

Background: JAK3, a tyrosine kinase associated to cytokine receptors, is frequently mutated in leukemia.
Results: JAK3L857P induces constitutive signaling independently of cytokine receptors and JAK1, in contrast to other JAK mutants.
Conclusion: Different JAK mutants signal through distinct mechanisms and show different sensitivity to JAK1- or JAK3-specific inhibitors.
Significance: Depending on the JAK residue mutated, patients will require different treatments.
Keywords: Janus kinase (JAK), leukemia, oncogene, signal transduction, tyrosine-protein kinase (tyrosine kinase), JAK inhibitor
Abstract
JAK1 and JAK3 are recurrently mutated in acute lymphoblastic leukemia. These tyrosine kinases associate with heterodimeric cytokine receptors such as IL-7 receptor or IL-9 receptor, in which JAK1 is appended to the specific chain, and JAK3 is appended to the common gamma chain. Here, we studied the role of these receptor complexes in mediating the oncogenic activity of JAK3 mutants. Although JAK3V674A and the majority of other JAK3 mutants needed to bind to a functional cytokine receptor complex to constitutively activate STAT5, JAK3L857P was unexpectedly found to not depend on such receptor complexes for its activity, which was induced without receptor or JAK1 co-expression. Introducing a mutation in the FERM domain that abolished JAK-receptor interaction did not affect JAK3L857P activity, whereas it inhibited the other receptor-dependent mutants. The same cytokine receptor independence as for JAK3L857P was observed for homologous Leu857 mutations of JAK1 and JAK2 and for JAK3L875H. This different cytokine receptor requirement correlated with different functional properties in vivo and with distinct sensitivity to JAK inhibitors. Transduction of murine hematopoietic cells with JAK3V674A led homogenously to lymphoblastic leukemias in BALB/c mice. In contrast, transduction with JAK3L857P induced various types of lymphoid and myeloid leukemias. Moreover, ruxolitinib, which preferentially blocks JAK1 and JAK2, abolished the proliferation of cells transformed by the receptor-dependent JAK3V674A, yet proved much less potent on cells expressing JAK3L857P. These particular cells were, in contrast, more sensitive to JAK3-specific inhibitors. Altogether, our results showed that different JAK3 mutations induce constitutive activation through distinct mechanisms, pointing to specific therapeutic perspectives.
Introduction
JAK3 is a member of the Janus kinase (JAK) family, which comprises three other tyrosine kinases: JAK1, JAK2, and TYK2. These intracellular kinases are associated with a wide variety of cytokine receptors that lack intrinsic kinase activity. Although the three other JAKs are ubiquitously expressed and can bind various receptor chains, JAK3 is exclusively associated with the common gamma chain (γc),5 and both genes are preferentially expressed in hematopoietic cells. γc partners with a specific alpha (or beta) chain that binds JAK1 to form the heterodimeric receptor complexes for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 cytokines, key players of lymphoid development. For the IL-2 receptor complex, IL-2 stimulation revealed JAK1 to be responsible for the phosphorylation of JAK3 and STAT5, with the role of JAK3 being to phosphorylate JAK1 (1).
The JAKs possess three functional domains that are well conserved. The kinase domain is the catalytic domain of the tyrosine kinases, located at the C terminus. Adjacent to this is the pseudokinase domain, which presents a kinase-like sequence yet lacks critical residues required for proper catalytic activity. This domain is supposed to prevent inappropriate activation of the kinase domain in the absence of cytokine stimulation (2). The N terminus contains the FERM (band-4.1, ezrin, radixin, moesin) domain. This domain mediates direct JAK interaction with the intracellular portion of cytokine receptors (3). Mutations disturbing the structural integrity of the extreme N terminus of the FERM domain are interfering with γc association, as illustrated, for example, by the Y100C mutation of JAK3 (4).
The JAK3Y100C mutation was described in patients suffering from severe combined immunodeficiency (SCID) (5). This germ line JAK3 inactivating mutation abrogates the γc-dependent cytokine signaling essential for lymphoid cell development, leading to a profound defect in T, B, and NK cells. In contrast to these inactivating mutations, acquired JAK3 activating point mutations have been described in various leukemia types, such as T cell acute lymphoblastic leukemia (T-ALL) (6), acute megakaryoblastic leukemia (7), or juvenile myelomonocytic leukemia (8), resulting in constitutive JAK activation and STAT phosphorylation.
The severe effect of JAK3 signaling impairment specifically impacting on lymphoid development in SCID patients has instigated the development of immunosuppressive drugs targeting this kinase. Tofacitinib, a type I JAK inhibitor targeting JAK3 (1), is currently an approved treatment for rheumatoid arthritis in the United States. This drug is being assessed for use in the treatment of T cell-specific autoimmune diseases, such as psoriasis, multiple sclerosis, and inflammatory bowel disease, as well as for the prevention of organ transplant rejection (9, 10). Although originally described as a selective JAK3 inhibitor, tofacitinib also inhibits JAK1, and its efficacy is likely due to combined inhibition of both kinases (1, 11). On the other hand, JAK inhibitors are also currently being investigated for the treatment of hematological malignancies caused by abnormal JAK activation. Ruxolitinib, a JAK1/2 type I inhibitor, is currently administered in the treatment for myeloproliferative neoplasms, which are frequently associated with the V617F activating mutation in JAK2 (12). In line with the results observed with JAK inhibitor use in myeloproliferative neoplasm treatment, these inhibitors represent an appealing therapeutic strategy for leukemia cases associated with JAK3 activation.
This study sought to gain more insight into the potential use of JAK inhibitors in mutated JAK3-driven malignancies. We therefore decided to first determine whether ALL-associated JAK3 mutants need to bind γc to be constitutively active and then to test the sensitivity of different mutants to JAK inhibitors.
Experimental Procedures
Cells, Cell Culture, and Cytokines
Ba/F3 pro-B cells were cultured in Iscove-Dulbecco's medium supplemented with 10% fetal bovine serum, 50 μm β-mercaptoethanol, 0.55 mm l-arginine, 0.24 mm l-asparagine, 1.25 mm l-glutamine and in the presence of murine IL-3. Autonomous Ba/F3 cells expressing JAK3 mutants or BCR-ABL were cultured without IL-3. BCR-ABL transduced Ba/F3 cells were a kind gift of Jean-Baptiste Demoulin (Université Catholique de Louvain, Brussels, Belgium). HEK293 human embryonic kidney cells and JAK1 deficient U4C cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. These cells do not express endogenous IL-9Rα, γc, and JAK3.
Plasmid Constructions
Human JAK3 mutants and double mutants cDNAs (Y100A, M511I, A572V, V674A, V674A/Y100A, L857P, L857A, L857E, and L857P/Y100A), murine JAK1 mutants and double mutants (V658F, K907A as kinase-dead mutation, L910P, and L910P/Y107A), and murine JAK2 mutants and double mutants (V617F, L884P, and L884P/Y114A) were generated using a QuikChange XL II site-directed mutagenesis kit (Stratagene, La Jolla, California) and subcloned into the pMX-IRES-CD4 (JAK3) or pMX-IRES-GFP (JAK1 and JAK2) biscistronic retroviral vectors upstream of the IRES. The WT, V674A, L857Q, L875H, P906S, and E958K human JAK3 pMSCV-GFP plasmids were described earlier (13). Mutagenesis was performed on the WT plasmid to create Y824A, T848A, L857P, and L857P/Y100A human JAK3 mutants. WT human JAK3 and human IL-9 receptor α cDNA were subcloned into the pMX-IRES-CD4 biscistronic retroviral vector. WT murine JAK1, WT murine JAK2, and human common γ chain cDNA were subcloned into the pMX-IRES-GFP biscistronic retroviral vector.
