

Background: Mechanisms that guide membrane protein folding in the endoplasmic reticulum membrane remain unresolved.
Results: During aquaporin-4 synthesis, extracellular peptides loops enter the endoplasmic reticulum lumen sequentially, whereas delivery of cytosolic loops is actively delayed.
Conclusion: The assembled ribosome translocon complex (RTC) shields large regions of the protein from the cytosol throughout synthesis.
Significance: Early membrane protein folding occurs in a proteinaceous environment provided by the RTC.
Keywords: aquaporin, endoplasmic reticulum (ER), membrane protein, protein folding, protein translocation, ribosome function, polytopic membrane protein, ribosome translocon complex, sec61 translocon
Abstract
Transmembrane topology of polytopic membrane proteins (PMPs) is established in the endoplasmic reticulum (ER) by the ribosome Sec61-translocon complex (RTC) through iterative cycles of translocation initiation and termination. It remains unknown, however, whether tertiary folding of transmembrane domains begins after the nascent polypeptide integrates into the lipid bilayer or within a proteinaceous environment proximal to translocon components. To address this question, we used cysteine scanning mutagenesis to monitor aqueous accessibility of stalled translation intermediates to determine when, during biogenesis, hydrophilic peptide loops of the aquaporin-4 (AQP4) water channel are delivered to cytosolic and lumenal compartments. Results showed that following ribosome docking on the ER membrane, the nascent polypeptide was shielded from the cytosol as it emerged from the ribosome exit tunnel. Extracellular loops followed a well defined path through the ribosome, the ribosome translocon junction, the Sec61-translocon pore, and into the ER lumen coincident with chain elongation. In contrast, intracellular loops (ICLs) and C-terminalresidues exited the ribosome into a cytosolically shielded environment and remained inaccessible to both cytosolic and lumenal compartments until translation was terminated. Shielding of ICL1 and ICL2, but not the C terminus, became resistant to maneuvers that disrupt electrostatic ribosome interactions. Thus, the early folding landscape of polytopic proteins is shaped by a spatially restricted environment localized within the assembled ribosome translocon complex.
Introduction
Polytopic membrane protein (PMP)2 folding is generally thought to occur in three physically distinct environments: the cytosol, the endoplasmic reticulum (ER) lumen, and the lipid bilayer (1–4). In cells, delivery of peptide domains into these cellular compartments is facilitated by a large macromolecular machine, referred to here as the ribosome translocon complex (RTC), which assembles when the translating ribosome is transferred from signal recognition particle to the Sec61 protein conducting channel (PCC) and its associated proteins at the ER membrane (5–9). The RTC performs several critical functions during PMP biogenesis. It provides a proteinaceous channel for peptide movement across the ER membrane and a lateral pathway for transmembrane segment (TM) integration into the bilayer (10–19). It processes the nascent polypeptide by cotranslationally cleaving N-terminal signal sequences (20) and attaching N-linked carbohydrates (21, 22). It forms a physical barrier that prevents the nascent chain from prematurely accessing the cytosol (17, 23–25). It also provides a protected space for nascent chain accumulation prior to compartmentalization (17, 25).
PMP transmembrane topology is generated by dynamic interactions between the RTC and topogenic determinants encoded within the nascent polypeptide that direct sequential cycles of translocation initiation, termination, and membrane integration (1, 2, 26). In the simplest case, signal or signal anchor (SA) sequences insert into the PCC (12), open the translocon pore, and direct peptide movement into the ER lumen. Subsequent synthesis of a stop transfer (ST) sequence terminates translocation and redirects the growing nascent polypeptide into the cytosol (1, 27–33). Certain PMPs, including aquaporin-4 (AQP4), utilize such a strategy and encode TMs with alternating SA and ST activities that establish topology of each successive TM as the nascent polypeptide emerges from the ribosome (34–36). Other proteins, however, lack a well ordered SA-ST-SA arrangement and, as a result, transiently acquire a non-native topology during their synthesis that is modified during subsequent stages of tertiary folding and helical packing (37–42). This latter process may involve subtle readjustment of TM boundaries (40, 43) or inversion of TMs or groups of TMs as native helical contacts are established (1, 38, 39, 41, 42, 44–46). It remains unknown, however, where these early folding events take place and how they might be influenced by the physical environment of the nascent polypeptide.
Two possible models for TM integration provide differing views of cellular mechanisms of PMP folding. One model predicts that TMs rapidly partition into the lipid bilayer through energetically favorable hydrophobic interactions (8, 11, 32, 47–49). Such a model supports a two-step folding process in which TMs first equilibrate with membrane lipids as they emerge from the lateral translocon gate, prior to forming native intramolecular TM-TM contacts (50, 51). Alternatively, biochemical, cross-linking, and FRET experiments have shown that TMs can be transiently retained, either within the translocon or adjacent to translocon components, before release (2, 28, 33, 36, 52–56). Although the precise mechanism of TM retention remains unknown, evidence suggests that charged residues and/or reduced TM hydrophobicity may delay release, thereby favoring protein-protein over protein-lipid interactions during early stages of folding (54, 55). These findings raise key questions regarding the role of the RTC in PMP biogenesis. Does the RTC act primarily as a passive channel for partitioning of TMs into a lipid environment (11), or does TM retention provide a specialized environment to influence early stages of helical packing and/or tertiary folding (1, 38, 54, 55)?
To address these issues, we monitored the cytosolic and lumenal accessibility of multiple peptide loops during cotranslational biogenesis of the six-spanning AQP4 water channel. Our results show that the RTC shields the nascent AQP4 peptide from the cytosol upon exiting the ribosome. Extracellular loops (ECLs) follow a direct pathway and sequentially enter the ER lumen coincident with nascent chain elongation. In contrast, intracellular loops (ICLs) accumulate beneath the ribosome and gain access to cytosol only after the nascent chain is released from the ribosome. These data indicate that the RTC provides a cytosolically restricted environment that simultaneously accommodates multiple hydrophilic peptide regions during early stages of biogenesis and folding.
Experimental Procedures
Plasmid Construction
Cysteine-less rat AQP4 cDNA (AQP4 Δ6Cys) was constructed from plasmid pSP64.MIWC (34) by replacing the six native cysteine (Cys) residues with alanine (Ala) using PCR with overlap extension (57). Using the same technique, single Cys codons were then reintroduced (Table 1) at amino acid positions indicated in Fig. 1, A and B. The sequence of all PCR amplified and cloned regions of DNA were verified by DNA sequencing.
