the Prex2V5His custom database using Mascot server 2.4, and data were analyzed using Scaffold 4 software. Peptides were reported with a false discovery rate threshold of 1%.
Modeling of the PREX2 PH Domain
For modeling, the software Modeler version 9v9 maintained by the Sali laboratory was used (44). The template for the PH domain of PREX2 was the crystal structure of the PH domain of CDC42 (Protein Data Bank code 2DFK, chain A). To explore the variations in query-template alignments, we used S4, a program that relies on information on secondary structure elements of the template (45). The alternative alignments generated by S4 were evaluated with statistical potential-based pG score and structure verification program Verify3D. The pG score is the posterior probability that the model has a correct three-dimensional conformation, given its normalized z-score (obtained using the program Prosa II (46)) and length. Verify3D assesses the compatibility of the segments of the sequence with their three-dimensional structures by plotting the average statistical preference scores in a window of 21 residues (47). The best model among alternative alignment had a pG score of 0.99. The minimum value of Verify3D profile window plot was 0.06.
V5 PREX2 Purification for the Rac-GEF Assay
V5 PREX2 was purified from HEK293 cells as described previously with small modifications (20). In short, 15-cm plates were transfected with V5 PREX2 and were harvested in 1.5 ml of lysis buffer per plate (25 mm Tris, pH 7.4, 150 mm NaCl, 0.1% Triton X-100, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, 1× eukaryotic protease inhibitor mixture). The cells were vortexed, sonicated, and then centrifuged for 1 h at 40,000 × g. The supernatant was incubated with V5-agarose (10 μl per plate) for 16 h rotating at 4 °C. The beads were then washed on a poly-prep column (Bio-Rad) four times with lysis buffer and four times with buffer containing 1× PBS, pH 7.5, 10% glycerol, 1 mm EGTA, and 1 mm DTT. Then 1.5-bed volumes of elution buffer (1× PBS, pH 7.5, 10% glycerol, 1 mm EGTA, 1 mm DTT, and 500 μg/ml V5 peptide (Sigma)) was added to the beads, and the column was incubated at 37 °C for 30 min. The column was then put into a 50-ml conical tube and centrifuged at 200 × g. This elution process was repeated a second time, and elutions were combined. Glycerol was added to 50%; bovine serum albumin (BSA) was added to 2 mg/ml; and the protein was snap-frozen and stored at −80 °C. Protein samples taken prior to the addition of BSA were quantified by SDS-PAGE followed by staining with Gel Code Blue (Thermo) in the presence of BSA standards.
Expression and Purification of Recombinant Gβ1His-γ2
Recombinant purified Gβγ dimers were produced as described elsewhere (48). Essentially, fall armyworm ovary cells (Sf9, from Gibco BRL, Eggenstein, Germany) were cultured in suspension with TNM-FH medium (Sigma, Deisenhofen, Germany) supplemented with 10% (v/v) fetal calf serum (Gibco), Lipid Medium Supplement (1:100; Sigma), penicillin (100 units/ml), and streptomycin (0.1 mg/ml). For protein expression, Sf9 cells (1.5 × 106 cells/ml) were infected with viruses encoding Gβ1 and N-terminally hexahistidine-tagged Gγ2. After 60 h of infection, cells were collected by centrifugation at 1,000 × g for 5 min and washed twice with PBS. Recombinantly expressed isoprenylated Gβ1His-γ2 complexes were isolated from the membrane fraction of Sf9 cells as detailed earlier (49, 50). Purified proteins were quantified by SDS-PAGE followed by Coomassie Blue staining with BSA standards and stored at −80 °C.
In Vitro Rac-GEF Assay
In vitro analysis of PIP3- and Gβγ-stimulated V5 PREX2 GEF activity toward GDP-loaded GST Rac1 was performed as described previously, except glutathione-Sepharose beads (GE Healthcare) were used to isolate the GST Rac1 after the incubation with V5 PREX2 (23, 51). The purification of GST Rac1 proteins was performed as described previously (14). After elution with glutathione, a 500-μl elution was combined in a 10,000 MWCO Amicon filter with 15 ml of buffer containing 40 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, and 1 mm DTT. The solution was concentrated to 1 ml, and this was repeated three more times. The solution was removed from the filter; GDP was added to 1 mm, and the solution was rotated at 4 °C for 1 h. MgCl2 was then added to 15 mm to stop loading, and the solution was added to a 10,000 MWCO Amicon filter with 15 ml of 40 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EGTA, 1 mm DTT, 5 mm MgCl2, and 10 μm GDP. The solution was concentrated to 1 ml, and this was repeated three more times. The final protein was snap-frozen and stored at −80 °C. For the in vitro GEF assay, PIP3 dipalmitoyl C16 was purchased from Echelon Biosciences and was incorporated into liposomes. Gβ1His-γ2 was purified from Sf9 insect cells. The final concentrations of GST Rac1 and V5 PREX2 in the reaction were 100 and 1 nm, respectively. Purified PREX2 and GST Rac1 were incubated in a final reaction volume of 10 μl with PIP3 or Gβγ, 5 μm cold GTPγS, and 1 μCi of [35S]GTPγS (PerkinElmer Life Sciences) for 10 min at 30 °C. GST Rac1 was isolated on glutathione-Sepharose beads, and the loading of [35S]GTPγS by GST Rac1 was measured by scintillation counting.
GST and PIP3 Bead Pulldowns
For pulldowns of V5 PREX2, HEK293 cells were transfected and then harvested in lysis buffer (20 mm HEPES, pH 7.4, 0.25% Nonidet P-40, 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, 1× eukaryotic protease inhibitor mixture). The lysate was then vortexed, sonicated, and centrifuged at 4 °C for 30 min. For pulldowns with GST-fused proteins, lysates were pre-cleared with GST-loaded glutathione-Sepharose beads for 1 h rotating at 4 °C. The supernatants were then incubated with 5 μg of GST PTEN, GST PP1α, or GST Rac1 loaded onto glutathione-Sepharose beads for 4 h rotating at 4 °C. Purification of GST proteins was performed as described previously (14). For PIP3 pulldowns, 10 μl of PIP3 bead slurry (Echelon) was incubated with lysates for 4 h rotating at 4 °C. The beads were washed five times in lysis buffer and resuspended in 50 μl of 2× Laemmli sample buffer. For PIP3 pulldowns of endogenous PREX2, the same protocol was followed except that the lysates from 15-cm plates of HEK293 cells were divided into two samples, one for the PIP3 pulldown and another for the immunoprecipitation of PREX2 with 5 μl of PREX2 antibody.
In Vitro GST PP1α and PP2A Dephosphorylation Assay
HEK293 cells were transfected with V5 PREX2, and cells were lysed in buffer containing 20 mm HEPES, pH 7.4, 0.25% Nonidet P-40, 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, and 1× eukaryotic protease inhibitor mixture. The lysate was then vortexed, sonicated, and centrifuged at 4 °C for 30 min. The supernatant was then incubated with V5-agarose beads for 4 h rotating at 4 °C. Next, the beads were washed four times in lysis buffer and once with either PP1α reaction buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 0.1 mm EDTA, 1 mm MnCl2, 5 mm DTT) for PP1α assays or protein phosphatase dilution buffer (catalog no. 20-169) from Millipore for PP2A assays. For PP1α dephosphorylation assays, 99 μl of PP1α reaction buffer was added to each tube of V5 PREX2 beads followed by either 1 μl of GST elution buffer (25 mm Tris, pH 8.0, 100 mm NaCl, 50 mm glutathione) for the mock treatment or 1 μl of GST PP1α (at a concentration of 1 μg/μl). For PP2A dephosphorylation assays, 45 μl of protein phosphatase dilution buffer was added to the V5 PREX2 beads, and then either 5 μl of protein phosphatase buffer or 0.5 units (5 μl) of purified PP2A (Millipore, catalog no. 14-111) was added to each reaction. The reactions were incubated for 60 min at 30 °C and stopped by removing the supernatant and resuspending the beads in 2× Laemmli sample buffer.
Gβγ-PREX2 Co-immunoprecipitation
HEK293 cells were transfected with V5 PREX2 alone or V5 PREX2 and FLAG/HA Gβγ together. Cells co-expressing PREX2 and Gβγ were either untreated or treated with 100 nm calyculin A for 30 min. Cells were lysed in buffer containing 25 mm Tris, pH 7.5, 0.2% Triton X-100, 200 mm KCl, 10% glycerol, 1 mm EDTA, 1 mm EGTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, and 1× eukaryotic protease inhibitor mixture. The lysates were rotated at 4 °C for 15 min, vortexed, and then spun down at 4 °C for 30 min. The protein concentrations of the lysates were measured using the Bio-Rad DC protein assay, and protein concentrations were equalized for all samples. The samples were then pre-cleared with protein A/G PLUS-agarose beads rotating at 4 °C for 1 h. Finally, samples were incubated with 10 μl of M2-FLAG-agarose affinity gel for 4 h rotating at 4 °C. The beads were then washed five times in lysis buffer and were resuspended in 2× Laemmli sample buffer.