Virus Production, Stable DNA Transfections, and Analysis of Transfected Cells
Retroviral supernatants were generated by transient transfection of HEK293T cells with the packaging plasmid pCL-Eco (coding for gag/pol/env) and biscistronic vectors encoding the gene of interest and human CD4 as marker and used for infection of 0.5 × 106 Ba/F3 cells. HEK293 cells were transfected with 3.75 μg of pCL-Eco and 3.75 μg of the different constructs with 22.5 μl of TransIT-LT1 transfection reagent (Mirus). The next day, supernatants were collected and added to Ba/F3 cells for the spin infection (2-h incubation at 1000 × g at room temperature). This last step was repeated the day after to increase the transfection level. Expression of the markers was analyzed by FACS (BD FACSCaliburTM flow cytometer), using a phycoerythrin-coupled antibody against hCD4 (catalog no. 555347; BD Pharmingen) diluted 1/30. For murine bone marrow cell transduction, mutated JAK3 pMSCV-GFP viral vectors were produced in HEK293T cells using an EcoPack packaging plasmid and TurboFect transfection reagent (Fermentas). Virus was harvested after 48 h.
Dual Luciferase Assay
STAT5 transcriptional activity was assessed by measurements of luciferase expression in HEK293 cells upon transient transfection of appropriate cDNA constructs and pLHRE-luc vector harboring tandem copies of the STAT5-inducible lactogenic hormone response element (LHRE) of the rat β-casein promoter, inserted upstream from a luciferase gene (14). Another reporter plasmid, Renilla luciferase (pRLTk; Promega), was co-transfected as an internal transfection control. Transient transfection of HEK293 cells by Lipofectamine (Invitrogen) was previously described (15). 24 h after transfection, luciferase assays were performed using the dual luciferase reporter assay system (Promega).
Western Blots
For Western blot analysis of JAK3 expression in HEK293 cells, 4 × 105 cells were transfected with 1 μg of different JAK3 constructs with Lipofectamine (Invitrogen) as previously described (15). Cell lysate preparation, gel electrophoresis, and transfer to nitrocellulose membranes were performed as previously described (16). Phosphorylation of signaling proteins was investigated with following phospho-specific antibodies from Cell Signaling Technology: anti-pY1034/1035 JAK1 (catalog no. 3331), anti-pY980/981 JAK3 (catalog no. 5031), anti-pY705 STAT3 (catalog no. 9131), anti-pY694 STAT5 (catalog no. 9351), and anti-pT202/Y204 ERK1/2 (catalog no. 9101). Blots were reprobed with anti-JAK1 (catalog no. 3332), anti-JAK3 (catalog no. 3775), anti-STAT3 (catalog no. 9132) (Cell Signaling Technology, Beverly, MA), anti-STAT5 (catalog no. SC-835; Santa Cruz Biotechnology), or anti-β-actin (catalog no. A5441; Sigma) antibodies, as a control.
Murine Bone Marrow Transplantation
Balb/c mice were purchased from Charles River Laboratories. Harvest of bone marrow cells from male donor mice, lineage negative cells enrichment, transduction with viral supernatant, and injection into irradiated female recipient mice was performed as previously described (13). After sacrifice of the mice, single-cell suspensions were prepared from peripheral blood, bone marrow, spleen, thymus, and lymph nodes. Cells were analyzed on a FACSCanto flow cytometer (BD Biosciences).
Proliferation Assays
For knockdown experiments, 1.5 × 106 IL-3-dependent control Ba/F3 cells or growth factor-independent Ba/F3 cells obtained after transduction with ALL-associated JAK3 mutants (V674A and L857P) and double mutant (L857P/Y100A) were resuspended in 400 μl of medium containing 200 nm siRNA duplexes (Silencer Select Il2rg mouse, Ambion catalog no. s68269; Stealth siRNA Jak1 mouse, Invitrogen catalog no. mss205625; Silencer Select Negative Control No. 1 siRNA, Ambion catalog no. 4390843; and Stealth RNAiTM siRNA Negative Control, Invitrogen catalog no. 12935–400) and then transferred to 4-mm cuvettes (Bio-Rad). Cells were electroporated (260 V, 90 Ω, 1500 microfarad) and seeded in 96-well plates at 10,000 cells/well. For JAK inhibitors treatment, Ba/F3 cells stably transduced with ALL-associated JAK3 mutants (V674A and L857P), double mutant (L857P/Y100A), or BCR-ABL were seeded in 96-well plates at 5,000 cells/well in the presence of increasing concentrations of ruxolitinib (INCB018424; Haoyuan Chemexpress Co), NIBR3048 (TSC21311; Tocris Bioscience) (17), or tofacitinib (CP-690550; Selleckchem). After 72 h, [methyl-3H]thymidine (Amersham Biosciences; catalog no. TRK120) was added to the cells for 4 h, and thymidine incorporation was measured with a Top Count microplate scintillation counter (Canberra-Packard, Meriden, CT).
JAK3 Structure
The three-dimensional structural model of the pseudokinase domain of JAK3 was obtained by employing the SWISS-MODEL server and was superimposed on TYK2 JH1-JH2 crystal structure (Protein Data Bank code 4OLI) as template, together with the JAK3 kinase domain crystal structure (Protein Data Bank code 1YVJ), using the PyMOL program.
Results
ALL-associated JAK3 Mutants Signal through a Functional Cytokine Receptor Complex, Except the L857P Mutant
More and more patients suffering from different types of leukemia, such as ALL, acute megakaryoblastic leukemia, or juvenile myelomonocytic leukemia, have been reported to exhibit mutations in the JAK3 gene (6–8, 18, 19). These mutations were often characterized as activating (7, 18–20), yet little is known about their activation mechanism. Of the other JAK kinases, JAK2 V617F, found in 95% of polycythemia vera cases, has been shown to be dependent on homodimeric receptors, like the erythropoietin receptor, to become active (20). Similar results were obtained for the activating JAK1 V658F and A634D mutants, which needed to bind to IL-9 receptor alpha or IL-2 receptor beta to form homo- or heterodimeric cytokine receptor complexes able to mediate constitutive signaling (21). Furthermore, the active JAK3A572V mutant requires heterodimeric complexes formed by IL-9 receptor alpha or IL-2 receptor beta and γc (22). We sought to determine whether ALL-associated JAK3 mutants shared similar receptor-dependent mechanisms of constitutive signaling. To this end, we studied four activating JAK3 mutants scattered throughout the kinase and pseudokinase domains, namely M511I, A572V, V674A, and L857P identified in patients with T-ALL.6 We took advantage of HEK293 cells that are deficient in JAK3 and γc, the only cytokine receptor chain known to bind JAK3. WT JAK3 and the four mutants were transiently transfected into HEK293 cells, in combination with a STAT5-responsive reporter construct. This transfection was carried out both in the absence of IL-9Rα or γc and in their presence to reconstitute a functional IL-9R complex. As shown in Fig. 1, the four tested JAK3 mutants induced constitutive STAT5 activation, although for the M511I, A572V, and V674A mutants, this was achieved only in the presence of a functional IL-9R complex (γc and IL-9Rα). Surprisingly, JAK3L857P induced STAT5 activation even without co-expression of any receptor chain. This observation suggested a receptor-independent activity that was specific to JAK3L857P. An alternative hypothesis would be the involvement of another cytokine receptor chain that could bind JAK3 and would be expressed in HEK293. To exclude the latter hypothesis, we tested the effect of a SCID mutation localized in the FERM domain of JAK3 (Y100C) that inactivates the JAK by disabling its binding to γc (4, 5). In fact, mutation of this conserved hydrophobic residue in JAK FERM domains disturbs the structural integrity of parts of this FERM domain, which plays an essential role in all JAK-cytokine receptor interactions, as also shown for the homologous mutations of JAK1 (Y107A) (3) and JAK2 (Y114A) (23). We introduced the Y100A mutation into a WT, V674A, and L857P JAK3 and evaluated the capacity of each mutant and double mutant to mediate constitutive signaling. As illustrated in Fig. 2A, the JAK3V674A activity in the presence of the IL-9R complex was abrogated by introducing the Y100A mutation, thereby confirming the essential role of Tyr100 for receptor binding. This observation contrasted with the results obtained for JAK3L857P for which introducing the Y100A mutation did not abrogate its activity. On the contrary, increased constitutive signaling in the absence of a functional receptor was observed. Notably, the expression level of each JAK3 mutant was similar under all experimental conditions (Fig. 2B).