TABLE 1.
cDNA encoding AQP4 Δ6Cys mutant was subjected to oligonucleotide-mediated, site-directed mutagenesis, creating a series of mutants in which single cysteines were incorporated into the peptide
Residue numbers refer to the amino acid numbers as reported in previous studies of AQP4 (36).
| Residue No. | Amino acid change | Codon change |
|---|---|---|
| 9 | Thr → Cys | ACT → TGC |
| 40 | Ser → Cys | TCA → TGC |
| 68 | Gly → Cys | GGC → TGC |
| 121 | Gly → Cys | GGA → TGC |
| 161 | Thr → Cys | ACT → TGC |
| 195 | Ser → Cys | TCC → TGC |
| 240 | Leu → Cys | CTA → TGC |
FIGURE 1.

AQP4 topology is unaffected by single cysteine (Cys) substitution and is accurately predicted by PEG-Mal accessibility. A, crystal structure of AQP4 monomer showing sites where single Cys residues (cyan) were incorporated into the AQP4 Δ6Cys mutant (modified from Protein Data Bank code 3gd8) (83). B, two-dimensional schematic showing topology of transmembrane segments and single Cys substitution as in panel A. C, AQP4 Δ6Cys mutant function as measured by osmotic water permeability. D, SDS-PAGE analysis of AQP4 Cys mutants treated with PEG-Mal 5,000. Residues predicted to be cytosolic were modified by PEG-Mal prior to permeabilization. Lumenal residues were pegylated upon addition of digitonin or melittin. Pegylated (P) and unpegylated (U) bands are indicated by single (*) and double asterisks (**), respectively. E, quantified pegylation efficiency from SDS-PAGE in panel D, showing fraction pegylated data calculated from pixel intensity of (P/U + P). F, data from panel E, showing fraction pegylated data normalized to PEG-Mal only.
In Vitro Transcription Translation
Truncated cDNA templates were generated by PCR amplification of single Cys plasmid constructs using a sense oligonucleotide complimentary to bp 2741 of the pSP64 vector (5′-CGTAGAGGATCTGGCTAGCG-3′) and antisense oligonucleotides complementary to downstream regions of the AQP4 coding sequence. For each truncation, the last translated codon was converted to valine to minimize spontaneous hydrolysis of the peptidyl-tRNA bond. Full-length AQP4 cDNA was generated using an antisense oligonucleotide complimentary to bp 51 of the pSP64 vector (5′-CACAGGAAACAGCTATGACC-3′).
PCR products were transcribed in vitro at 40 °C for 1 h with SP6 RNA polymerase in the presence of the RNA cap analog diguanosine triphosphate (500 μm), as described previously (58). Capped mRNA was then added directly to a rabbit reticulocyte lysate translation reaction supplemented with [35S]methionine and canine pancreatic microsomes (A280 = 5.0, unless otherwise stated) at 24 °C for 1 h, as described elsewhere (58). Where indicated nascent chains were released from the ribosome via incubation with 50 ng/μl of RNase for 10 min at room temperature (59).
Mass Tagging with PEG-Mal 5,000
ER microsomes containing newly synthesized AQP4 polypeptides were isolated from translation reactions following dilution in an equal volume of buffer A (50 mm HEPES-KOH (pH 7.5), 100 mm KCl, 5 mm MgCl2), by pelleting through buffer A containing 0.7 m sucrose supplemented with 1 mm DTT at 175,000 × g at 4 °C for 10 min. Pelleted microsomes were then suspended in an equal starting volume of buffer A containing 0.1 m sucrose and 50 μm DTT. Ribosome nascent chain complexes (RNCs), prepared in the absence of ER microsomes, were pelleted by layering translation reactions over buffer A containing 0.7 m sucrose supplemented with 1 mm DTT and centrifuging at 386,000 × g at 4 °C for 1 h. Pelleted RNCs were suspended in equal starting volume of buffer A in 0.1 m sucrose and 50 μm DTT.
RNCs or microsomes prepared as described above were incubated on ice for 1 h with 2 mm methoxypolyethylene glycol maleimide 5,000 Da (PEG-Mal 5,000) (Sigma) after addition of 0.5 m NaCl, 20 μm melittin (Sigma), or 2% (w/v) digitonin (Calbiochem, Darmstadt, Germany), where indicated. Aliquots were then quenched with 0.2 m DTT at room temperature for 15 min, solubilized in 4 volumes of Laemmli sample buffer (8% (w/v) SDS and 0.1 m Tris-HCl (pH 8.9)) with 1 m DTT, and run on 10–18% continuous gradient SDS-PAGE gels. Bands were quantified by phosphorimaging using a Personal FX PhosphorImager and QuantityOne software (Bio-Rad). Pegylation efficiency was calculated from pegylated and unpegylated bands using the formula,
where P = band intensity of pegylated protein and U = band intensity of unpegylated protein. To quantify accessibility of nascent ribosome-attached polypeptides, pegylation efficiency was determined from bands corresponding to the intact peptidyl-tRNA species. Pegylation of full-length AQP4 and RNase-released polypeptides was determined by quantification of bands corresponding to the chain-terminated peptides.
AQP4 Functional Studies
Xenopus laevis oocytes were surgically harvested under tricaine anesthesia and digested for 3 h at room temperature with 0.2 units/ml of Blendzyme III (Roche Applied Sciences) in Ca2+-free MBSH (88 mm NaCl, 1 mm KCl, 24 mm NaHCO3, 0.82 mm MgSO4, 10 mm HEPES-KOH (pH 7.4)) supplemented with 0.41 mm CaCl2. Stage VI oocytes were then collected and injected with 50 nl of RNA transcript and incubated at 17 °C in MBSH plus 100 units/ml of penicillin, 100 μg/ml of streptomycin sulfate, and 50 μg/ml of gentamicin. Osmotic water permeability was measured 48 h after injection by transferring individual oocytes into 24-well plates containing a 10-fold dilution of MBSH in distilled water, and collecting digitized images at 1-s intervals by phase-contrast microscopy using IP-Labs 3.5 Morphometric software (BD Bioimaging). Total oocyte volume at each time point was calculated based on the two-dimensional projected area (average diameter), assuming a spherical shape, as reported previously by us and others (39, 60). Osmotic water permeability was calculated from the initial swelling rate (first 30 s) using the following equation,
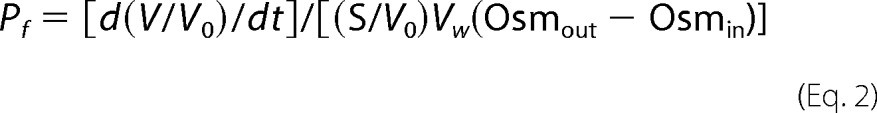 |
where d(V/V0)/dt is the initial rate of swelling, S/V0 ≈ 50 cm−1, Vw = 18 cm3 mol−1, and Osmout − Osmin = 180 mosmol. Four to seven oocytes were analyzed for each construct, and each experiment was independently repeated 3 to 6 times. Average Pf values obtained in a given experiment were normalized to wild-type AQP4 evaluated in the same experiment. Results are presented as mean ± S.E. of 3–6 independent experiments.