Cell Fractionation
Either untransfected (for endogenous PREX2 analysis) or V5 PREX2-transfected HEK293 cells were harvested in lysis buffer without detergents (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 100 nm calyculin A, 1× eukaryotic protease inhibitor mixture), and the lysates were passed through a 25-gauge needle 10 times. The nuclear fraction was removed by centrifuging the lysate at 720 × g for 5 min. The supernatant was centrifuged again at 17,000 × g for 20 min, followed by another spin at 17,000 × g for 10 min. The membrane fraction was separated by ultracentrifugation at 100,000 × g for 60 min. The supernatant (cytosolic fraction) was saved, and the pellet (membrane fraction) was resuspended in 400 μl of lysis buffer without detergents and re-centrifuged at 100,000 × g for 45 min. For V5 PREX2, the pellet was resuspended in 100 μl of lysis buffer, including 1% Nonidet P-40 and 0.25% sodium deoxycholate. For the endogenous fractionation, the membrane pellet was resuspended in 400 μl of lysis buffer plus detergents, and GST PTEN beads (5 μg of GST PTEN per sample) were incubated with both the cytosolic and membrane fraction for 4 h rotating at 4 °C to concentrate the endogenous PREX2 to be visualized by Western blot.
Purification of His6-PAK2
The His6-PAK2 plasmid was transformed in to BL-21(DE3) pLysE chemically competent cells (Invitrogen). Single colonies were picked and grown in a small culture overnight, and this was used to inoculate a 500-ml culture of LB broth until the A600 was 0.3–0.5 when protein expression was induced by the addition of 0.1 mm isopropyl β-d-1-thiogalactopyranoside (Sigma) for 4 h at 23 °C. The bacterial pellet was then resuspended in 10 ml of lysis buffer (50 mm Tris, pH 7.2, 400 mm NaCl, 1% Triton X-100, 1 mm EDTA). The lysate was sonicated and then centrifuged for 1 h at 40,000 × g. The lysate was loaded onto a HisTrap column (GE Healthcare) using an AKTA-FPLC (Running Buffer: 500 mm NaCl, 25 mm Tris, pH 7.6, 20 mm imidazole), and the protein was then eluted off the column with Elution Buffer containing 500 mm NaCl, 25 mm Tris, pH 7.6, and 500 mm imidazole. The eluate was combined in a 30,000 MWCO Amicon filter with 15 ml of buffer containing 25 mm Tris, pH 7.5, 150 mm NaCl, and 1 mm DTT. The solution was concentrated to 500 μl, and this was repeated three more times. 20% glycerol was added to the final protein, which was then snap-frozen and stored at −80 °C. Protein samples were quantified by SDS-PAGE followed by staining with Gel Code Blue in the presence of BSA standards.
In Vitro PAK2 Kinase Assays
V5 PREX2- or V5 CRAF(306–648)-expressing HEK293 cells were starved and then treated with 500 nm GDC0941 and 10 μm PF-3758309 for 30 min. One 15-cm plate was transfected for every three kinase reactions in a given experiment. Each plate was harvested in 1.5 ml of high salt lysis buffer containing 25 mm Tris, pH 7.5, 0.1% Triton X-100, 1 m NaCl, and 1× eukaryotic protease inhibitor mixture, and the lysates were then vortexed, sonicated, and centrifuged for 30 min at 4 °C. The lysates were combined and then incubated with V5-agarose beads for 16 h rotating at 4 °C. The V5 PREX2 and V5 CRAF(306–648) beads were washed three times in lysis buffer followed by three washes with 1× phospho buffer containing 50 mm HEPES, pH 7.5, 25 mm NaCl, 1.25 mm MgCl2, and 1.25 mm MnCl2. The beads were then divided equally into the appropriate number of tubes and were used in a 30-μl reaction consisting of the indicated amount of His6-PAK2, 100 μm cold ATP, and 10 μCi of [γ-32P]ATP, all diluted in 1× phospho buffer. The reactions were incubated at 30 °C for 45 min. After the reactions, the beads were then washed two times in kinase buffer and resuspended in 2× Laemmli sample buffer. Incorporation of [γ-32P]ATP was detected by gel electrophoresis. For non-radioactive kinase assays, the same protocol was followed except [γ-32P]ATP was not included in the reaction, and the beads were not washed as 2× Laemmli sample buffer was added directly to each 30-μl reaction.
Results
PREX2 Is Phosphorylated upon Insulin Stimulation by a PI3K-dependent Mechanism
Given the role of PREX2 in insulin signaling, we tested whether insulin treatment altered post-translational modifications of PREX2. When V5 PREX2 was expressed in HEK293 cells that were treated with insulin, enrichment of a slower migrating species of PREX2 was detected relative to starved cells after gel electrophoresis (Fig. 1A). Treatment of V5 PREX2 with λ-phosphatase, a nonspecific protein phosphatase, eliminated the insulin-induced upper band, suggesting that this mobility shift was caused by phosphorylation. When endogenous PREX2 was immunoprecipitated from HEK293 cells that had been treated with insulin, an electrophoretic mobility shift was also observed that was sensitive to λ-phosphatase (Fig. 1B). It is important to note that the endogenous PREX2 is almost completely shifted upon insulin treatment (which is not seen with V5 PREX2), suggesting that under physiological conditions PREX2 is stoichiometrically phosphorylated by a signal activated by insulin. Interestingly, although IGF1 also caused phosphorylation of PREX2, treatment with EGF did not (Fig. 1C). Although ERK signaling was induced by EGF in these cells, AKT signaling was induced only slightly, indicating that PI3K was probably not significantly activated under these conditions. This suggested that PREX2 phosphorylation could be PI3K-dependent. Using pharmacological inhibitors, we found that inhibition of PI3K with the pan class I PI3K inhibitor GDC0941 eliminated insulin-induced phosphorylation of PREX2, whereas inhibitors of AKT, PDK1, and mammalian target of rapamycin, three kinases activated by PIP3 downstream of insulin and PI3K, had no effect on PREX2 phosphorylation (Fig. 1D). Furthermore, in PTEN-null U87MG glioblastoma cells, expression of two tumor hot spot mutants that constitutively activate the catalytic PI3K subunit p110α, E545K and H1047R, induced PREX2 phosphorylation (Fig. 1E). These data show that PREX2 is phosphorylated after insulin stimulation through a PI3K-dependent mechanism.
FIGURE 1.

Insulin and PI3K induce phosphorylation of PREX2. A, Western blot analysis of V5 PREX2 that was isolated from starved or insulin-treated (5 μg/ml for 30 min) HEK293 cells and then treated with λ-phosphatase. B, Western blot analysis of PREX2 that was immunoprecipitated from starved or insulin-treated HEK293 cell lysates and then treated with λ-phosphatase. C, Western blot analysis of V5 PREX2-expressing HEK293 cells, which were starved and treated with insulin, 50 ng/ml IGF, or 50 ng/ml EGF for the indicated times. D, Western blot analysis of V5 PREX2-expressing HEK293 cells, which were starved and treated with DMSO or 500 nm GDC0941 (PI3Ki), 500 nm MK2206 (AKTi), 5 μm GSK2334470 (PDK1i), or 20 nm Rad001 (mTORC1i) followed by treatment with insulin. E, Western blot analysis of starved V5 PREX2-, p110α E545K-, or H1047R-expressing U87MG cells. EV, empty vector. F, mass spectrometry analysis was performed on V5 PREX2 isolated from HEK293 cells that were unstimulated (starved and treated with 500 nm GDC0941) or stimulated (insulin alone or combined with 100 nm calyculin A). Residues in blue are those only identified in one of the stimulated conditions. G, Western blot analysis of HEK293 cells expressing either V5 PREX2 WT or S1107A. H, Western blot analysis of V5 PREX2-expressing HEK293 cells that were starved and then treated with DMSO or 500 nm GDC0941 followed by treatment with insulin. I, Western blot analysis using indicated anti-PREX2 antibodies of GST or GST PTEN pulldowns from DBTRG cells that were starved and then treated with DMSO or 500 nm GDC0941 followed by insulin treatment. Antibodies used for Western analysis are indicated to the right of each panel. Phospho-specific antibodies to AKT (Thr-308 and Ser-473), ERK1/2 (Thr-202/Tyr-204), S6 (Ser-235/Ser-236), and PREX2 (Ser-1107) were used to detect alterations downstream of receptor tyrosine kinase ligands, small molecule inhibitors, and PI3K activation.