FIGURE 1.
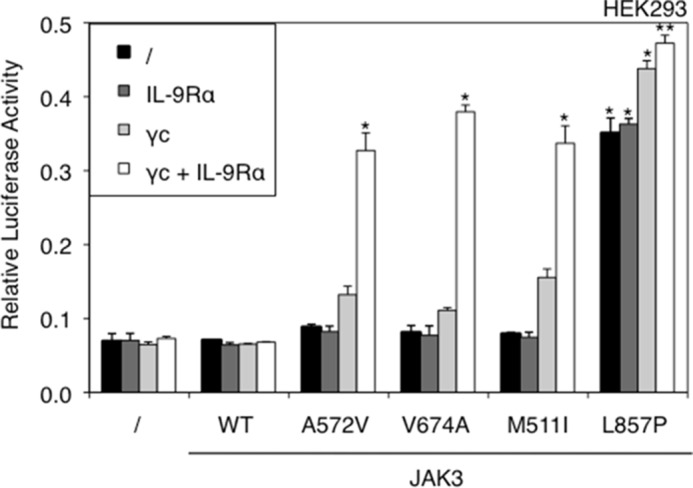
STAT5 activation by JAK3 activating mutants is receptor-dependent, except for the L857P mutant. JAK3- and γc-deficient HEK293 cells were transiently co-transfected with JAK3WT or ALL-associated A572V, V674A, M511I, and L857P JAK3 mutants and γc and/or IL-9Rα, in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of triplicates. Similar results were obtained in three independent experiments. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the control condition without JAK3 and the WT or mutant forms of JAK3 for each condition (with or without IL-9R complex). *, p < 0.05; **, p < 0.01.
FIGURE 2.
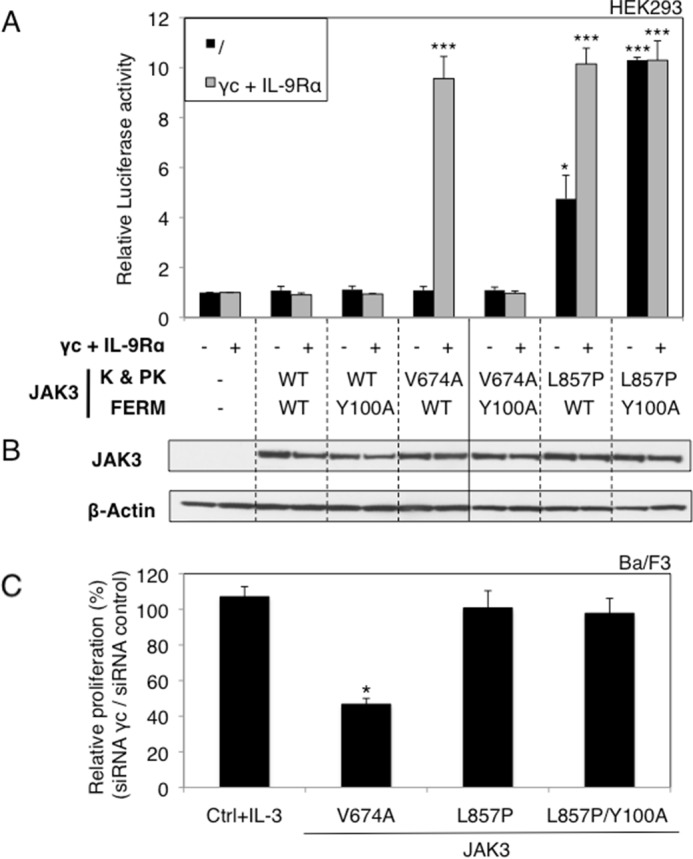
JAK3 FERM domain integrity is required for the activity of the V674A mutant, but not for the L857P mutant. A, HEK293 cells were transiently co-transfected either with JAK3WT, or different JAK3 mutants (Y100A, V674A, and L857P) or double mutants (V674A/Y100A and L857P/Y100A) together with γc and IL-9Rα or not, in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the control condition without JAK3 and the WT or mutant forms of JAK3 for each condition (with or without IL-9R complex). *, p < 0.05; ***, p < 0.001. K, kinase domain; PK, pseudokinase domain. B, in parallel of the luciferase assay, transfected HEK293 cells were lysed 24 h post-transfection and subjected to Western blot analysis using an anti-JAK3 antibody and an anti-β-actin antibody as loading control. C, relative proliferation of IL-3-dependent Ba/F3 cells or autonomous Ba/F3 cells obtained after transduction with ALL-associated JAK3 mutants (V674A and L857P) and double mutant (L857P/Y100A) after knockdown of endogenous γc compared with the proliferation observed with an irrelevant control siRNA. After 72 h, tritiated thymidine incorporation was measured. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the IL-3-dependent control Ba/F3 cells and the transformed Ba/F3 cells. *, p < 0.05.
To confirm the lack of γc requirement for constitutive JAK3L857P signaling in hematopoietic cells, we down-regulated the expression of endogenous murine γc in growth factor-independent Ba/F3 cells obtained after stable transduction with JAK3V674A, JAK3L857P, and JAK3L857P/Y100A or in IL-3-dependent control Ba/F3 cells. Fig. 2C shows that siRNA-mediated knockdown of γc resulted in a significant decrease of the proliferation of Ba/F3 cells transformed with JAK3V674A compared with the proliferation observed after control siRNA treatment, whereas no decrease in proliferation was observed for cells transformed by JAK3L857P or JAK3L857P/Y100A. These results confirm the unique capacity of JAK3L857P to induce cytokine-receptor independent constitutive signaling.
Cytokine receptor complexes are formed of two receptor chains, each associated with one JAK. In γc-associated receptor complexes, such as IL-7 or IL-9 receptor, JAK3 and JAK1 are believed to interact and cross-phosphorylate each other to enable signaling. To assess the role of JAK1 in mutant JAK3-induced signaling, we took advantage of JAK1-deficient U4C cells. As shown in Fig. 3A, in the absence of JAK1, the JAK3V674A was unable to activate STAT5, even when cytokine receptor chains were expressed. This activity was restored by co-expression with JAK1WT, but not with a kinase-dead (KD) JAK1. The results obtained for JAK3L857P, shown in Fig. 3B, indicated that the activity of this mutant is entirely independent of JAK1, once again supporting the hypothesis of a cytokine receptor-independent activation mode.
FIGURE 3.