Results
The AQP4 water channel is an attractive model PMP with a highly conserved six-membrane spanning topology and well defined tertiary structure (Fig. 1, A and B). Previously we showed that TMs 1, 3, and 5 encode SA activity that initiates polypeptide translocation into the ER lumen, whereas TMs 2, 4, and 6 encode ST activity that cotranslationally terminates translocation and redirects the growing polypeptide into the cytosol (34–36, 39). These results predict that topology of each TM segment is acquired during synthesis as the RTC dynamically coordinates movement of sequential peptide regions into their appropriate compartment.
Defining Peptide Accessibility by Cysteine Modification
In the current study, native AQP4 Cys residues were substituted with Ala and single Cys residues were systematically introduced at the N and C termini and within each ICL and ECL (Fig. 1, A and B). Importantly, AQP4 Δ6Cys retained water channel activity (∼60% of wild type), indicating that folding in the membrane is not grossly perturbed (Fig. 1C). To establish baseline accessibility of peptide loops, full-length Cys-substituted constructs were translated in the presence of ER-derived canine rough microsomes (CRMs) and subjected to covalent modification by the aqueous, membrane-impermeable reagent, PEG-Mal 5,000 as described under “Experimental Procedures” (17, 25, 61). Although the extent of pegylation varied for different sites, residues predicted to reside in the cytosol (Cys-9, Cys-68, Cys-161, and Cys-240) were modified in intact microsomes (0.3–0.7 fraction pegylated) and exhibited minimal or no further increase in pegylation upon microsome permeabilization (Fig. 1, E and F). Lumenal residues (Cys-40s, Cys-121, and Cys-195) exhibited low basal reactivity (0.06–0.12; Fig. 1D), and a 4–7-fold increase in pegylation following membrane permeabilization (Fig. 1, E and F). Thus, PEG-Mal modification accurately reports on the topology of in vitro synthesized AQP4 in the ER membrane.
AQP4 Targeting to the ER and RTC Formation
Like most PMPs, AQP4 encodes an N-terminal type II SA that targets the RNC to the ER membrane (25, 34, 35). To determine the minimum chain length required for targeting, AQP4 mRNA was truncated and translated in the presence of CRMs (Fig. 2A). Membrane-bound nascent chains were recovered by ultracentrifugation, analyzed by SDS-PAGE (Fig. 2B), and plotted as a function of chain length (Fig. 2C). Half-maximal targeting was observed when the nascent chain reached a length of ∼65 aa and targeting was essentially complete at a length of 80 aa (Fig. 2C). Membrane binding also became resistant to high salt conditions at a truncation length of ∼90 amino acids, presumably due to hydrophobic interactions between Sec61 and the SA sequence (25).
FIGURE 2.
AQP4 targeting to the ER membrane. A, schematic indicating truncation site relative to position of transmembrane segments in ribosome-attached intermediates. B, SDS-PAGE of AQP4 Δ6Cys truncated integration intermediates showing total translation products (T), supernatant (S), and membrane pellet (P) fractions. Peptidyl-tRNA bands are indicated by the downward arrow (▾). C, quantified fraction of nascent chains in Panel B that remained associated with the microsomes after pelleting ±NaCl, as indicated. In the absence of high salt (open circles), the nascent chain was targeted to the membrane at 72 aa.
RTC Assembly Controls Nascent Chain Access to Cytosol
To understand how the RTC facilitates localization of PMP domains, we determined the accessibility of cytosolic and lumenal loops in truncated AQP4 polypeptides that were cotranslationally captured within intact RTCs. Programmed integration intermediates were synthesized from truncated RNA transcripts to generate uniform populations of nascent polypeptides that remain attached to the ribosome via a covalent peptidyl-tRNA bond (Fig. 3A). Thus, each truncation site provides a static snapshot of the equilibrated nascent chain conformation in the context of native biosynthetic machinery (25, 36, 62–65). Nascent chain accessibility was then determined at defined stages of synthesis, by PEG-Mal modification of Cys residues that were engineered into cytosolic and lumenal peptide loops (25, 43, 61, 66).
FIGURE 3.

The ribosome shields membrane-targeted AQP4. A, schematic showing organization of RNC complexes and membrane-bound RTCs containing the nascent chain with the Cys probe (cyan). RNCs and RTCs were generated from truncated mRNAs in the absence and presence of CRMs, respectively. B, pegylation of each single Cys mutant RNC was plotted as a function of Cys probe distance from the PTC. C-I, cysteines were inaccessible to PEG-Mal in the presence of microsomes. In addition, pegylation of full-length, membrane-targeted AQP4 polypeptide was plotted (dotted line).
When translation was performed in the absence of CRMs (Fig. 3A), all peptide regions examined became accessible to PEG-Mal as the tether from the peptidyl-transferase center (PTC) reached a length of 30–50 residues, consistent with the predicted span of the ribosome exit tunnel (Fig. 3B). Following ribosome binding to the ER membrane, however, fractional pegylation was reduced 5–10-fold for all sites examined (Fig. 3, C–I). Surprisingly, the entire polypeptide, except the N terminus, remained inaccessible to cytosolic PEG-Mal throughout translation even when the ribosome had reached the last residue prior to the termination codon, which is ∼70 residues from the C terminus of TM6 (Fig. 3, C–I). Thus, ribosome binding to the translocon restricts AQP4 cytosolic exposure throughout synthesis of the entire polypeptide, excepting the N terminus.