To identify PREX2 phosphorylation sites, we analyzed immunoprecipitated V5 PREX2 from HEK293 cells by mass spectrometry. This revealed a total of 19 serine and threonine phosphorylation sites, 12 of which were only identified on PREX2 after stimulation (cells treated with insulin alone or in combination with calyculin A, a PP1 and PP2A phosphatase inhibitor) (Fig. 1F). Using the Scansite tool under medium stringency, none of our phosphorylated residues fit a consensus motif of a kinase that is known to be activated by insulin and is included in the Scansite database, including AKT, GSK3β, PDK1, and ERK (52). Ser-1107 was of particular interest because it was phosphorylated in the stimulated condition, and the homologous site on PREX1 has previously been identified as a growth factor-dependent phosphorylation site. We developed an antibody to Ser-1107 and confirmed that the antibody could not recognize PREX2 when Ser-1107 was mutated to alanine, suggesting that the antibody is specific for the phosphorylated form of PREX2 at Ser-1107 (Fig. 1G). Importantly, phosphorylation of Ser-1107 on V5 PREX2 was increased after insulin treatment and was ablated following inhibition of PI3K (Fig. 1H). It is also important to note that although Ser-1107 was significantly enriched in the upper band of PREX2 compared with the total protein, the lower band was also phosphorylated to a small degree. If Ser-1107 were the sole site responsible for the shift, then only the upper band should be recognized by the phospho-specific antibody. This suggests that other insulin-dependent phosphorylation sites exist, and given that our mass spectrometry experiments revealed numerous phosphorylated residues in the stimulated condition in addition to Ser-1107, it is possible that some of these other residues could be involved. To verify that endogenous PREX2 is also phosphorylated at Ser-1107 and to examine the phosphorylation of PREX2 in another cell line, DBTRG glioblastoma cells, which express high levels of endogenous PREX2 and are PTEN null, were treated with insulin and GDC0941 (Fig. 1I) (20). A GST PTEN pulldown was performed to efficiently isolate endogenous PREX2, and phosphorylation at Ser-1107 was found to be increased after insulin treatment and was reduced upon PI3K inhibition. Taken together, we show that global phosphorylation of PREX2 and phosphorylation at Ser-1107 can be increased by insulin and requires PI3K activation.
PREX2 Is Dephosphorylated by the Protein Phosphatases PP1α and PP2A
As described previously, mass spectrometry revealed only serine and threonine phosphorylation sites on PREX2, many of which were detected with a PP1 inhibitor. It has also been reported that PP1α binds a fragment of PREX2 that contains a consensus PP1α-binding site, and furthermore, dephosphorylation of PREX1 by PP1α activates its GEF activity (53, 54). These data led us to test the hypothesis that PP1α could dephosphorylate PREX2. First, we showed that PREX2 could bind to GST PP1α, confirming the previous reports that these two proteins interact (Fig. 2A) (53, 54). Additionally, the PREX2-PP1α interaction was ablated by mutating two critical residues within the PP1α consensus binding site, Val-1085 and Phe-1087, to alanine. Treatment of WT PREX2 with calyculin A stimulated phosphorylation of PREX2 even in the absence of insulin and resulted in a maximally phosphorylated form of PREX2 (Fig. 2B). Conversely, overexpression of PP1α eliminated the insulin-induced phosphorylation of PREX2, including phosphorylation at Ser-1107, suggesting that it is a PP1α-regulated site. Furthermore, the 1085/87A mutant was insensitive to dephosphorylation by PP1α. To test whether PREX2 is a direct substrate of PP1α, we performed an in vitro phosphatase assay and found that GST PP1α can dephosphorylate PREX2 (Fig. 2C).
FIGURE 2.

PREX2 is dephosphorylated by PP1α and PP2A. A, Western blot analysis of GST or GST PP1α pulldowns from HEK293 cells expressing either V5 PREX2 WT or 1085/87A. EV, empty vector. B, Western blot analysis of HEK293 cells expressing V5 PREX2 WT or 1085/87A with or without co-expression of V5 PP1α. Cells were starved and then treated with DMSO or 100 nm calyculin A, followed by treatment with insulin. C, ability of GST PP1α to dephosphorylate V5 PREX2 isolated from HEK293 cells was assessed and analyzed by Western blot. D, Western blot analysis of starved V5 PREX2 WT or 1085/87A-expressing HEK293 cells that were treated with the indicated concentrations of okadaic acid for 30 min. E, ability of purified PP2A to dephosphorylate V5 PREX2 isolated from HEK293 cells was assessed and analyzed by Western blot. F, Western blot analysis of HEK293 cells expressing V5 PREX2 with or without co-expression of V5 PP1α or HA PP2A (catalytic subunit). Cells were starved and then treated with insulin.
Because the 1085/1087A mutant does not bind PP1α, it was surprising that it was not always in the maximally shifted form that is seen after calyculin A treatment (Fig. 2B). One reason for this could be that PP2A, which is also inhibited by calyculin A, is able to compensate for the absence of PP1α regulation by dephosphorylating PREX2. To probe this, we used okadaic acid, which inhibits both PP1α and PP2A but is much more potent toward PP2A at low doses (55). When treated with okadaic acid, phosphorylation of PREX2 was increased, suggesting PP2A may have a role in PREX2 dephosphorylation (Fig. 2D). The 1085/1087A mutant was phosphorylated more than wild type PREX2 at all doses, indicating that PP1α contributes to PREX2 dephosphorylation when PP2A is inhibited. We then performed an in vitro PP2A phosphatase assay to show that PP2A dephosphorylates PREX2 (Fig. 2E). Finally, overexpression of the PP2A catalytic subunit reduced phosphorylation of PREX2, including at Ser-1107, although not nearly as efficiently as PP1α (Fig. 2F). From these data it is clear that phosphatases are important in regulating PREX2 phosphorylation, potentially acting in opposition of a kinase downstream of insulin stimulation.
Phosphorylation of PREX2 Downstream of Insulin Requires Binding to PIP3
We next sought to understand how PREX2 phosphorylation was regulated after insulin stimulation to better inform our search for a PREX2 kinase. Our data from Fig. 1 suggest that PREX2 is either phosphorylated by PI3K or by a kinase downstream of PIP3 generation. We found that expression of PTEN in U87MG cells eliminated PREX2 phosphorylation and that the lipid phosphatase activity of PTEN is required as the lipid phosphatase dead G129E mutant had no effect (Fig. 3A). Because PTEN counteracts PI3K by reducing PIP3 levels rather than directly regulating PI3K, this result demonstrated that PREX2 phosphorylation is dependent on a PIP3-regulated kinase. To further support the hypothesis that PREX2 phosphorylation is PIP3-dependent, we sought to mutate PREX2 in its PH domain to interfere with PIP3 binding and then determine the effect on phosphorylation. Previous studies have identified a PIP3 consensus binding sequence within the PH domains, which consists of a lysine at the second to last position in the PH domain β1 strand and an arginine or lysine at the second and fourth position from the beginning of the β2 strand (56, 57). Although there is no known structure of PREX2, we created a model of the PREX2 PH domain, and the potential residues that compose the β1β2 loop were identified (Fig. 3, B and C). The β1β2 loop of PREX2 from our model was aligned with the β1β2 loop of two well known PIP3-binding proteins, AKT1 and PDK1. Critical residues for binding of AKT1 and PDK1 to PIP3 have been identified, and these residues are highlighted in Fig. 3D (58, 59). According to our model, PREX2 contains the conserved lysine in the β1 strand (Lys-254) and has an arginine at the third position of the β2 strand (Arg-263), and we hypothesized that these two residues could be critical for PREX2 binding to PIP3. Mutation of either Lys-254 or Arg-263 to glutamate was sufficient to block the binding of PREX2 to PIP3 beads, and the R263E mutation blocked PIP3-mediated stimulation of PREX2 GEF activity (Lys-254 was not tested) (Fig. 3, D and E). Importantly, both of these mutations also resulted in significantly reduced phosphorylation of PREX2 after insulin stimulation (Fig. 3F). These data confirm that insulin-dependent phosphorylation of PREX2 requires PIP3 binding to the PH domain and that residues Lys-254 and Arg-263 surrounding the β1β2 loop are necessary for this interaction, and thus are critical for phosphorylation.
FIGURE 3.

Phosphorylation of PREX2 downstream of insulin requires PIP3 binding. A, Western blot analysis of U87MG cells expressing V5 PREX2 and either PTEN WT or the lipid phosphatase dead mutant PTEN G129E. EV, empty vector. B, ribbon diagram of the PREX2 PH domain model with the putative PIP3 binding region identified. C, predicted secondary structures of the PREX2 PH domain from model. D, top, Western blot analysis of PIP3 pulldowns from HEK293 cells expressing either V5 PREX2 WT, K254E, or R263E. Bottom, diagram showing the β1β2 loop region of the PREX2 PH domain model aligned with the same region on PDK1 and AKT1. E, V5 PREX2 WT or R263E was purified from HEK293 cell lysates, and the ability of increasing doses of PIP3 to stimulate PREX2 GEF activity toward GST Rac1 was assessed in an in vitro GEF assay. Data are mean ± S.D. for at least two experiments with samples done in duplicate at each PIP3 concentration, and * represents p < 0.001 by t test. F, Western blot analysis of starved or insulin-treated HEK293 cells expressing V5 PREX2 WT, K254E, or R263E.