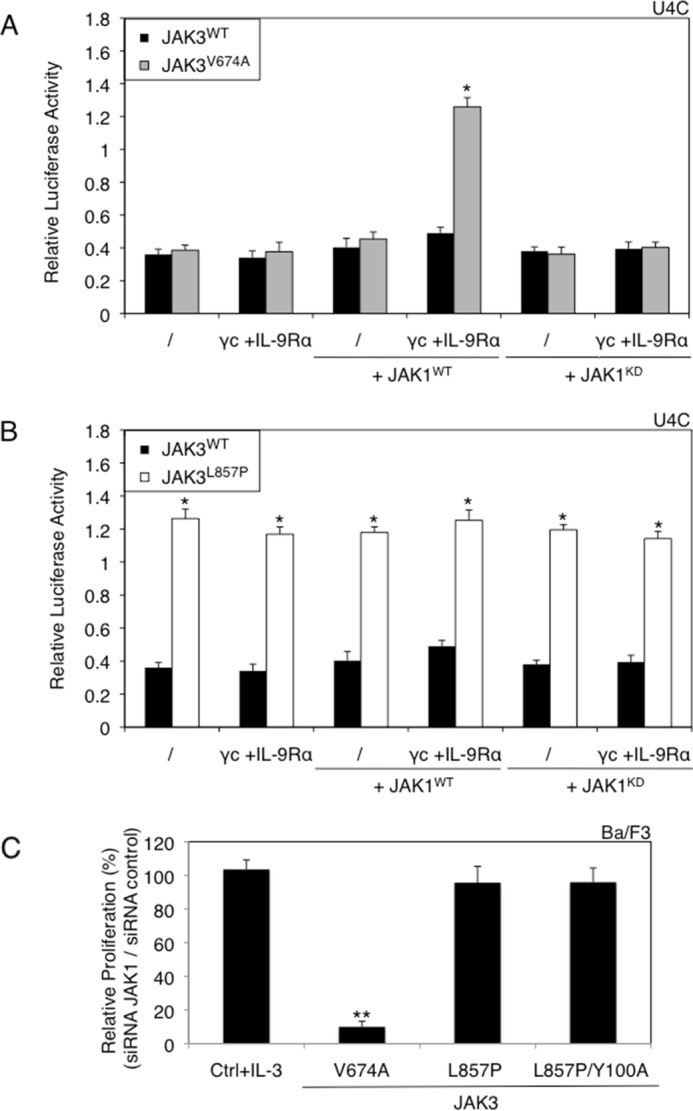
JAK1 expression is required for the activity of the V674A mutant, but not for the L857P mutant. A, JAK1-deficient U4C cells were transiently co-transfected with JAK3WT or JAK3V674A, γc, and IL-9Rα with or without JAK1WT or JAK1KD, in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values. *, p < 0.05. B, JAK1-deficient U4C cells were transiently co-transfected with JAK3WT or JAK3L857P, γc, and IL-9Rα with or without JAK1WT or JAK1KD, in addition to the STAT5-responsive luciferase reporter pLHRE-luc and the pRLTK plasmid as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values. *, p < 0.05. C, relative proliferation of IL-3-dependent Ba/F3 cells or autonomous Ba/F3 cells obtained after transduction with ALL-associated JAK3 mutants (V674A and L857P) and double mutant (L857P/Y100A) after knockdown of endogenous JAK1 compared with the proliferation observed with an irrelevant control siRNA. After 72 h, tritiated thymidine incorporation was measured. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the IL-3-dependent control Ba/F3 cells and the transformed Ba/F3 cells. **, p < 0.01.
To confirm the lack of JAK1 requirement for constitutive JAK3L857P signaling in hematopoietic cells, endogenous JAK1 knockdown was performed as described for γc (Fig. 3C). This knockdown decreased the proliferation of Ba/F3 cells transformed with JAK3V674A, whereas it did not alter the proliferation of cells transformed by JAK3L857P or JAK3L857P/Y100A. Taken together, these results revealed JAK3L857P as possessing a unique characteristic: the capacity to activate signaling in the absence of binding to a cognate cytokine receptor complex. This contrasts with the other tested JAK3 mutants, as well as all JAK1 and JAK2 activating mutants described in the literature (20, 21).
Given that the leucine 857 of JAK3 was conserved among all JAK kinases, we sought to determine whether mutations of the homologous residue in JAK1 (Leu910) and JAK2 (Leu884) could confer a similar receptor-independent constitutive signaling capacity. Those mutations were introduced together with the FERM domain mutations Y107A in JAK1 or Y114A in JAK2 (homologous to Y100A in JAK3). As illustrated in Fig. 4, upon transient expression in HEK293 cells, JAK1L910P/Y107A and JAK2L884P/Y114A were capable of activating STAT5, demonstrating that the activating mutations of a leucine residue conserved among JAK kinases share the capacity of inducing STAT5 activation independently of the receptor-FERM domain interaction.
FIGURE 4.
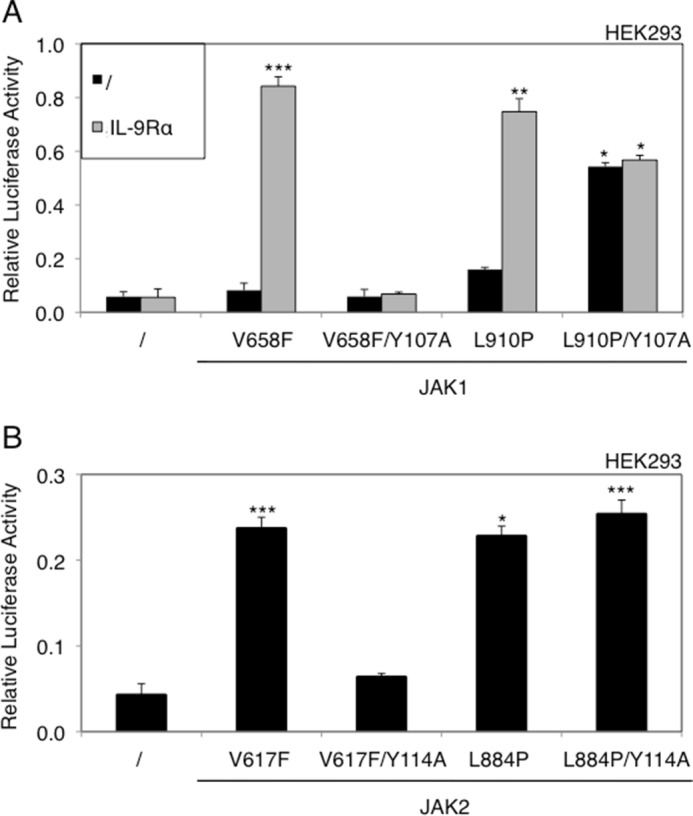
Integrity of the FERM domain is not required for activation of JAK1 and JAK2 through homologous mutation of JAK3L857P. A, HEK293 cells were transiently co-transfected with ALL-associated JAK1V658F, JAK1L910P (homologous to JAK3 L857P), or JAK1L910P/Y107A, with or without IL-9Rα, in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of means of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the control condition without JAK1 and the mutant forms of JAK1 for each condition (with or without IL-9Rα). *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, HEK293 cells were transiently co-transfected with myeloproliferative neoplasm-associated JAK2V617F, JAK2L884P (homologous to JAK3 L857P), or JAK2L884P/Y114A, in addition to the STAT5-responsive luciferase reporter pLHRE-luc and the pRLTK plasmid as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The results are means ± standard deviation of means of triplicates of three different experiments. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the control condition without JAK2 and the mutant forms of JAK2. *, p < 0.05; ***, p < 0.001.
To gain more insight about potential mechanisms enabling receptor-independent constitutive activation of JAK3L857P, we substituted Leu857 with different residues and tested the receptor-independent activity of these JAK3 mutants in HEK293 cells, using JAK3 cloned in either pMX-IRES-CD4 (Fig. 5A) or pMSCV-GFP (Fig. 5B). Receptor-independent constitutive STAT5 activation was observed after replacing the leucine with a proline, alanine, or glutamine, but not with a glutamic acid, probably because of this residue's negative charge, even if the mutant was still active in presence of IL-9 (data not shown).
FIGURE 5.