Lumenal Loops Follow a Prescribed Path through the RTC
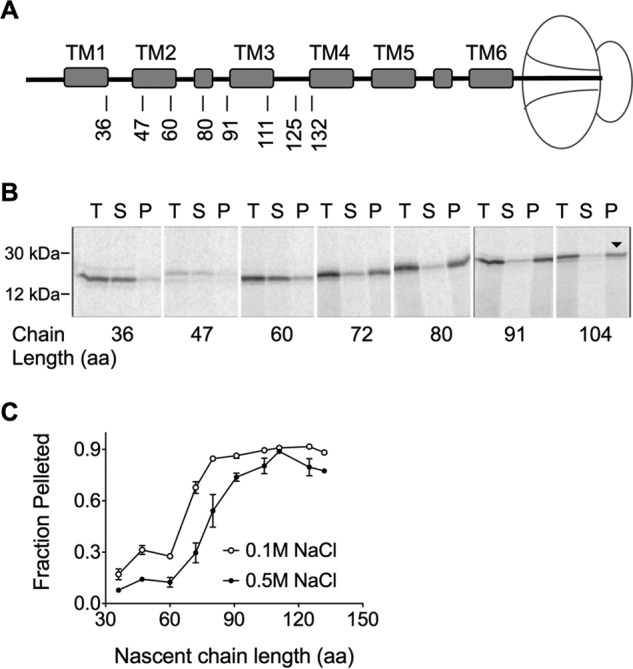
We next determined when lumenal loops move into the ER lumen. Because high salt (0.5 m NaCl) disrupts electrostatic interactions between the ribosome and Sec61 (25, 67, 68), an increase in pegylation following NaCl treatment indicates that the probe site has exited the ribosome tunnel and resides near the ribosome translocon junction (Fig. 4A) (9, 17, 25). In contrast, an increase in pegylation following membrane permeabilization indicates that the probe resides in the ER lumen (Fig. 4A). To follow the movement of the nascent chain, pegylation profiles were determined for Cys-40, Cys-121, and Cys-195, and plotted as a function of chain length (Fig. 4, B–D). Each peptide loop became sensitive to high salt as the tether length between the Cys probe and the PTC was increased from 30 to 60 aa (Fig. 4, B–D). Upon further elongation, ECLs underwent a coordinated transition characterized by loss of salt sensitivity and increased sensitivity to melittin, which plateaued at levels similar to those observed for full-length AQP4 (Figs. 4, B–D). As expected, melittin did not affect pegylation within the salt-sensitive junctional space. Thus, all three ECLs followed an orderly progression into the ER lumen. Movement of each loop occurred sequentially and independently of subsequent loops, whereas the nascent polypeptide remained fully shielded from the cytosol; first by the ribosome exit tunnel, then the ribosome-translocon junction, and finally by the physical barrier of the ER membrane (Fig. 4A).
FIGURE 4.
Progressive translocation of AQP4 ECLs into the ER lumen. A, schematic showing location of the Cys probe (cyan) within the assembled RTC and effect of high salt (NaCl) on ribosome translocon junction versus melittin on ER membrane integrity. B-D, pegylation of the indicated Cys residues plotted as a function of nascent chain length. For each sample, pegylation was performed in intact microsomes (blue), following the addition of 0.5 m NaCl (green) or melittin (black). Data show that ECL1, ECL2, and ECL3 remain inaccessible to bulk cytosol as they traverse the RTC into the ER lumen.
The Ribosome Simultaneously Shields Multiple ICLs throughout Synthesis
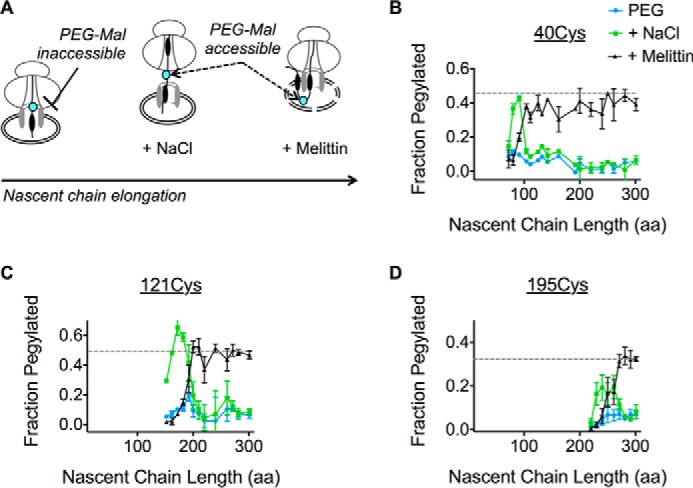
We next examined movement of ICLs. Our previous studies demonstrated that AQP4 TM1 undergoes head-first insertion and inversion when expressed as a fusion protein from which downstream TMs have been removed (25). In the presence of downstream TMs, however, it was difficult to distinguish distinct melittin or salt-sensitive phases for the N terminus (Cys-9) during the early stages of synthesis (Fig. 5A). A modest melittin sensitivity was observed upon membrane targeting (<100 aa length), and gave way to a weak salt sensitivity at nascent chain lengths of ∼150 aa. As the nascent chain was elongated to ∼180 aa, pegylation efficiency gradually increased to a level similar to full-length AQP4, suggesting the N terminus had moved into the cytosol.
FIGURE 5.
The ribosome continuously shields multiple ICLs throughout synthesis. A–D, pegylation profiles for Cys residues in the presence of PEG-Mal only (blue), with the addition of high salt (green) or melittin (black). Relative pegylation of the full-length AQP4 with PEG-Mal only is also shown (dotted line).
On other hand, ICL1, ICL2, and the C terminus exhibited salt-sensitive pegylation when the Cys probes were ∼40–60 residues from the PTC (Fig. 5, B–D). Residue Cys-68 (in ICL1) also exhibited a transient increase in melittin sensitivity for unclear reasons that may reflect brief exposure of ICL1 to lumenal contents. Salt-sensitive pegylation of ICL1 and ICL2 decreased severalfold as synthesis continued beyond 180 and 230 aa, respectively, and remained below that observed for full-length AQP4 throughout translation (Fig. 5, B and C). In contrast, the C terminus remained inaccessible to cytosol, but in a salt-sensitive environment.
Ribosomes Shield the Nascent Chain in a Salt-resistant Manner
Ribosome binding to Sec61 is mediated primarily through electrostatic interactions between the exit tunnel and L8 and C-terminal residues of Sec61 (68). To determine whether the persistent cytosolic shielding observed for ICL1 and ICL2 was ribosome independent, microsomes were treated with RNase prior to incubation with PEG-Mal. RNase digestion restored pegylation for both ICL1 and ICL2 to a level similar to full-length, membrane-integrated AQP4 (Fig. 6, A and B). As expected, RNase treatment had no additional effect on the C terminus pegylation (Fig. 6C). Thus cytosolic regions of the nascent AQP4 polypeptide are continuously shielded from the cytosol by the RTC in at least two biochemically distinguishable environments (Fig. 6D).
FIGURE 6.
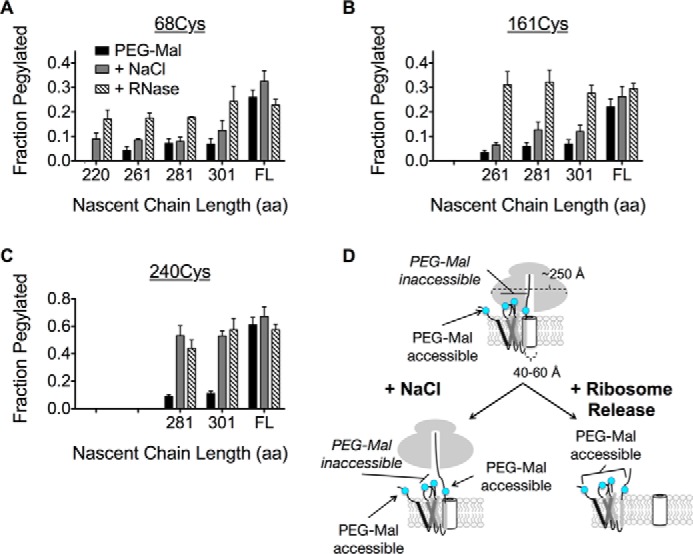
Salt-insensitive shielding of ICL1 and ICL2 is ribosome dependent. A and B, pegylation of ICL1 (68Cys) and ICL2 (161Cys) at longer truncations following ribosome removal with RNase restores pegylation to a level similar to full-length (FL), membrane-integrated AQP4. C, the C terminus exhibits salt-sensitive pegylation that is not further effected by RNase treatment. D, schematic of salt-insensitive ICL1 and ICL2 shielding and C terminus salt-sensitive shielding by the RTC, which supports a model of TM retention within or near the translocon until translation termination.