Expression of Gβγ Stimulates Phosphorylation of PREX2
Given that PREX2 is activated by PIP3 and requires PIP3 for phosphorylation, we hypothesized that phosphorylation of PREX2 may be associated with activation of PREX2 GEF activity. Overexpression of the second messenger Gβγ, another activator of PREX2 GEF activity, induced the phosphorylation of PREX2, further suggesting a link between PREX2 activation and phosphorylation (Fig. 4A). Interestingly, Gβγ-induced phosphorylation of PREX2 was only mildly sensitive to PI3K inhibition by GDC0941 at a concentration where insulin-induced phosphorylation of PREX2 and AKT was completely eliminated, suggesting that phosphorylation mediated by Gβγ occurs largely through a PI3K-independent mechanism (Fig. 4B). Gβγ activates PI3Kγ and PI3Kβ independently of insulin receptor activation, and GDC0941 has a relatively high IC50 toward PI3Kγ, so we tested a more effective PI3Kγ inhibitor, BEZ235 (60). This drug had very little effect on Gβγ-induced phosphorylation of PREX2, while reducing insulin-dependent phosphorylation to baseline levels (Fig. 4C). The mild effect of PI3K inhibitors on Gβγ-induced phosphorylation of PREX2 could be a result of eliminating the basal PIP3-dependent phosphorylation that exists in starved cells or there could be some cross-talk between Gβγ and certain PI3K isoforms. Furthermore, as was the case with insulin-dependent phosphorylation, PP1α expression eliminated phosphorylation of PREX2 that was stimulated by Gβγ (Fig. 4D). From these data, it is clear that increasing levels of intracellular Gβγ is another mechanism to stimulate phosphorylation of PREX2.
FIGURE 4.

Gβγ stimulates phosphorylation of PREX2. A, Western blot analysis of starved HEK293 cells where V5 PREX2 was expressed with or without FLAG/HA Gβγ. B, Western blot analysis of HEK293 cells where V5 PREX2 was expressed with or without FLAG/HA Gβγ. Cells were then starved and treated with DMSO or 500 nm GDC0941 followed by treatment with insulin. C, Western blot analysis of HEK293 cells where V5 PREX2 was expressed with or without FLAG/HA Gβγ. Cells were then starved and treated with DMSO or either 500 nm or 5 μm BEZ235 followed by treatment with insulin. D, Western blot analysis of starved HEK293 cells expressing V5 PREX2 with FLAG/HA Gβγ and V5 PP1α.
Phosphorylation of PREX2 Blocks Binding to the Cellular Membrane, PIP3, and Gβγ
To investigate the effect of phosphorylation on PREX2 function, we first analyzed the ability of phosphorylated PREX2 to localize to the cellular membrane. Rac1, the target of PREX2 GEF activity, is at the membrane, and furthermore, membrane-bound PREX1 has been shown to be more active than cytosolic PREX1, suggesting that alterations in membrane binding could have important functional implications for PREX2 (61). To probe this, V5 PREX2-expressing cells were subjected to cellular fractionation. The cytoplasmic fraction clearly showed that after insulin and Gβγ stimulation, both the phosphorylated and dephosphorylated forms of PREX2 existed in the cell (Fig. 5A). Importantly, only the dephosphorylated form of PREX2 was present in the membrane fraction, suggesting that the phosphorylated form of PREX2 was unable to bind to the membrane. The same localization pattern was observed with endogenous PREX2 after pulling down endogenous PREX2 from both the membrane and cytosolic fractions with GST PTEN (Fig. 5B).
FIGURE 5.

Phosphorylation of PREX2 prevents binding to the membrane, PIP3, and Gβγ. A, Western blot analysis of cytosolic and membrane fractions from starved or insulin-treated HEK293 cells expressing V5 PREX2 with or without co-expression of FLAG/HA Gβγ. B, Western blot analysis of GST PTEN pulldowns of endogenous PREX2 from cytosolic and membrane fractions of HEK293 cells. Cells were starved and then treated with insulin for indicated times. C, Western blot analysis of PIP3 pulldowns from starved or insulin-treated HEK293 cells expressing V5 PREX2 with or without FLAG/HA Gβγ. D, Western blot analysis of endogenous PREX2 isolated from HEK293 cells with either PIP3 beads (top) or PREX2 antibody (bottom). Cells were starved and then treated with DMSO or 500 nm GDC0941 followed by treatment with insulin. E, Western blot analysis of FLAG immunoprecipitations from HEK293 cells co-expressing FLAG/HA Gβγ and V5 PREX2 with and without 100 nm calyculin A treatment. F, Western blot analysis of pulldowns with GST PTEN, GST PP1α, GST Rac1, or PIP3 beads from HEK293 cells expressing V5 PREX2 with or without FLAG/HA Gβγ. The cells were starved and treated with insulin or 100 nm calyculin A. U, upper band; L, lower band.
Both PIP3 and Gβγ are membrane-associated, so we hypothesized that phosphorylated PREX2 may have reduced affinity for PIP3 and Gβγ, resulting in the localization patterns we observed. When cells expressing V5 PREX2 were stimulated with insulin or Gβγ and a PIP3 pulldown was performed, the phosphorylated form of PREX2, although present in the input sample, was completely absent in the pulldown (Fig. 5C). The effect of insulin stimulation on PIP3 binding was even more striking with endogenous PREX2, as insulin led to stoichiometric phosphorylation of PREX2, which then profoundly reduced the ability of PREX2 to bind to PIP3, an effect that was reversed with PI3K inhibition (Fig. 5D). Next, we tested whether phosphorylation of PREX2 affected its ability to bind to Gβγ by performing a co-immunoprecipitation of V5 PREX2 and FLAG-Gβγ. Although the effect was not quite as striking as with PIP3, an immunoprecipitation with a FLAG antibody showed that the dephosphorylated and faster migrating species of PREX2 preferentially bound to Gβγ (Fig. 5E). Furthermore, calyculin A treatment reduced the overall binding of PREX2 to Gβγ. To determine whether phosphorylation prevented binding of PREX2 to all of its known interacting partners, we performed pulldowns with GST-tagged PTEN, PP1α, and Rac1. Although PIP3 only bound the dephosphorylated form of PREX2, Rac1 and PTEN did not discriminate between the forms of PREX2 (Fig. 5F). The dephosphorylated form of PREX2 was somewhat enriched in the PP1α pulldowns (although not as much as in the PIP3 pulldowns), but this is likely because PP1α is dephosphorylating PREX2. These findings suggest that phosphorylation selectively blocks binding of PREX2 to PIP3 and Gβγ. Collectively, these data show that phosphorylation of PREX2 functions to block binding to PIP3 and Gβγ, preventing PREX2 from being on the membrane where it can stimulate Rac1.
Phosphorylation Inhibits PIP3- and Gβγ-stimulated GEF Activity of PREX2 as Part of Rac1-dependent Negative Feedback
Given that phosphorylation blocked PREX2 binding to PIP3 and Gβγ, it was then critical to determine whether phosphorylation prevented PIP3- and Gβγ-dependent activation of GEF activity. V5 PREX2 was purified from HEK293 cells after being treated with either GDC0941, insulin in combination with Gβγ co-expression, or calyculin A to achieve relatively dephosphorylated, partially phosphorylated, and maximally phosphorylated states of PREX2, respectively. When compared with dephosphorylated PREX2, partially phosphorylated PREX2 showed decreased activation of GEF activity at multiple PIP3 concentrations, whereas activation of the maximally phosphorylated form of PREX2 was reduced even further (Fig. 6A). In addition, Gβγ-dependent stimulation of PREX2 GEF activity was similarly affected by phosphorylation across many Gβγ concentrations (Fig. 6B). Furthermore, upon insulin treatment of HEK293 cells, the phosphorylation of both endogenous PREX2 and transfected V5 PREX2 was low at early time points (30 s to 2 min), and it continued to increase throughout the 30-min time course (Fig. 6, C and D). This is in contrast to phosphorylation of AKT, which was highly phosphorylated within the first 30 s of insulin treatment. This delayed time course of PREX2 phosphorylation, in combination with our data showing that phosphorylation reduces GEF activity, supports a model where phosphorylation occurs after PREX2 activation by PIP3 and Gβγ to attenuate its function and the Rac1 signal.
FIGURE 6.

Phosphorylation reduces PREX2 GEF activity and is dependent on Rac1. A and B, V5 PREX2 was expressed in HEK293 cells with or without co-expression of FLAG/HA Gβγ and then was purified after being treated with either 500 nm GDC0941, insulin (with co-expression of FLAG/HA Gβγ), or 100 nm calyculin A. The ability of increasing doses of PIP3 in A or Gβγ in B to stimulate PREX2 GEF activity toward GST Rac1 was assessed in an in vitro GEF assay. Data at each PIP3 and Gβγ concentration are means ± S.D. for three experiments, and # represents p < 0.05 for GDC0941 versus Gβγ/insulin, and * represents p < 0.005 for GDC0941 versus calyculin A by t test. C, Western blot analysis of endogenous PREX2 that was immunoprecipitated (IP) from HEK293 cells that were starved and treated with insulin for the indicated times. D, Western blot analysis of V5 PREX2-expressing HEK293 cells that were starved and then treated with insulin for the indicated times. For quantification of phosphorylation in C and D, intensity of the band was normalized to total protein of that sample. The highest value was set to 1, and the remaining samples were normalized to this value. E, Western blot analysis of starved or insulin-treated HEK293 cells expressing V5 PREX2 with or without MYC Rac1 WT or the dominant negative mutant T17N. F, Western blot analysis of starved or insulin-treated HEK293 cells expressing V5 PREX2 WT or the GEF dead E30A/N212A mutant.