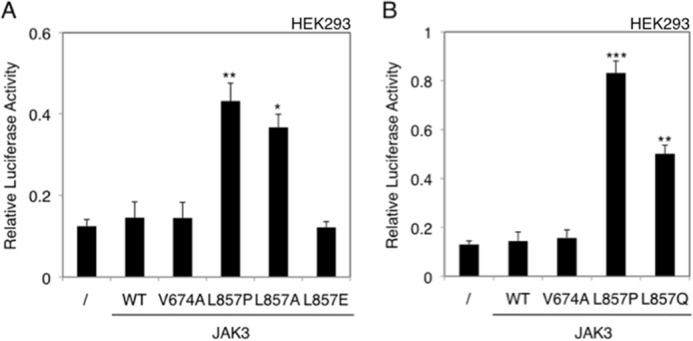
Substitution of Leu857 of JAK3 with different residues can confer receptor-independent constitutive activity. HEK293 cells were transiently transfected either with WT JAK3, or different JAK3 mutants: JAK3WT, JAK3V674A, JAK3L857P, JAK3L857A, and JAK3L857E in pMX-IRES-CD4 (A) and JAK3WT, JAK3V674A, JAK3L857P, and JAK3L857Q (B) in pMSCV-GFP. The STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) were co-transfected. 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of means of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the control condition without JAK3 and the WT or mutant forms of JAK3 for each condition. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Another Kinase Domain Mutant, JAK3L875H, Exhibits the Same Receptor-independent Activating Capacity as JAK3L857P
Among the JAK3 mutants studied here, L857P was the only mutant involving the kinase domain, raising the hypothesis that cytokine receptor independence could be explained solely by the mutation's location in the kinase domain, compared with the pseudokinase mutations that would be receptor-dependent. To address this question, other activating mutations of the kinase domain were investigated. These consisted of one JAK3 mutation associated with T cell pro-lymphocytic leukemia (Y824D) (24), three ALL-associated mutants (L875H, P906S, and E958K),7 and one mutant described in a murine T cell line (T848A) (16). As illustrated in Fig. 6A, four of these five new mutants were dependent on the co-expression of IL-9Rα and γc for their activity in HEK293 cells. In contrast, JAK3L875H behaved like JAK3L857P, exhibiting a receptor-independent activating capacity. The close proximity of L857P and L875H mutants to the α-helix C of the kinase domain is illustrated in the predicted protein structure of JAK3 kinase domain, represented in Fig. 6B. By contrast, the receptor-dependent mutants map to the interface between pseudokinase and kinase domains, as previously described (16).
FIGURE 6.
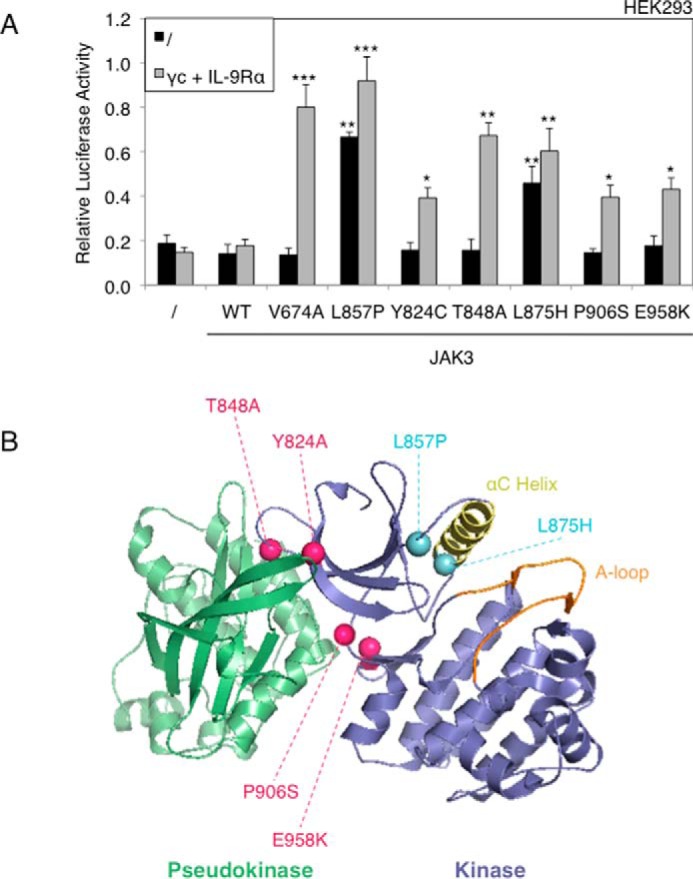
The majority of kinase domain mutants are receptor-dependent, except JAK3L857P and JAK3L875H. A, HEK293 cells were transiently co-transfected either with JAK3WT or different JAK3 mutants (V674A, L857P, Y824A, T848A, L875H, P906S, and E958K), in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of means of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values between the control condition without JAK3 and the WT or mutant forms of JAK3 for each condition. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, localization of ALL-associated JAK3 mutants of the kinase domain. The figure represents the kinase and pseudo-kinase domains of JAK3 based on the recent TYK2 JH1/JH2 crystal structure (33). The kinase domain is shown in indigo, with the αC helix in yellow and the activation loop in orange. The adjacent pseudokinase domain is shown in green. Mutated residues close to the pseudokinase domain are indicated with pink balls, and mutated residues close to the αC helix are indicated with light blue balls.
The Difference in Cytokine Receptor Requirement Correlates to a Distinct JAK Inhibitor Specificity
Given that JAK3L857P and the other JAK3 mutants presented different receptor and JAK1 requirements, we sought to determine whether they would exhibit the same sensitivity to ruxolitinib, a JAK1–2 inhibitor. To assess this, we treated growth factor-independent Ba/F3 cells obtained after stable transduction with JAK3V674A, JAK3L857P, and JAK3L857P/Y100A, with increasing ruxolitinib concentrations and measured their proliferation using a tritiated thymidine incorporation assay. We employed Ba/F3 cells that had been stably transduced with the chronic myeloid leukemia-associated BCR-ABL fusion gene as a negative control, choosing this particular oncogene for its lack of sensitivity to JAK inhibitors. Fig. 7A illustrates that Ba/F3 cells transformed with JAK3V674A were at least 10-fold more sensitive to ruxolitinib than those transformed with JAK3L857P and JAK3L857P/Y100A. The calculated IC50 values are shown in Table 1. This difference in ruxolitinib sensitivity between the V674A and L857P/Y100A JAK3 mutants was reproduced in transiently transfected HEK293 cells treated with 0.5 μm of ruxolitinib, with STAT5 transcriptional activity being used as readout (Fig. 7B). To complement these observations, we monitored the phosphorylation status of STAT5, JAK1, and JAK3 in growth factor-independent JAK3V674A and JAK3L857P/Y100A Ba/F3 cells treated with 0.5, 1, and 2 μm of ruxolitinib (Fig. 7C). This JAK1–2 inhibitor abrogated STAT5 phosphorylation induced by the V674A mutant, whereas it had a marginal effect on the L857P/Y100A double mutant, thus confirming the difference in ruxolitinib sensitivity. Although able to inhibit STAT5 phosphorylation by JAK3V674A, ruxolitinib did not inhibit JAK1 phosphorylation, probably because it is a type I kinase inhibitor, blocking the kinase in an active conformation that can still be phosphorylated by JAK3. JAK3 phosphorylation was only observed in the double mutant cells, with no ruxolitinib-induced inhibition observed, in line with the absence of effect seen with STAT5. Altogether, these results demonstrate that the activity of JAK3V674A can be blocked with a JAK1-specific inhibitor, whereas JAK1-independent JAK3L857P is much less sensitive to ruxolitinib.
FIGURE 7.