Discussion
This study describes cotranslational delivery of a native PMP into its appropriate cellular compartments during repetitive cycles of translocation initiation and termination. By quantifying PEG-Mal accessibility of engineered Cys residues in sequentially truncated integration intermediates, we were able to reconstruct movement of each peptide segment (excepting the N terminus, see above) through the RTC at sequential stages of synthesis. Results show that following ribosome binding to the ER membrane, nascent AQP4 became shielded from the cytosol and remained cytosolically inaccessible throughout its synthesis. Extracellular peptide loops, ECL1, ECL2, and ECL3, passed directly from the ribosome exit tunnel, through the ribosome translocon junction, the translocon pore, and into the ER lumen coincident with nascent chain elongation. Movement along this pathway corresponds to the predicted length of a relatively extended polypeptide tethered to the ribosome peptidyl-transferase center. After membrane targeting, the N terminus was briefly accessible to the lumen and became cytosolically exposed as the nascent chain was lengthened, consistent with previous studies (25). Unexpectedly, intracellular peptide loops, ICL1, ICL2, and the C terminus remained shielded from the cytosol by the RTC until translation was terminated and the nascent polypeptide was released from the ribosome. In addition, these peptide regions were retained in biochemically distinct environments. ICL1 and ICL2 passed from a salt-sensitive to a salt-insensitive location, whereas the C terminus remained salt-sensitive until synthesis was completed. Thus, the assembled RTC simultaneously restricts cytosolic access of at least three distinct peptide loops for a prolonged period of AQP4 synthesis.
These results provide insight into how the cytosolic loops and their associated TMs disengage from the RTC during early stages of helical packing and folding. Previous studies analyzing TM integration by cryo-electron microscopy (16, 18), chemical cross-linking, site-specific photocross-linking (36, 64), and FRET (56) have suggested two scenarios. One model proposed that TMs rapidly sample the hydrophobic membrane environment upon entering Sec61, and spontaneously partition through the lateral PCC gate into membrane lipids (10–12, 18, 69) based on the effective tether length and partitioning free energy of the TM segment (49, 70). An alternative model suggests that TMs remain adjacent to the translocon and/or its associated proteins, and are released into the bilayer via mechanistically triggered events, i.e. synthesis of downstream TMs or translation termination (28, 33, 36, 52, 54–56, 71). If TMs integrated rapidly into the lipid bilayer, then the length of the nascent chain tethering the TM segment to the PTC would limit diffusion in the membrane. In that case, cytosolic loops would be expected to sequentially move out from beneath the ribosome as the tether length from the exit tunnel to the C terminus of the TM segment reached ∼70 Å. Given the length of AQP4 ICLs (10–30 amino acids), release of TMs into the bilayer would minimally occur for ICL1 and ICL2 at or near truncation positions 166 and 259, respectively. However, neither ICL1, ICL2, nor the C terminus were cytosolically accessible during AQP4 synthesis, suggesting that TMs accumulate within or in very close proximity to the fully assembled RTC during synthesis. This finding is consistent with evidence that multiple AQP4 TMs (TM1, -3, -4, and -5) can simultaneously cross-link Sec61α (36).
Our results are also consistent with free energies of partitioning (ΔGapp) as calculated by Hessa et al. (70), where TMs with ΔGapp < 0 were found to promote insertion. The ΔGapp values calculated for AQP4 varied from −0.3 kcal mol−1 (TM1 and -3) to greater than +1.0 kcal mol−1 (TM4 and -5), with an overall average of +0.482 kcal mol−1 (49). These values are similar to TM helices in PMPs of known three-dimensional structure (49). It has also been proposed that many native PMPs require neighboring TM helices for proper partitioning into the membrane (38, 39, 49, 70). Therefore, TM retention by the RTC may reflect more energetically favorable early protein-protein interactions over less favorable protein-lipid interactions (28, 36, 56, 71–74).
Although we are currently unable to determine precisely when AQP4 acquires native tertiary structure, the proposed ribosome shielding likely provides a proteinaceous environment for the nascent polypeptide to sample non-native conformations without the ensuing complications of early cytosolic contact. Taken together, our results lend support to a model in which the RTC may impact the folding landscape by minimizing potential off-pathway folding events (1), in a manner analogous to cytosolic chaperonins such as the Escherichia coli GroEL/ES complex (75) and eukaryotic TRiC (76), and allow nascent proteins to access folding intermediates that would otherwise be energetically unfavorable (i.e. TM boundary adjustment, helical packing, or even TM inversion (25, 38, 42, 77, 78)). Because integration intermediates examined here reflect an equilibrated conformation, we cannot assess whether additional kinetic intermediates might exist in cells where translation occurs continuously (79). Examination of different substrates and conditions may be needed to resolve this issue, particularly because TM integration can be influenced by cooperative interactions between adjacent TMs (37, 47, 80) and protein-protein interactions with the translocon (33, 36).
Another unexpected result is that cytosolic loops are retained in an environment that is both ribosome-dependent and salt-insensitive for synthesis of ∼80 residues. Biochemical and structural investigations have shown that ribosomes bind principally to the cytoplasmic loop 8 and C terminus of Sec61α (or homolog SecY) (15, 68) and engage ribosomal RNA helices H50-H53-H59 and H6-H24-H50 in the exit vestibule (12, 16) via salt-sensitive electrostatic interactions (12, 81, 82). This raises an interesting question about the nature of interactions responsible for salt resistance and their role in early PMP folding. One possibility is that salt-insensitive pegylation could reflect ribosome effects on PEG-Mal reactivity by altering the pKa of the thiol side chain. Alternately, additional ribosome interactions, perhaps with other translocon components such as Sec62/63 (9), may stabilize the RTC complex in a salt-resistant manner during accumulation of PMP cytosolic peptide loops.