Based on this model, we predicted that higher Rac1 activity within the cell would initiate this negative feedback and increase phosphorylation of PREX2. Indeed, expression of Rac1 in starved HEK293 cells induced phosphorylation of PREX2 even in the absence of insulin (Fig. 6E). In contrast, the dominant negative T17N Rac1 mutant did not increase PREX2 phosphorylation in starved cells, and furthermore, the T17N mutant blocked the insulin-stimulated phosphorylation, indicating that Rac1 is downstream of PIP3 generation. In addition, a GEF dead PREX2 mutant (E30A/N212A) was not phosphorylated following insulin treatment, confirming that PREX2 must be active to initiate this pathway (Fig. 6F) (30). These results suggest that the kinase or kinases that are phosphorylating PREX2 are downstream of Rac1 activation.
PAK Kinases Mediate the Phosphorylation of PREX2 Downstream of Rac1
The PAK family of kinases, consisting of group I PAKs (PAK1–3) and group II PAKs (PAK4–6), are important mediators of signaling downstream of RHO family GTPases. The regulation of group I PAKs is best understood, and their kinase activity is activated upon binding to GTP-bound Rac1, making them candidate kinases for PREX2 (7). Furthermore, PAK1 kinase activity is activated downstream of insulin activation, a context where we see phosphorylation of PREX2 (62). Inhibition of both groups of PAKs with the inhibitor PF-3758309 blocked the insulin- and Gβγ-induced phosphorylation of PREX2 (Fig. 7, A and B). Furthermore, overexpression of PAK1 stimulated phosphorylation of PREX2 in both the starved and insulin-treated conditions (Fig. 7C). In addition, the kinase dead and dominant negative PAK1 mutant (K299R) completely eliminated any phosphorylation of PREX2 in either the starved or insulin-treated contexts. As we saw in the case of insulin and Gβγ-induced phosphorylation in Fig. 6, PAK1-induced phosphorylation also affected PREX2 function (Fig. 7D). PREX2 that was purified from cells co-expressing wild type PAK1 had lower GEF activity toward Rac1 over multiple PIP3 concentrations compared with PREX2 purified from cells co-expressing dominant negative PAK1. Furthermore, the GEF activity of PREX2 that was dephosphorylated by co-expression of PAK1 K299R was not different from that of PREX2 that was dephosphorylated by PI3K inhibition. These data show that PAKs are important regulators of PREX2 phosphorylation and PREX2 GEF activity following insulin and Gβγ stimulation.
FIGURE 7.

PAKs mediate the phosphorylation of PREX2. A, Western blot analysis of V5 PREX2-expressing HEK293 cells that were starved and then treated with DMSO or varying concentrations of the pan-PAK inhibitor PF-3758309 followed by insulin treatment. B, Western blot analysis of HEK293 cells expressing V5 PREX2 with or without Gβγ. Cells were starved and treated with DMSO or varying concentrations of PF-3758309. C, Western blot analysis of starved or insulin-treated HEK293 cells expressing V5 PREX2 with or without MYC PAK1 WT or the kinase dead mutant K299R. D, PREX2 was purified from HEK293 cells co-expressing either MYC PAK1 WT or K299R. The ability of increasing doses of PIP3 to stimulate GEF activity toward GST Rac1 was assessed in an in vitro GEF assay. Data are means ± S.D. for two experiments with samples done in triplicate at each PIP3 concentration. Bar graph at right compares PREX2 purified from GDC0941-treated HEK293 cells to PREX2 from HEK293 cells co-expressing either MYC PAK1 WT or K299R. Data are means ± S.D. for one representative experiment with samples done in triplicate at each PIP3 concentration. For both panels, * represents p < 0.005 by t test. E, in vitro kinase assay with purified His PAK2 and V5 PREX2 or CRAF(306–648) isolated on V5-agarose beads. Incorporation of [γ-32P]ATP was analyzed. F, Western blot analysis of an in vitro kinase assay with purified His PAK2 and V5 PREX2 or CRAF(306–648) isolated on V5-agarose beads using antibodies to detect phosphorylation as indicated. G, in vitro kinase assay with purified His PAK2 and V5 PREX2 WT or S1107A isolated on V5-agarose beads. Incorporation of [γ-32P]ATP was analyzed.
We then asked whether PREX2 was a direct substrate of PAK by performing in vitro kinase assays. For these assays, we used recombinant PAK2, which has previously been shown to be constitutively active when produced in bacteria (63). PREX2 was phosphorylated in a dose-dependent manner by PAK2 to a similar degree as a fragment of CRAF, a known substrate of PAKs (Fig. 7E) (64). Importantly, PAK2 phosphorylation caused a complete mobility shift in PREX2, suggesting high levels of phosphorylation and further implicating PAKs as the primary PREX2 kinases. PAK2 also resulted in clear phosphorylation at Ser-1107; however, an alanine mutation at Ser-1107 did not significantly reduce overall phosphorylation of PREX2 (Fig. 7, F and G). This is further evidence that Ser-1107 is one of multiple residues on PREX2 that is regulated by PAKs downstream of PIP3 and Gβγ. It is important to note that the PREX2 was purified from mammalian cells for these in vitro kinase assays, and we cannot completely discount the possibility that other kinases co-precipitating with PREX2 are activated by PAK. However, it is clear from these data that a signal activated by PAKs is sufficient for full phosphorylation of PREX2, providing further evidence that PAKs initiate negative feedback to reduce Rac1 activity downstream of PIP3 or Gβγ activation of PREX2.
Discussion
Rac1 is a critical mediator of insulin signaling that is activated after insulin binding through a PIP3-dependent mechanism (16). A current model of Rac1 regulation involves GTP loading and activation by GEFs, such as PREX2, followed by GAP- or GDI-mediated inactivation. However, Rac1 inactivation after insulin stimulation is not well understood, and our study provides further understanding of this process by showing that Rac1 inactivation occurs via feedback initiated by its own effectors. We have identified a signaling pathway downstream of insulin receptor activation of PREX2 where the Rac1-regulated PAKs phosphorylate PREX2 to reduce PREX2 GEF activity. We also found that activation of PREX2 with Gβγ initiates the same pathway. Altogether, we propose a model in which PREX2 is dephosphorylated in an unstimulated cell, and then upon stimulation with insulin or a GPCR agonist, PIP3 or Gβγ levels increase, and dephosphorylated PREX2 is primed to bind these molecules on the membrane where it then activates Rac1 (Fig. 8). Higher levels of GTP-bound Rac1 lead to activation of PAKs, which phosphorylate PREX2 to release it from the membrane, decreasing the Rac1 signal. Importantly, we found in multiple cell lines that most of the endogenous PREX2 in the cell is phosphorylated downstream of insulin, demonstrating that this is a major mechanism of regulation for PREX2 GEF activity. Additionally, phosphatase activity by either PP1α or PP2A can complete the circuit by dephosphorylating PREX2, priming it for future activation by PIP3 or Gβγ. Altogether, this model allows for tight regulation of the length and amplitude of Rac1 activation, resulting in a transient Rac1 signal following ligand binding.
FIGURE 8.
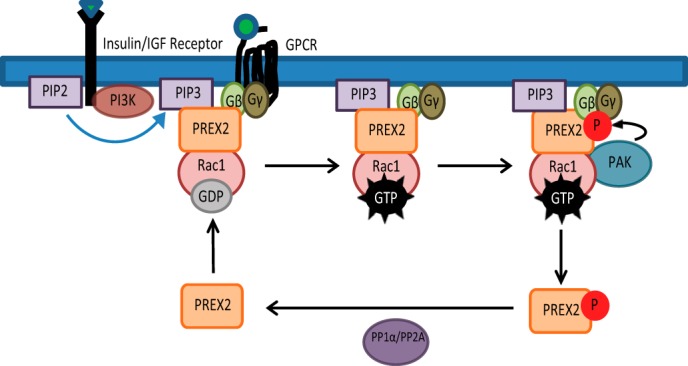
Model for PAK-mediated regulation of PREX2 GEF activity and localization. Upon receptor stimulation, PIP3 and Gβγ recruit PREX2 to the cell membrane and activate PREX2 GEF activity, which promotes GTP loading on Rac1. Rac1-GTP then activates PAKs to propagate the Rac1 signal. PAKs also phosphorylate PREX2, reducing the ability of PREX2 to bind to PIP3, Gβγ, and the membrane, resulting in a decrease in PREX2 GEF activity toward Rac1. PREX2 can then be dephosphorylated by phosphatases such as PP1α and PP2A to complete the circuit and prime PREX2 for future activation by PIP3 or Gβγ. In this model, chronic stimulation of PIP3 or Gβγ could lead to ineffective signaling of PREX2 to Rac1.