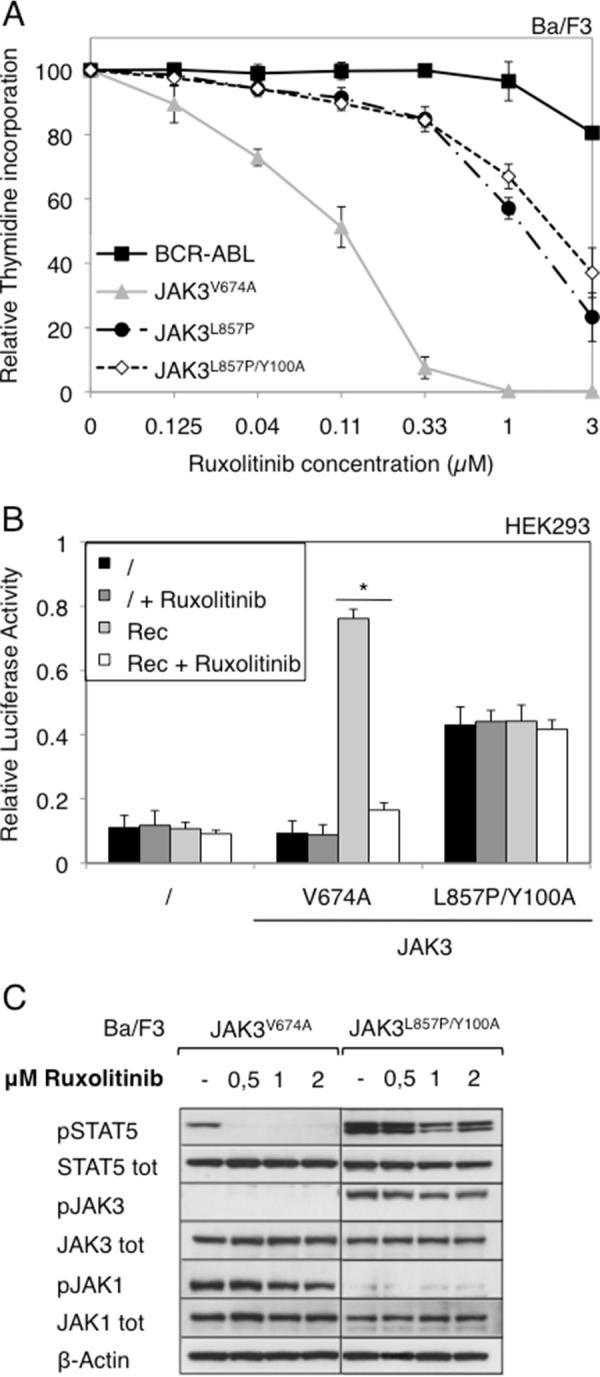
Cells expressing JAK3V674A mutant are more sensitive to ruxolitinib than cells expressing JAK3L857P or JAK3L857P/Y100A. A, autonomous Ba/F3 cells stably transduced with ALL-associated JAK3 mutants (V674A and L857P) and double mutant (L857P/Y100A) or BCR-ABL as a control were treated with increasing concentrations of JAK1/JAK2 inhibitor ruxolitinib (0–3 μm). After 72 h, tritiated thymidine incorporation was measured. The results are means ± standard deviation of three different experiments, each performed in triplicate, represented as percentages of the proliferation of the respective untreated cells. B, HEK293 cells were transiently co-transfected either with JAK3V674A or JAK3L857P/Y100A, with a receptor complex (Rec = γc, IL-9Rα, and JAK1WT), in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. Cells were treated with ruxolitinib (1 μm). 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values. *, p < 0.05. C, 106 autonomous Ba/F3 cells stably transduced with ALL-associated JAK3 mutant V674A or double mutant L857P/Y100A were treated with increasing concentration of ruxolitinib (0–2 μm). Two hours after treatment, the cells were lysed and subjected to Western blot analysis. Phosphorylation of STAT5, JAK3, and JAK1 was detected using specific anti-pY694 STAT5, anti-pY980/81 JAK3, and anti-pY1034/35 JAK1 antibodies. Membranes were reprobed with anti-STAT5, anti-JAK3, anti-JAK1, and anti-β-actin antibodies as loading controls.
TABLE 1.
Calculated IC50 values of NIBR3049, ruxolitinib, and tofacitinib in autonomous Ba/F3 cells
The table shows the calculated IC50 values (means ± S.E.) of autonomous Ba/F3 cells stably transduced with ALL-associated JAK3 mutants (V674A and L857P) or double mutant (L857P/Y100A), treated with ruxolitinib, NIBR3049, and tofacitinib. The IC50 mean and S.E. values were calculated as means of triplicate cultures from three different experiments.
| IC50 |
|||
|---|---|---|---|
| JAK1/2 inhibitor Ruxolitinib | JAK3 inhibitor NIBR3049 | JAK1/3 inhibitor Tofacitinib | |
| μm | |||
| Ba/F3 JAK3V674A | 0.1 ± 0.03 | 3.42 ± 0.22 | 0.17 ± 0.05 |
| Ba/F3 JAK3L857P | 1.26 ± 0.2 | 1.15 ± 0.21 | 0.32 ± 0.03 |
| Ba/F3 JAK3L857P/Y100A | 2.33 ± 0.21 | 1.31 ± 0.24 | 0.44 ± 0.05 |
We next tested the effect of a JAK3-specific inhibitor (NIBR3049) (17) on the different JAK3 mutants by performing the same experiments as for ruxolitinib. As illustrated in Fig. 8A, the Ba/F3 cells transformed with JAK3L857P and JAK3L857P/Y100A proved more sensitive to NIBR3049 than those transformed with JAK3V674A. The calculated IC50 values are shown in Table 1. Similar results were obtained for STAT5 activity in HEK293 cells (Fig. 8B). In Ba/F3 cells, the NIBR3049 inhibitor completely blocked STAT5 and JAK3 phosphorylation in the JAK3L857P/Y100A cells, confirming their sensitivity to this molecule (Fig. 8C). By contrast, NIBR3049 barely affected STAT5 and JAK1 phosphorylation in JAK3V674A cells and even increased JAK3 phosphorylation because this type I inhibitor blocked JAK3 in the active conformation without affecting the kinase activity of the JAK1 partner. Altogether, these results show that JAK3L857P activity can be blocked by a JAK3-specific inhibitor, whereas the JAK3V674A is much less sensitive to NIBR3049. Finally, when an inhibitor active against both JAK1 and JAK3 was used (tofacitinib), both receptor-dependent and -independent mutants exhibited a similar sensitivity, as illustrated by a decrease in the proliferation of Ba/F3 cells and in STAT5 activity in HEK293 cells (Fig. 9).
FIGURE 8.