An obvious question therefore is how much cytosolic polypeptide can be accommodated beneath the ribosome in a cytosolically protected state. Similarly, what additional components (if any) participate in delivery of cytosolic domains into the cytosol? Unfortunately, current structural models provide little guidance into these questions, as most studies to date have examined relatively simple proteins in the context of a single Sec61-PCC and ribosome. Our findings suggest that the RTC is a dynamic complex that acts not only as a translocation and integration conduit, but also to influence and orchestrate early PMP folding events. Structural studies of PMP intermediates in assembled RTCs containing native accessory proteins (OST, TRAM, TRAP, Sec62, and Sec63) will likely be needed to fully resolve these issues.
Author Contributions
M. A. P. designed and performed the majority of experiments, obtained the majority of data shown, and contributed to writing the manuscript and figure preparation. A. B. designed and conceived experiments, helped develop the experimental system, and obtained preliminary data needed for the study. P. K. D. initiated the project, performed experiments, and developed the experimental pegylation protocols. J. W. performed the oocyte water channel experiments collected and analyzed this data with L. R. L. R. performed oocyte water channel experiments, supervised J. W., and analyzed and formatted the data and contributed to the conceptual development of the project. Z. Y. designed and synthesized cDNA plasmid constructs and verified sequencing data. W. R. S. conceived the project, supervised all aspects of the project, contributed to writing and figure preparation, and data analysis. All authors analyzed results and approved the final version of the manuscript.
Acknowledgments
We thank current members of the Skach laboratory and Dr. Brian Conti for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants DK51818 (to W. R. S.), GM53457 (to W. R. S.), and T32 HL083808 (to M. A. P.), and Cystic Fibrosis Foundation Therapeutics Inc. Grant SKACH05X0 (to W. R. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- PMP
- polytopic membrane protein
- ER
- endoplasmic reticulum
- RTC
- ribosome translocon complex
- AQP4
- aquaporin-4
- ICL
- intracellular loop
- PCC
- protein-conducting channel
- TM
- transmembrane segment
- SA
- signal anchor sequence
- ST
- stop transfer sequence
- ECL
- extracellular loop
- RNC
- ribosome nascent chain complex
- PEG-Mal 5,000
- methoxypolyethylene glycol maleimide 5,000 daltons
- CRMs
- canine rough microsomes
- PTC
- peptidyl-tRNA transferase center
- aa
- amino acid.
References
- 1.Skach W. R. (2009) Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 16, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alder N. N., and Johnson A. E. (2004) Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J. Biol. Chem. 279, 22787–22790 [DOI] [PubMed] [Google Scholar]
- 3.Hegde R. S., and Lingappa V. R. (1997) Membrane protein biogenesis: regulated complexity at the endoplasmic reticulum. Cell 91, 575–582 [DOI] [PubMed] [Google Scholar]
- 4.White S. H., and von Heijne G. (2004) The machinery of membrane protein assembly. Curr. Opin. Struct. Biol. 14, 397–404 [DOI] [PubMed] [Google Scholar]
- 5.Walter P., and Lingappa V. R. (1986) Mechanism of protein translocation across the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 2, 499–516 [DOI] [PubMed] [Google Scholar]
- 6.Johnson A. E., and van Waes M. A. (1999) The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15, 799–842 [DOI] [PubMed] [Google Scholar]
- 7.Daniel C. J., Conti B., Johnson A. E., and Skach W. R. (2008) Control of translocation through the Sec61 translocon by nascent polypeptide structure within the ribosome. J. Biol. Chem. 283, 20864–20873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park E., and Rapoport T. A. (2012) Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 41, 21–40 [DOI] [PubMed] [Google Scholar]
- 9.Conti B. J., Devaraneni P. K., Yang Z., David L. L., and Skach W. R. (2015) Cotranslational stabilization of Sec62/63 within the ER Sec61 translocon is controlled by distinct substrate-driven translocation events. Mol. Cell 58, 269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martoglio B., Hofmann M. W., Brunner J., and Dobberstein B. (1995) The protein-conducting channel in the membrane of the endoplasmic reticulum is open laterally toward the lipid bilayer. Cell 81, 207–214 [DOI] [PubMed] [Google Scholar]
- 11.Heinrich S. U., Mothes W., Brunner J., and Rapoport T. A. (2000) The Sec61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell 102, 233–244 [DOI] [PubMed] [Google Scholar]
- 12.Van den Berg B., Clemons W. M. Jr., Collinson I., Modis Y., Hartmann E., Harrison S. C., and Rapoport T. A. (2004) X-ray structure of a protein-conducting channel. Nature 427, 36–44 [DOI] [PubMed] [Google Scholar]
- 13.Cannon K. S., Or E., Clemons W. M. Jr., Shibata Y., and Rapoport T. A. (2005) Disulfide bridge formation between SecY and a translocating polypeptide localizes the translocation pore to the center of SecY. J. Cell Biol. 169, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osborne A. R., Rapoport T. A., and van den Berg B. (2005) Protein translocation by the Sec61/SecY channel. Annu. Rev. Cell Dev. Biol. 21, 529–550 [DOI] [PubMed] [Google Scholar]
- 15.Becker T., Bhushan S., Jarasch A., Armache J. P., Funes S., Jossinet F., Gumbart J., Mielke T., Berninghausen O., Schulten K., Westhof E., Gilmore R., Mandon E. C., and Beckmann R. (2009) Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 326, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frauenfeld J., Gumbart J., Sluis E. O., Funes S., Gartmann M., Beatrix B., Mielke T., Berninghausen O., Becker T., Schulten K., and Beckmann R. (2011) Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat. Struct. Mol. Biol. 18, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti B. J., Elferich J., Yang Z., Shinde U., and Skach W. R. (2014) Cotranslational folding inhibits translocation from within the ribosome-Sec61 translocon complex. Nat. Struct. Mol. Biol. 21, 228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gogala M., Becker T., Beatrix B., Armache J. P., Barrio-Garcia C., Berninghausen O., and Beckmann R. (2014) Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506, 107–110 [DOI] [PubMed] [Google Scholar]
- 19.Voorhees R. M., Fernández I. S., Scheres S. H., and Hegde R. S. (2014) Structure of the mammalian ribosome-Sec61 complex to 3.4-Å resolution. Cell 157, 1632–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans E. A., Gilmore R., and Blobel G. (1986) Purification of microsomal signal peptidase as a complex. Proc. Natl. Acad. Sci. U.S.A. 83, 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruiz-Canada C., Kelleher D. J., and Gilmore R. (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., and Förster F. (2014) Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 5, 3072. [DOI] [PubMed] [Google Scholar]