We also identified an insulin-, Gβγ-, and PAK-dependent phosphorylation site at Ser-1107. Given that phosphorylation of PREX2 reduces binding to PIP3 and Gβγ, both of which appear to interact with PREX2 through the PH domain, it is interesting that phosphorylation of the IP4P domain could affect binding to these second messengers. One possible explanation is that phosphorylation events in the IP4P domain affect intramolecular interactions within PREX2, altering the accessibility of the PH domain to reduce PIP3 and Gβγ binding. Our data also show that Ser-1107 is not the only site that is phosphorylated by PAK, so it is possible that phosphorylation events in other domains of PREX2 are cooperating with Ser-1107 phosphorylation to affect intramolecular interactions. Further phosphorylation events may even occur within the PH domain where they can sterically block PIP3 and Gβγ binding. Our mass spectrometry data revealed a total of 12 phosphorylation events that were specific to the stimulated condition, and it is possible that in addition to Ser-1107 some of these residues are also PAK-dependent sites. Interestingly, phosphorylation has previously been shown to affect intramolecular interactions of the PREX2 homolog, PREX1. PKA phosphorylation reduces PREX1 binding to Gβγ and also reduces Gβγ-induced PREX1 GEF activity by disrupting an intramolecular interaction between the IP4P domain and a region comprising the second DEP and first PDZ domains (37).
Given the role of PAKs in propagating Rac1-dependent processes, such as lamellipodia formation and glucose uptake, it is unexpected for PAKs to mediate negative feedback to turn off Rac1 activity (65). However, there is precedent for this in lower eukaryotes. In the yeast Saccharomyces cerevisiae, the Pak family member Cla4 phosphorylates Cdc24, a GEF for the Cdc42 GTPase, and phosphorylation reduces GEF activity in vitro and reduces levels of Cdc42 at the front of a cell during polarization (66–68). Furthermore, in the fungus Ustilago maydis, where Cdc24 is also a Rac1 GEF, Cla4 phosphorylates Cdc24, causing its degradation and a reduction in Rac1 activation (69). Here, we show that this type of PAK-mediated negative regulation of GEFs occurs in higher eukaryotes as well.
This type of regulation, where a pathway evolves a mechanism to turn a signal off after it has been activated, has been reported in many contexts within mammalian cells, including the insulin-signaling pathway. For example, mTORC1, an important effector of insulin signaling, also initiates negative feedback to reduce activation of the pathway. Insulin activation of mTORC1 leads to the stimulation of various downstream kinases to regulate protein synthesis. However, mTORC1 activation also results in serine phosphorylation of insulin receptor substrate (IRS) proteins, reducing their stability and decreasing the insulin signal. IRS protein levels can then be restored upon pathway inactivation (70–73). Importantly, mTORC1-dependent regulation of IRS stability appears to be a critical consideration for therapeutics given that inhibition of mTORC1 leads to increased AKT phosphorylation in cancer cell lines and human patients, which may partially explain the modest effects of these inhibitors in cancer patients (74).
The role of mTORC1 in insulin signaling is very similar to the model we propose for PAKs, where PAK can both propagate the insulin-dependent Rac1 signal and negatively regulate it through PREX2. Furthermore, similar to mTORC1-dependent negative feedback, PAK phosphorylation of PREX2 could have important therapeutic implications. PAK inhibitors are currently being evaluated as cancer therapeutics, and if PAK negative regulation of PREX2 is playing a significant role in insulin signaling, or more generally in Rac1 activation, then it may be important to consider in the context of certain cancers (75). For example, a tumor with PREX2 overexpression may not respond well to PAK inhibitors, which could increase PREX2 GEF activity toward Rac1. Furthermore, given the role of PREX2 in cancer, it will be important to study PAK-mediated inactivation of PREX2 not only in the context of resistance to therapeutics but in tumorigenesis. Many PREX2 mutations have been identified in different types of cancer, and it is possible that some of these could disrupt the PAK-dependent negative feedback and activate Rac1 signaling.
PREX2 has previously been identified as an important mediator of insulin signaling, and our data showing that PREX2 function is tightly regulated by insulin-induced phosphorylation further supports this idea (14). Future experiments should focus on the effect of this feedback circuit on both PREX2- and Rac1-dependent physiological outputs downstream of insulin receptor activation, including glucose metabolism. It will also be of interest to study this signaling pathway in the context of insulin-related disease, such as diabetes. PREX2 protein expression appears to be lower in adipose tissue of insulin-resistant human patients, suggesting that reducing PREX2 activity to prevent glucose uptake is a possible mechanism for insulin resistance (14). In addition, the importance of Rac1 in insulin resistance is highlighted by the fact that Rac1 activity can be decreased in muscle tissue of insulin-resistant mice and humans (76, 77). Our data present the possibility that a reduction in PREX2 GEF activity and Rac1 activation could also be achieved through PAK-mediated phosphorylation and that phosphorylation of PREX2 could be altered in diabetes and could potentially be a cause of insulin resistance. Interestingly, the activation of feedback mechanisms in insulin resistance has been identified previously, including mTORC1-dependent ribosomal S6 kinase phosphorylation of IRS1 (78, 79).
Our findings for PREX2 could also have important implications for PREX1. Interestingly, the residue on PREX1 (Ser-1169) that is homologous to Ser-1107 on PREX2 is phosphorylated following treatment with neuregulin and IGF (26, 35). PREX1 is also dephosphorylated by PP1α, and PP1α dephosphorylation activates PREX1 GEF activity, which is consistent with our model for PREX2 (53). In addition, fractionation experiments in Sf9 insect cells show that PI3K and Gβγ co-expression leads to an electrophoretic mobility shift of PREX1, and the slower migrating species, presumably a phosphorylated form, is mostly excluded from the membrane fraction (61). The similarities between phosphorylation events on PREX1 and PREX2 suggest that it is certainly possible that PREX1 is also regulated by PAK phosphorylation.
Collectively, our data represent a novel paradigm for maintaining a tightly regulated Rac1 signal by blocking the stimulation of the specific GEF that was activated, in this case PREX2. This type of regulation adds a layer of complexity to the current model where Rac1 is activated by GEFs and inactivated by GAPs and GDIs. If PAKs regulate other GEFs the same way, then this signaling pathway could have implications in many aspects of cellular physiology.
Author Contributions
D. B. designed experiments, performed experiments, analyzed data, and wrote the paper. R. P. designed the studies, analyzed data, and wrote the paper. S. M. S. and C. H. contributed new reagents/analytic tools. A. Silkov and B. H. created the PREX2 PH domain model. A. C. developed the phospho-Ser-1107 PREX2 antibody. A. Shymanets and B. N. purified the Gβγ for in vitro GEF assays in Fig. 6. J. M. A. designed, performed, and analyzed the mass spectrometry experiments shown in Fig. 1. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank all the members of the Parsons laboratory for reading the manuscript and providing great feedback. We also thank Min Yuan for help with mass spectrometry experiments.
This work was supported in part by National Institutes of Health Grants R01CA155117 (to R. P.), R01CA184016 (to R. P.), 5P01CA120964 (to J. M. A.), 1S10OD010612 (to J. M. A.), and R01GM030518 (to B. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- PIP3
- phosphatidylinositol-3,4,5-trisphosphate
- GEF
- guanine nucleotide exchange factor
- PAK
- p21-activated kinase
- GPCR
- G-protein-coupled receptor
- GDI
- guanine nucleotide dissociation inhibitor
- PH
- pleckstrin homology
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- GAP
- GTPase-activating protein
- PTEN
- phosphatase and tensin homolog
- MWCO
- molecular weight cutoff
- IRS
- insulin receptor substrate.