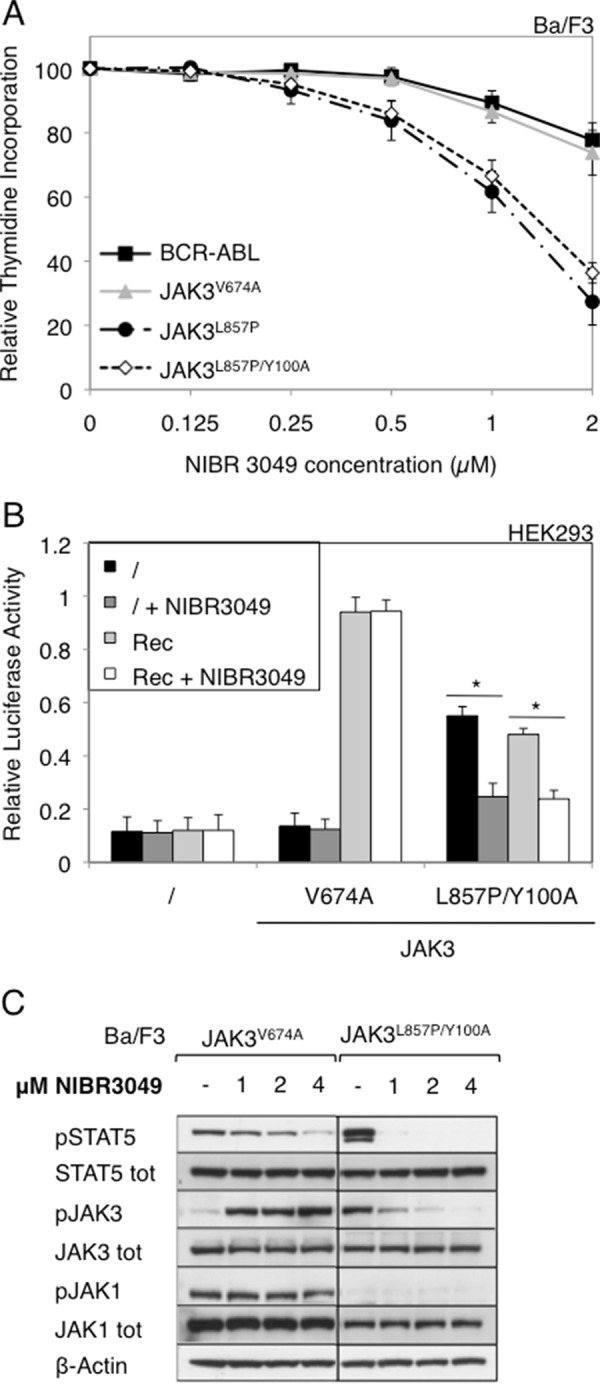
Cells expressing JAK3 L857P or L857P/Y100A mutants are more sensitive to NIBR3049 than cells expressing JAK3 V674A. A, autonomous Ba/F3 cells stably transduced with ALL-associated JAK3 mutants (V674A and L857P) and double mutant (L857P/Y100A) or BCR-ABL as a control were with increasing concentrations of JAK3 inhibitor NIBR3049 (0–2 μm). After 72 h, tritiated thymidine incorporation was measured. The results are means ± standard deviation of three different experiments, each performed in triplicate, represented as percentages of the proliferation of the respective untreated cells. B, HEK293 cells were transiently co-transfected either with JAK3V674A or JAK3L857P/Y100A, with a receptor complex (Rec = γc, IL-9Rα, and JAK1WT), in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. Cells were treated with NIBR3049 (0.5 μm). 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values. *, p < 0.05. C, 106 autonomous Ba/F3 cells stably transduced with ALL-associated JAK3 mutant V674A or double mutant L857P/Y100A were treated with increasing concentration of NIBR3049 (0–4 μm). Two hours after treatment, the cells were lysed and subjected to Western blot analysis. Phosphorylation of STAT5, JAK3, and JAK1 was detected using specific anti-pY694 STAT5, anti-pY980/81 JAK3, and anti-pY1034/35 JAK1 antibodies. Membranes were reprobed with anti-STAT5, anti-JAK3, anti-JAK1, and anti-β-actin antibodies as loading controls.
FIGURE 9.
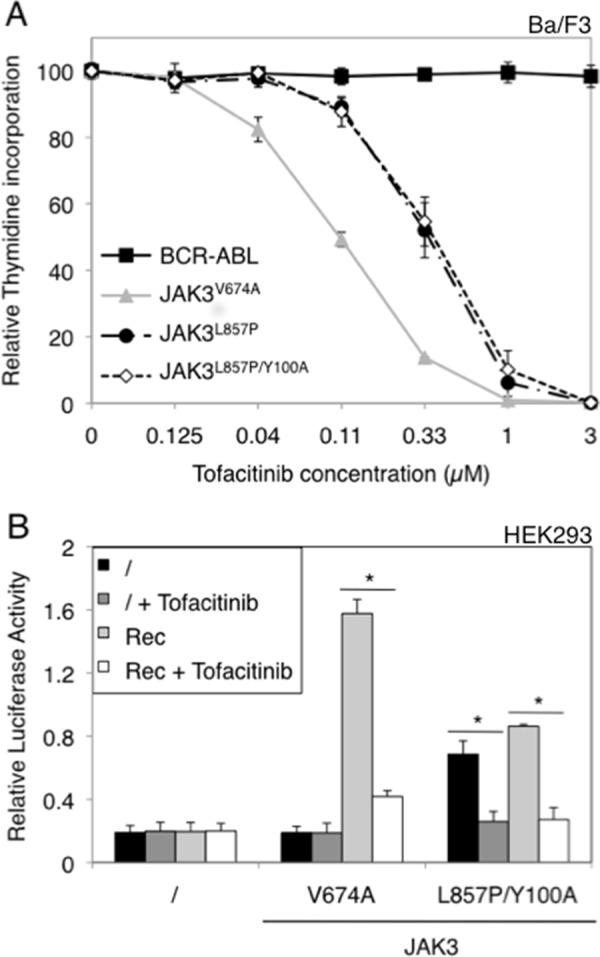
Cells expressing JAK3 V674A, L857P, and L857P/Y100A mutants are sensitive to tofacitinib. A, autonomous Ba/F3 cells stably transduced with ALL-associated JAK3 mutants (V674A and L857P) and double mutant (L857P/Y100A) or BCR-ABL as a control were treated with increasing concentrations of JAK1/JAK3 inhibitor tofacitinib (0–3 μm). After 72 h, tritiated thymidine incorporation was measured. The results are means ± standard deviation of three different experiments, each performed in triplicate, represented as percentages of the proliferation of the respective untreated cells. B, HEK293 cells were transiently co-transfected either with JAK3V674A or JAK3L857P/Y100A, with a receptor complex (Rec = γc, IL-9Rα, and JAK1WT), in addition to the STAT5-responsive luciferase reporter pLHRE-luc (firefly luciferase) and the pRLTK plasmid (Renilla luciferase) as transfection control. Cells were treated with tofacitinib (0.5 μm). 24 h post-transfection, the cells were subjected to a luciferase assay. The relative luciferase activity corresponds to the firefly luciferase light emission values divided by the Renilla luciferase light emission values. The results are means ± standard deviation of three different experiments, each performed in triplicate. A Kruskal-Wallis test with Dunn correction was performed to determine p values. *, p < 0.05.
JAK3 Mutants Cause Distinct Leukemia Phenotypes in a Murine Hematopoietic Stem Cell Transplant Model
Degryse et al. (13) reported that mice transplanted with bone marrow progenitor cells expressing T-ALL-associated JAK3 mutants developed a disease resembling T-ALL, characterized by an accumulation of immature CD8 positive T cells. This phenotype can be explained as the result of constitutive activation of γc-associated receptors, which are essential for T cell lymphoproliferation. Given that JAK3L857P and JAK3L857P/Y100A exhibit no cytokine receptor restriction in vitro, we wondered whether their expression in hematopoietic stem cells would be leukemogenic and produce the same phenotype in vivo. Hematopoietic progenitor cells were transduced with retroviral vectors expressing JAK3V674A, JAK3L857P, or JAK3L857P/Y100A, together with GFP, and injected into irradiated Balb/c mice. As shown in Table 2, almost all the mice exhibited increased white blood cell counts ranging from 20,000 to 300,000 cells/microliter at sacrifice, within 50–200 days post-transplant. JAK3V674A mice displayed an accumulation of CD8 single positive cells in the spleen and bone marrow (Table 2, bracket A, and Fig. 10A), as well as in the peripheral blood, thymus, and lymph nodes (data not shown). These mice homogenously developed a disease resembling T-ALL, similar to the one described by Degryse et al. By contrast, JAK3L857P and JAK3L857P/Y100A were found to induce at least four different types of lympho- or myeloproliferative diseases, characterized by an accumulation of either CD4/8 double positive T-lymphocytes (Table 2, bracket B, and Fig. 10B), Gr1 positive myeloid cells (Table 2, bracket C, and Fig. 10C), CD8 single positive T-lymphocytes (Table 2, bracket D, and Fig. 10D), or B220 positive B-lymphocytes (Table 2, bracket E, and Fig. 10E). These results indicated that JAK3L857P and JAK3L857P/Y100A exhibit an oncogenic potential in vivo but induced a more heterogeneous phenotype compared with cytokine receptor-restricted JAK mutants.
TABLE 2.
Expression of JAK3 mutants in bone marrow cells of Balb/c mice leads to distinct leukemia phenotypes
The table shows the days post-transplant at sacrifice, the white blood cells count at sacrifice, and the diagnosis of Balb/c mice receiving a bone marrow transplantation of lineage negative cells expressing JAK3V674A, JAK3L857P or JAK3L857P/Y100A. Five different diagnosis were observed: CD8+ T-lymphoproliferation in JAK3V674A mice (Bracket A), CD4/8+ T-lymphoproliferation in JAK3L857P and JAK3L857P/Y100A mice (Bracket B), Gr1+ myeloproliferation in JAK3L857P and JAK3L857P/Y100A mice (Bracket C), another type of CD8+ T-lymphoproliferation in JAK3L857P and JAK3L857P/Y100A mice (Bracket D), and B220+ B-lymphoproliferation in a JAK3L857P mice (Bracket E).