- 23.Crowley K. S., Reinhart G. D., and Johnson A. E. (1993) The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell 73, 1101–1115 [DOI] [PubMed] [Google Scholar]
- 24.Crowley K. S., Liao S., Worrell V. E., Reinhart G. D., and Johnson A. E. (1994) Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell 78, 461–471 [DOI] [PubMed] [Google Scholar]
- 25.Devaraneni P. K., Conti B., Matsumura Y., Yang Z., Johnson A. E., and Skach W. R. (2011) Stepwise insertion and inversion of a type II signal anchor sequence in the ribosome-Sec61 translocon complex. Cell 146, 134–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blobel G. (1980) Intracellular protein topogenesis. Proc. Natl. Acad. Sci. U.S.A. 77, 1496–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yost C. S., Hedgpeth J., and Lingappa V. R. (1983) A stop transfer sequence confers predictable transmembrane orientation to a previously secreted protein in cell-free systems. Cell 34, 759–766 [DOI] [PubMed] [Google Scholar]
- 28.Do H., Falcone D., Lin J., Andrews D. W., and Johnson A. E. (1996) The cotranslational integration of membrane proteins into the phospholipid bilayer is a multistep process. Cell 85, 369–378 [DOI] [PubMed] [Google Scholar]
- 29.Haigh N. G., and Johnson A. E. (2002) A new role for BiP: closing the aqueous translocon pore during protein integration into the ER membrane. J. Cell Biol. 156, 261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.High S., Flint N., and Dobberstein B. (1991) Requirements for the membrane insertion of signal-anchor type proteins. J. Cell Biol. 113, 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.High S., and Dobberstein B. (1992) Mechanisms that determine the transmembrane disposition of proteins. Curr. Opin. Cell Biol. 4, 581–586 [DOI] [PubMed] [Google Scholar]
- 32.Mothes W., Heinrich S. U., Graf R., Nilsson I., von Heijne G., Brunner J., and Rapoport T. A. (1997) Molecular mechanism of membrane protein integration into the endoplasmic reticulum. Cell 89, 523–533 [DOI] [PubMed] [Google Scholar]
- 33.McCormick P. J., Miao Y., Shao Y., Lin J., and Johnson A. E. (2003) Cotranslational protein integration into the ER membrane is mediated by the binding of nascent chains to translocon proteins. Mol. Cell 12, 329–341 [DOI] [PubMed] [Google Scholar]
- 34.Shi L. B., Skach W. R., Ma T., and Verkman A. S. (1995) Distinct biogenesis mechanisms for the water channels MIWC and CHIP28 at the endoplasmic reticulum. Biochemistry 34, 8250–8256 [DOI] [PubMed] [Google Scholar]
- 35.Foster W., Helm A., Turnbull I., Gulati H., Yang B., Verkman A. S., and Skach W. R. (2000) Identification of sequence determinants that direct different intracellular folding pathways for aquaporin-1 and aquaporin-4. J. Biol. Chem. 275, 34157–34165 [DOI] [PubMed] [Google Scholar]
- 36.Sadlish H., Pitonzo D., Johnson A. E., and Skach W. R. (2005) Sequential triage of transmembrane segments by Sec61α during biogenesis of a native multispanning membrane protein. Nat. Struct. Mol. Biol. 12, 870–878 [DOI] [PubMed] [Google Scholar]
- 37.Skach W. R., Calayag M. C., and Lingappa V. R. (1993) Evidence for an alternate model of human P-glycoprotein structure and biogenesis. J. Biol. Chem. 268, 6903–6908 [PubMed] [Google Scholar]
- 38.Lu Y., Turnbull I. R., Bragin A., Carveth K., Verkman A. S., and Skach W. R. (2000) Reorientation of aquaporin-1 topology during maturation in the endoplasmic reticulum. Mol. Biol. Cell 11, 2973–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buck T. M., Wagner J., Grund S., and Skach W. R. (2007) A novel tripartite motif involved in aquaporin topogenesis, monomer folding and tetramerization. Nat. Struct. Mol. Biol. 14, 762–769 [DOI] [PubMed] [Google Scholar]
- 40.Virkki M., Boekel C., Illergård K., Peters C., Shu N., Tsirigos K. D., Elofsson A., von Heijne G., and Nilsson I. (2014) Large tilts in transmembrane helices can be induced during tertiary structure formation. J. Mol. Biol. 426, 2529–2538 [DOI] [PubMed] [Google Scholar]
- 41.Bogdanov M., Heacock P. N., and Dowhan W. (2002) A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. EMBO J. 21, 2107–2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cymer F., and von Heijne G. (2013) Cotranslational folding of membrane proteins probed by arrest-peptide-mediated force measurements. Proc. Natl. Acad. Sci. U.S.A. 110, 14640–14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carveth K., Buck T., Anthony V., and Skach W. R. (2002) Cooperativity and flexibility of cystic fibrosis transmembrane conductance regulator transmembrane segments participate in membrane localization of a charged residue. J. Biol. Chem. 277, 39507–39514 [DOI] [PubMed] [Google Scholar]
- 44.Dowhan W., and Bogdanov M. (2009) Lipid-dependent membrane protein topogenesis. Annu. Rev. Biochem. 78, 515–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ojemalm K., Halling K. K., Nilsson I., and von Heijne G. (2012) Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol. Cell 45, 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Öjemalm K., Watson H. R., Roboti P., Cross B. C., Warwicker J., von Heijne G., and High S. (2013) Positional editing of transmembrane domains during ion channel assembly. J. Cell Sci. 126, 464–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heinrich S. U., and Rapoport T. A. (2003) Cooperation of transmembrane segments during the integration of a double-spanning protein into the ER membrane. EMBO J. 22, 3654–3663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hessa T., White S. H., and von Heijne G. (2005) Membrane insertion of a potassium-channel voltage sensor. Science 307, 1427. [DOI] [PubMed] [Google Scholar]
- 49.Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S. H., and von Heijne G. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 [DOI] [PubMed] [Google Scholar]
- 50.Popot J. L., and Engelman D. M. (1990) Membrane protein folding and oligomerization: the two-stage model. Biochemistry 29, 4031–4037 [DOI] [PubMed] [Google Scholar]
- 51.Popot J. L., and Engelman D. M. (2000) Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69, 881–922 [DOI] [PubMed] [Google Scholar]
- 52.Ismail N., Crawshaw S. G., Cross B. C., Haagsma A. C., and High S. (2008) Specific transmembrane segments are selectively delayed at the ER translocon during opsin biogenesis. Biochem. J. 411, 495–506 [DOI] [PubMed] [Google Scholar]
- 53.Cross B. C., and High S. (2009) Dissecting the physiological role of selective transmembrane-segment retention at the ER translocon. J. Cell Sci. 122, 1768–1777 [DOI] [PubMed] [Google Scholar]