References
- 1.Donald S., Hill K., Lecureuil C., Barnouin R., Krugmann S., John Coadwell W., Andrews S. R., Walker S. A., Hawkins P. T., Stephens L. R., and Welch H. C. (2004) P-Rex2, a new guanine-nucleotide exchange factor for Rac. FEBS Lett. 572, 172–176 [DOI] [PubMed] [Google Scholar]
- 2.Rosenfeldt H., Vázquez-Prado J., and Gutkind J. S. (2004) P-REX2, a novel PI-3-kinase sensitive Rac exchange factor. FEBS Lett. 572, 167–171 [DOI] [PubMed] [Google Scholar]
- 3.Cherfils J., and Zeghouf M. (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 4.Abo A., Pick E., Hall A., Totty N., Teahan C. G., and Segal A. W. (1991) Activation of the NADPH oxidase involves the small GTP-binding protein p21rac1. Nature 353, 668–670 [DOI] [PubMed] [Google Scholar]
- 5.Chen Z., Borek D., Padrick S. B., Gomez T. S., Metlagel Z., Ismail A. M., Umetani J., Billadeau D. D., Otwinowski Z., and Rosen M. K. (2010) Structure and control of the actin regulatory WAVE complex. Nature 468, 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fritsch R., de Krijger I., Fritsch K., George R., Reason B., Kumar M. S., Diefenbacher M., Stamp G., and Downward J. (2013) RAS and RHO families of GTPases directly regulate distinct phosphoinositide 3-kinase isoforms. Cell 153, 1050–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manser E., Leung T., Salihuddin H., Zhao Z. S., and Lim L. (1994) A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 367, 40–46 [DOI] [PubMed] [Google Scholar]
- 8.Knaus U. G., Morris S., Dong H. J., Chernoff J., and Bokoch G. M. (1995) Regulation of human leukocyte p21-activated kinases through G protein-coupled receptors. Science 269, 221–223 [DOI] [PubMed] [Google Scholar]
- 9.Wertheimer E., Gutierrez-Uzquiza A., Rosemblit C., Lopez-Haber C., Sosa M. S., and Kazanietz M. G. (2012) Rac signaling in breast cancer: a tale of GEFs and GAPs. Cell. Signal. 24, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoyanov B., Volinia S., Hanck T., Rubio I., Loubtchenkov M., Malek D., Stoyanova S., Vanhaesebroeck B., Dhand R., and Nürnberg B. (1995) Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science 269, 690–693 [DOI] [PubMed] [Google Scholar]
- 11.Kurosu H., Maehama T., Okada T., Yamamoto T., Hoshino S., Fukui Y., Ui M., Hazeki O., and Katada T. (1997) Heterodimeric phosphoinositide 3-kinase consisting of p85 and p110β is synergistically activated by the βγ subunits of G proteins and phosphotyrosyl peptide. J. Biol. Chem. 272, 24252–24256 [DOI] [PubMed] [Google Scholar]
- 12.Auger K. R., Serunian L. A., Soltoff S. P., Libby P., and Cantley L. C. (1989) PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 57, 167–175 [DOI] [PubMed] [Google Scholar]
- 13.Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 14.Hodakoski C., Hopkins B. D., Barrows D., Mense S. M., Keniry M., Anderson K. E., Kern P. A., Hawkins P. T., Stephens L. R., and Parsons R. (2014) Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 111, 155–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khayat Z. A., Tong P., Yaworsky K., Bloch R. J., and Klip A. (2000) Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 113 Pt 2, 279–290 [DOI] [PubMed] [Google Scholar]
- 16.JeBailey L., Rudich A., Huang X., Di Ciano-Oliveira C., Kapus A., and Klip A. (2004) Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol. Endocrinol. 18, 359–372 [DOI] [PubMed] [Google Scholar]
- 17.Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., and Hemmings B. A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 18.Taniguchi C. M., Emanuelli B., and Kahn C. R. (2006) Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 [DOI] [PubMed] [Google Scholar]
- 19.Ruderman N. B., Kapeller R., White M. F., and Cantley L. C. (1990) Activation of phosphatidylinositol 3-kinase by insulin. Proc. Natl. Acad. Sci. U.S.A. 87, 1411–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fine B., Hodakoski C., Koujak S., Su T., Saal L. H., Maurer M., Hopkins B., Keniry M., Sulis M. L., Mense S., Hibshoosh H., and Parsons R. (2009) Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science 325, 1261–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z., Paik J. H., Paik J. H., Wang Z., Hla T., and Wu D. (2005) Role of guanine nucleotide exchange factor P-Rex-2b in sphingosine 1-phosphate-induced Rac1 activation and cell migration in endothelial cells. Prostaglandins Other Lipid Mediat. 76, 95–104 [DOI] [PubMed] [Google Scholar]
- 22.Dong X., Mo Z., Bokoch G., Guo C., Li Z., and Wu D. (2005) P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr. Biol. 15, 1874–1879 [DOI] [PubMed] [Google Scholar]
- 23.Welch H. C., Coadwell W. J., Ellson C. D., Ferguson G. J., Andrews S. R., Erdjument-Bromage H., Tempst P., Hawkins P. T., and Stephens L. R. (2002) P-Rex1, a PtdIns(3,4,5)P3- and Gβγ-regulated guanine-nucleotide exchange factor for Rac. Cell 108, 809–821 [DOI] [PubMed] [Google Scholar]
- 24.Welch H. C., Condliffe A. M., Milne L. J., Ferguson G. J., Hill K., Webb L. M., Okkenhaug K., Coadwell W. J., Andrews S. R., Thelen M., Jones G. E., Hawkins P. T., and Stephens L. R. (2005) P-Rex1 regulates neutrophil function. Curr. Biol. 15, 1867–1873 [DOI] [PubMed] [Google Scholar]
- 25.Sosa M. S., Lopez-Haber C., Yang C., Wang H., Lemmon M. A., Busillo J. M., Luo J., Benovic J. L., Klein-Szanto A., Yagi H., Gutkind J. S., Parsons R. E., and Kazanietz M. G. (2010) Identification of the Rac-GEF P-Rex1 as an essential mediator of ErbB signaling in breast cancer. Mol. Cell 40, 877–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montero J. C., Seoane S., Ocaña A., and Pandiella A. (2011) P-Rex1 participates in Neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 30, 1059–1071 [DOI] [PubMed] [Google Scholar]
- 27.Campbell A. D., Lawn S., McGarry L. C., Welch H. C., Ozanne B. W., and Norman J. C. (2013) P-Rex1 cooperates with PDGFRbeta to drive cellular migration in 3D microenvironments. PLoS One 8, e53982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balamatsias D., Kong A. M., Waters J. E., Sriratana A., Gurung R., Bailey C. G., Rasko J. E., Tiganis T., Macaulay S. L., and Mitchell C. A. (2011) Identification of P-Rex1 as a novel Rac1-guanine nucleotide exchange factor (GEF) that promotes actin remodeling and GLUT4 protein trafficking in adipocytes. J. Biol. Chem. 286, 43229–43240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berger M. F., Hodis E., Heffernan T. P., Deribe Y. L., Lawrence M. S., Protopopov A., Ivanova E., Watson I. R., Nickerson E., Ghosh P., Zhang H., Zeid R., Ren X., Cibulskis K., Sivachenko A. Y., et al. (2012) Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485, 502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mense S. M., Barrows D., Hodakoski C., Steinbach N., Schoenfeld D., Su W., Hopkins B. D., Su T., Fine B., Hibshoosh H., and Parsons R. (2015) PTEN inhibits PREX2-catalyzed activation of RAC1 to restrain tumor cell invasion. Sci. Signal. 8, ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X., Pan M., Han L., Lu H., Hao X., and Dong Q. (2013) miR-338-3p suppresses neuroblastoma proliferation, invasion and migration through targeting PREX2a. FEBS Lett. 587, 3729–3737 [DOI] [PubMed] [Google Scholar]
- 34.Guo B., Liu L., Yao J., Ma R., Chang D., Li Z., Song T., and Huang C. (2014) miR-338-3p suppresses gastric cancer progression through a PTEN-AKT axis by targeting P-REX2a. Mol. Cancer Res. 12, 313–321 [DOI] [PubMed] [Google Scholar]
- 35.Montero J. C., Seoane S., and Pandiella A. (2013) Phosphorylation of P-Rex1 at serine 1169 participates in IGF-1R signaling in breast cancer cells. Cell. Signal. 25, 2281–2289 [DOI] [PubMed] [Google Scholar]
- 36.Mayeenuddin L. H., and Garrison J. C. (2006) Phosphorylation of P-Rex1 by the cyclic AMP-dependent protein kinase inhibits the phosphatidylinositol (3,4,5)-trisphosphate and Gβγ-mediated regulation of its activity. J. Biol. Chem. 281, 1921–1928 [DOI] [PubMed] [Google Scholar]
- 37.Urano D., Nakata A., Mizuno N., Tago K., and Itoh H. (2008) Domain-domain interaction of P-Rex1 is essential for the activation and inhibition by G protein βγ subunits and PKA. Cell. Signal. 20, 1545–1554 [DOI] [PubMed] [Google Scholar]
- 38.López-Lago M., Lee H., Cruz C., Movilla N., and Bustelo X. R. (2000) Tyrosine phosphorylation mediates both activation and down-modulation of the biological activity of Vav. Mol. Cell. Biol. 20, 1678–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aghazadeh B., Lowry W. E., Huang X. Y., and Rosen M. K. (2000) Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell 102, 625–633 [DOI] [PubMed] [Google Scholar]
- 40.Fleming I. N., Elliott C. M., Buchanan F. G., Downes C. P., and Exton J. H. (1999) Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J. Biol. Chem. 274, 12753–12758 [DOI] [PubMed] [Google Scholar]