- 54.Oberdorf J., Pitonzo D., and Skach W. R. (2005) An energy-dependent maturation step is required for release of the cystic fibrosis transmembrane conductance regulator from early endoplasmic reticulum biosynthetic machinery. J. Biol. Chem. 280, 38193–38202 [DOI] [PubMed] [Google Scholar]
- 55.Pitonzo D., Yang Z., Matsumura Y., Johnson A. E., and Skach W. R. (2009) Sequence-specific retention and regulated integration of a nascent membrane protein by the endoplasmic reticulum Sec61 translocon. Mol. Biol. Cell 20, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hou B., Lin P. J., and Johnson A. E. (2012) Membrane protein TM segments are retained at the translocon during integration until the nascent chain cues FRET-detected release into bulk lipid. Mol. Cell 48, 398–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 58.Matsumura Y., Rooney L., and Skach W. R. (2011) In vitro methods for CFTR biogenesis. Methods Mol. Biol. 741, 233–253 [DOI] [PubMed] [Google Scholar]
- 59.Kelkar D. A., Khushoo A., Yang Z., and Skach W. R. (2012) Kinetic analysis of ribosome-bound fluorescent proteins reveals an early, stable, cotranslational folding intermediate. J. Biol. Chem. 287, 2568–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verkman A. S., Shi L. B., Frigeri A., Hasegawa H., Farinas J., Mitra A., Skach W., Brown D., Van Hoek A. N., and Ma T. (1995) Structure and function of kidney water channels. Kidney Int. 48, 1069–1081 [DOI] [PubMed] [Google Scholar]
- 61.Lu J., and Deutsch C. (2001) Pegylation: a method for assessing topological accessibilities in Kv1.3. Biochemistry 40, 13288–13301 [DOI] [PubMed] [Google Scholar]
- 62.Johnson A. E. (2005) The co-translational folding and interactions of nascent protein chains: a new approach using fluorescence resonance energy transfer. FEBS Lett. 579, 916–920 [DOI] [PubMed] [Google Scholar]
- 63.Lin P. J., Jongsma C. G., Liao S., and Johnson A. E. (2011) Transmembrane segments of nascent polytopic membrane proteins control cytosol/ER targeting during membrane integration. J. Cell Biol. 195, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.High S., Martoglio B., Görlich D., Andersen S. S., Ashford A. J., Giner A., Hartmann E., Prehn S., Rapoport T. A., and Dobberstein B. (1993) Site-specific photocross-linking reveals that Sec61p and TRAM contact different regions of a membrane-inserted signal sequence. J. Biol. Chem. 268, 26745–26751 [PubMed] [Google Scholar]
- 65.Sato Y., Sakaguchi M., Goshima S., Nakamura T., and Uozumi N. (2002) Integration of Shaker-type K+ channel, KAT1, into the endoplasmic reticulum membrane: synergistic insertion of voltage-sensing segments, S3-S4, and independent insertion of pore-forming segments, S5-P-S6. Proc. Natl. Acad. Sci. U.S.A. 99, 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fons R. D., Bogert B. A., and Hegde R. S. (2003) Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 160, 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raden D., and Gilmore R. (1998) Signal recognition particle-dependent targeting of ribosomes to the rough endoplasmic reticulum in the absence and presence of the nascent polypeptide-associated complex. Mol. Biol. Cell 9, 117–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raden D., Song W., and Gilmore R. (2000) Role of the cytoplasmic segments of Sec61α in the ribosome-binding and translocation-promoting activities of the Sec61 complex. J. Cell Biol. 150, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rapoport T. A., Goder V., Heinrich S. U., and Matlack K. E. (2004) Membrane-protein integration and the role of the translocation channel. Trends Cell Biol. 14, 568–575 [DOI] [PubMed] [Google Scholar]
- 70.Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., and von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 [DOI] [PubMed] [Google Scholar]
- 71.Meacock S. L., Lecomte F. J., Crawshaw S. G., and High S. (2002) Different transmembrane domains associate with distinct endoplasmic reticulum components during membrane integration of a polytopic protein. Mol. Biol. Cell 13, 4114–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thrift R. N., Andrews D. W., Walter P., and Johnson A. E. (1991) A nascent membrane protein is located adjacent to ER membrane proteins throughout its integration and translation. J. Cell Biol. 112, 809–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saurí A., Saksena S., Salgado J., Johnson A. E., and Mingarro I. (2005) Double-spanning plant viral movement protein integration into the endoplasmic reticulum membrane is signal recognition particle-dependent, translocon-mediated, and concerted. J. Biol. Chem. 280, 25907–25912 [DOI] [PubMed] [Google Scholar]
- 74.Kida Y., Morimoto F., and Sakaguchi M. (2007) Two translocating hydrophilic segments of a nascent chain span the ER membrane during multispanning protein topogenesis. J. Cell Biol. 179, 1441–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fenton W. A., and Horwich A. L. (2003) Chaperonin-mediated protein folding: fate of substrate polypeptide. Q. Rev. Biophys. 36, 229–256 [DOI] [PubMed] [Google Scholar]
- 76.Leroux M. R., and Hartl F. U. (2000) Protein folding: versatility of the cytosolic chaperonin TRiC/CCT. Curr. Biol. 10, R260–264 [DOI] [PubMed] [Google Scholar]
- 77.Kida Y., Kume C., Hirano M., and Sakaguchi M. (2010) Environmental transition of signal-anchor sequences during membrane insertion via the endoplasmic reticulum translocon. Mol. Biol. Cell 21, 418–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seppälä S., Slusky J. S., Lloris-Garcerá P., Rapp M., and von Heijne G. (2010) Control of membrane protein topology by a single C-terminal residue. Science 328, 1698–1700 [DOI] [PubMed] [Google Scholar]
- 79.Cheng Z., and Gilmore R. (2006) Slow translocon gating causes cytosolic exposure of transmembrane and lumenal domains during membrane protein integration. Nat. Struct. Mol. Biol. 13, 930–936 [DOI] [PubMed] [Google Scholar]
- 80.Lin J., and Addison R. (1995) A novel integration signal that is composed of two transmembrane segments is required to integrate the Neurospora plasma membrane H+-ATPase into microsomes. J. Biol. Chem. 270, 6935–6941 [DOI] [PubMed] [Google Scholar]
- 81.Beckmann R., Spahn C. M., Eswar N., Helmers J., Penczek P. A., Sali A., Frank J., and Blobel G. (2001) Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell 107, 361–372 [DOI] [PubMed] [Google Scholar]
- 82.Ménétret J. F., Hegde R. S., Heinrich S. U., Chandramouli P., Ludtke S. J., Rapoport T. A., and Akey C. W. (2005) Architecture of the ribosome-channel complex derived from native membranes. J. Mol. Biol. 348, 445–457 [DOI] [PubMed] [Google Scholar]
- 83.Ho J. D., Yeh R., Sandstrom A., Chorny I., Harries W. E., Robbins R. A., Miercke L. J., and Stroud R. M. (2009) Crystal structure of human aquaporin 4 at 1.8 Å and its mechanism of conductance. Proc. Natl. Acad. Sci. U.S.A. 106, 7437–7442 [DOI] [PMC free article] [PubMed] [Google Scholar]