- 41.Fleming I. N., Elliott C. M., Collard J. G., and Exton J. H. (1997) Lysophosphatidic acid induces threonine phosphorylation of Tiam1 in Swiss 3T3 fibroblasts via activation of protein kinase C. J. Biol. Chem. 272, 33105–33110 [DOI] [PubMed] [Google Scholar]
- 42.Sells M. A., Knaus U. G., Bagrodia S., Ambrose D. M., Bokoch G. M., and Chernoff J. (1997) Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7, 202–210 [DOI] [PubMed] [Google Scholar]
- 43.Xu J., Xu Z., Zhou J. Y., Zhuang Z., Wang E., Boerner J., and Wu G. S. (2013) Regulation of the Src-PP2A interaction in tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J. Biol. Chem. 288, 33263–33271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Webb B., and Sali A. (2014) Comparative protein structure modeling using modeller. Curr. Protoc. Bioinformatics 47, 5.6.1–5.6.32 [DOI] [PubMed] [Google Scholar]
- 45.Kuziemko A., Honig B., and Petrey D. (2011) Using structure to explore the sequence alignment space of remote homologs. PLoS Comput. Biol. 7, e1002175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sippl M. J. (1993) Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362 [DOI] [PubMed] [Google Scholar]
- 47.Lüthy R., Bowie J. U., and Eisenberg D. (1992) Assessment of protein models with three-dimensional profiles. Nature 356, 83–85 [DOI] [PubMed] [Google Scholar]
- 48.Shymanets A., Prajwal, Bucher K., Beer-Hammer S., Harteneck C., and Nürnberg B. (2013) p87 and p101 subunits are distinct regulators determining class IB phosphoinositide 3-kinase (PI3K) specificity. J. Biol. Chem. 288, 31059–31068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shymanets A., Ahmadian M. R., and Nürnberg B. (2009) Gβγ-copurified lipid kinase impurity from Sf9 cells. Protein Pept. Lett. 16, 1053–1056 [DOI] [PubMed] [Google Scholar]
- 50.Shymanets A., Ahmadian M. R., Kössmeier K. T., Wetzker R., Harteneck C., and Nürnberg B. (2012) The p101 subunit of PI3Kβγ restores activation by Gβ mutants deficient in stimulating p110γ. Biochem. J. 441, 851–858 [DOI] [PubMed] [Google Scholar]
- 51.Hill K., and Welch H. C. (2006) Purification of P-Rex1 from neutrophils and nucleotide exchange assay. Methods Enzymol. 406, 26–41 [DOI] [PubMed] [Google Scholar]
- 52.Obenauer J. C., Cantley L. C., and Yaffe M. B. (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barber M. A., Hendrickx A., Beullens M., Ceulemans H., Oxley D., Thelen S., Thelen M., Bollen M., and Welch H. C. (2012) The guanine-nucleotide-exchange factor P-Rex1 is activated by protein phosphatase 1alpha. Biochem. J. 443, 173–183 [DOI] [PubMed] [Google Scholar]
- 54.Hendrickx A., Beullens M., Ceulemans H., Den Abt T., Van Eynde A., Nicolaescu E., Lesage B., and Bollen M. (2009) Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol. 16, 365–371 [DOI] [PubMed] [Google Scholar]
- 55.Cohen P., Klumpp S., and Schelling D. L. (1989) An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 250, 596–600 [DOI] [PubMed] [Google Scholar]
- 56.Isakoff S. J., Cardozo T., Andreev J., Li Z., Ferguson K. M., Abagyan R., Lemmon M. A., Aronheim A., and Skolnik E. Y. (1998) Identification and analysis of PH domain-containing targets of phosphatidylinositol 3-kinase using a novel in vivo assay in yeast. EMBO J. 17, 5374–5387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park W. S., Heo W. D., Whalen J. H., O'Rourke N. A., Bryan H. M., Meyer T., and Teruel M. N. (2008) Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol. Cell 30, 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas C. C., Deak M., Alessi D. R., and van Aalten D. M. (2002) High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr. Biol. 12, 1256–1262 [DOI] [PubMed] [Google Scholar]
- 59.Komander D., Fairservice A., Deak M., Kular G. S., Prescott A. R., Peter Downes C., Safrany S. T., Alessi D. R., and van Aalten D. M. (2004) Structural insights into the regulation of PDK1 by phosphoinositides and inositol phosphates. EMBO J. 23, 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maira S. M., Stauffer F., Brueggen J., Furet P., Schnell C., Fritsch C., Brachmann S., Chène P., De Pover A., Schoemaker K., Fabbro D., Gabriel D., Simonen M., Murphy L., Finan P., Sellers W., and García-Echeverría C. (2008) Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863 [DOI] [PubMed] [Google Scholar]
- 61.Barber M. A., Donald S., Thelen S., Anderson K. E., Thelen M., and Welch H. C. (2007) Membrane translocation of P-Rex1 is mediated by G protein βγ subunits and phosphoinositide 3-kinase. J. Biol. Chem. 282, 29967–29976 [DOI] [PubMed] [Google Scholar]
- 62.Tsakiridis T., Taha C., Grinstein S., and Klip A. (1996) Insulin activates a p21-activated kinase in muscle cells via phosphatidylinositol 3-kinase. J. Biol. Chem. 271, 19664–19667 [DOI] [PubMed] [Google Scholar]
- 63.De la Mota-Peynado A., Chernoff J., and Beeser A. (2011) Identification of the atypical MAPK Erk3 as a novel substrate for p21-activated kinase (Pak) activity. J. Biol. Chem. 286, 13603–13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dummler B., Ohshiro K., Kumar R., and Field J. (2009) Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 28, 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chiang Y. T., and Jin T. (2014) p21-Activated protein kinases and their emerging roles in glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 306, E707–E722 [DOI] [PubMed] [Google Scholar]
- 66.Bose I., Irazoqui J. E., Moskow J. J., Bardes E. S., Zyla T. R., and Lew D. J. (2001) Assembly of scaffold-mediated complexes containing Cdc42p, the exchange factor Cdc24p, and the effector Cla4p required for cell cycle-regulated phosphorylation of Cdc24p. J. Biol. Chem. 276, 7176–7186 [DOI] [PubMed] [Google Scholar]
- 67.Gulli M. P., Jaquenoud M., Shimada Y., Niederhäuser G., Wiget P., and Peter M. (2000) Phosphorylation of the Cdc42 exchange factor Cdc24 by the PAK-like kinase Cla4 may regulate polarized growth in yeast. Mol. Cell 6, 1155–1167 [DOI] [PubMed] [Google Scholar]
- 68.Kuo C. C., Savage N. S., Chen H., Wu C. F., Zyla T. R., and Lew D. J. (2014) Inhibitory GEF phosphorylation provides negative feedback in the yeast polarity circuit. Curr. Biol. 24, 753–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Frieser S. H., Hlubek A., Sandrock B., and Bölker M. (2011) Cla4 kinase triggers destruction of the Rac1-GEF Cdc24 during polarized growth in Ustilago maydis. Mol. Biol. Cell 22, 3253–3262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haruta T., Uno T., Kawahara J., Takano A., Egawa K., Sharma P. M., Olefsky J. M., and Kobayashi M. (2000) A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol. Endocrinol. 14, 783–794 [DOI] [PubMed] [Google Scholar]
- 71.Takano A., Usui I., Haruta T., Kawahara J., Uno T., Iwata M., and Kobayashi M. (2001) Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol. Cell. Biol. 21, 5050–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harrington L. S., Findlay G. M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N. R., Cheng S., Shepherd P. R., Gout I., Downes C. P., and Lamb R. F. (2004) The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 166, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Simpson L., Li J., Liaw D., Hennessy I., Oliner J., Christians F., and Parsons R. (2001) PTEN expression causes feedback upregulation of insulin receptor substrate 2. Mol. Cell. Biol. 21, 3947–3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O'Reilly K. E., Rojo F., She Q. B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L., Baselga J., and Rosen N. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baker N. M., Yee Chow H., Chernoff J., and Der C. J. (2014) Molecular pathways: targeting RAC-p21-activated serine-threonine kinase signaling in RAS-driven cancers. Clin. Cancer Res. 20, 4740–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sylow L., Jensen T. E., Kleinert M., Højlund K., Kiens B., Wojtaszewski J., Prats C., Schjerling P., and Richter E. A. (2013) Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes 62, 1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sylow L., Kleinert M., Pehmøller C., Prats C., Chiu T. T., Klip A., Richter E. A., and Jensen T. E. (2014) Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 26, 323–331 [DOI] [PubMed] [Google Scholar]
- 78.Um S. H., Frigerio F., Watanabe M., Picard F., Joaquin M., Sticker M., Fumagalli S., Allegrini P. R., Kozma S. C., Auwerx J., and Thomas G. (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205 [DOI] [PubMed] [Google Scholar]
- 79.Pende M., Kozma S. C., Jaquet M., Oorschot V., Burcelin R., Le Marchand-Brustel Y., Klumperman J., Thorens B., and Thomas G. (2000) Hypoinsulinaemia, glucose intolerance and diminished β-cell size in S6K1-deficient mice. Nature 408, 994–997 [DOI] [PubMed] [Google Scholar]