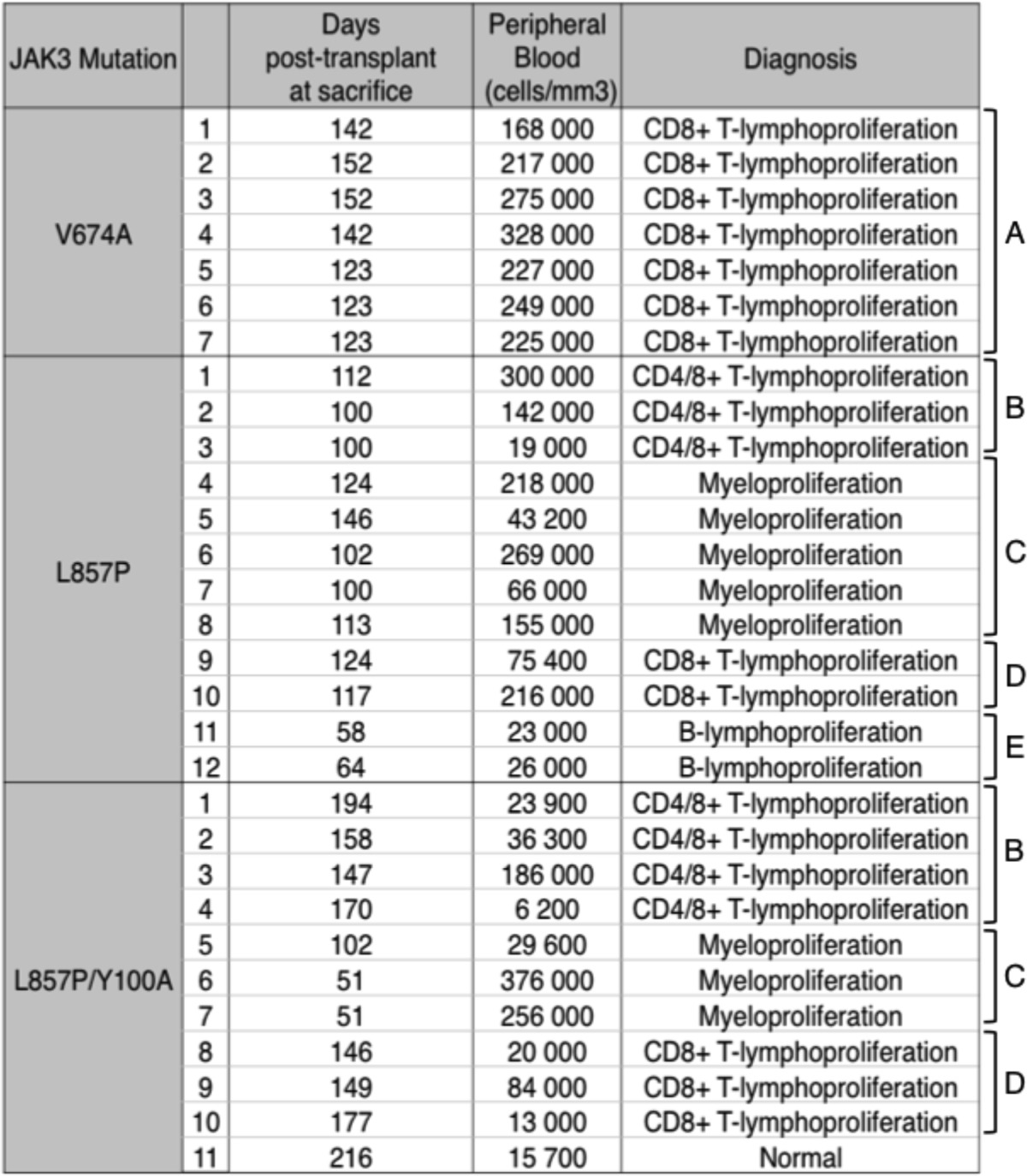
FIGURE 10.

Expression of JAK3 mutants in bone marrow cells of Balb/c mice leads to distinct leukemia phenotypes. Spleen and bone marrow cells of diseased animals were analyzed by flow cytometry with anti-CD4, anti-CD8, anti-Gr1, and anti-B220 antibodies. Cells were gated for GFP expression. FACS analysis representative of a CD8+ T-lymphoproliferation in a JAK3V674A mouse (A), a CD4/8+ T-lymphoproliferation in a JAK3L857P mouse (B), a Gr1+ myeloproliferation in a JAK3L857P/Y100A mouse (C), a CD8+ T-lymphoproliferation in a JAK3L857P/Y100A mouse (D), and a B220+ B-lymphoproliferation in a JAK3L857P mouse (E) are shown, either as dot plots (spleen) or as stacked column charts representing the percentage of cells positive for the different markers (bone marrow).
Discussion
The originality of our observations resides in the demonstration that cancer-associated activating mutations of different residues in the same JAK kinase induce different functional properties, both in vitro and in vivo. In vitro, different ALL-associated JAK3 mutants were associated with distinct sensitivity to JAK inhibitors, whereas the in vivo transduction of these mutants in hematopoietic precursors produced distinct leukemia phenotypes. This can, for the most part, be explained by the unexpected behavior of JAK3L857P, which did not require cytokine receptor binding to be active. These findings contrast sharply with what has previously been published about other cancer-associated JAK mutants (20, 21), including JAK3V674A, which required binding to a functional receptor complex to be active.
The different phenotypes observed in vivo can also be explained by the different cytokine receptor requirements of the various mutations tested. The transduction of murine hematopoietic cells with JAK3V674A led to homogenous lymphoblastic leukemias. Because the oncogenic activity of this mutant is restricted to cells expressing γc, downstream constitutive signaling mimics the signaling induced by γc-associated receptors like IL-2 receptor, IL-7 receptor, or IL-9 receptor, which are essential for lymphoproliferation. In contrast, transduction with JAK3L857P induced various types of lymphoid and myeloid leukemias in mice, revealing the existence of a broader oncogenic potential resulting from the absence of cytokine receptor restriction.
The second key observation reported here is that different activating mutations of JAK3 dictate specific pattern of sensitivity to JAK inhibitors. Such different sensitivities to JAK inhibitors could be explained by the model presented in Fig. 11. Cytokine receptor complexes are formed of two chains, each associated with one JAK, namely JAK1 and JAK3 for the γc cytokine receptor family (Fig. 11A). JAK3L857P, although still able to bind to γc, does not require this binding to be active. This explains its JAK1-independent activity and accounts for the inefficacy of JAK1 inhibitors, such as ruxolitinib (Fig. 11B). In contrast, a JAK1 inhibitor blocks the activity of classical JAK3 mutants, such as JAK3V674A, because these mutants are dependent on a functional cytokine receptor complex for signaling. A JAK3-specific inhibitor, such as NIBR3049, effectively inhibits the activity of JAK3L857P (Fig. 11C). Its relative inefficacy against JAK3V657A is in line with the role of the kinase activity of JAK3 in the context of IL-2 signaling, as reported by Haan et al. (1). Indeed, these authors demonstrated that although signal transduction by the IL-2 receptor complex required JAK3 expression, it was not abrogated by a kinase-defective JAK3, suggesting that JAK1 kinase activity was sufficient for the complex to be active, with JAK3 serving as a scaffold protein. One hypothesis is therefore that classical mutants, such as JAK3V674A, modify the conformation of JAK3 as a scaffold in the receptor complex to enable JAK1 constitutive kinase activity.
FIGURE 11.
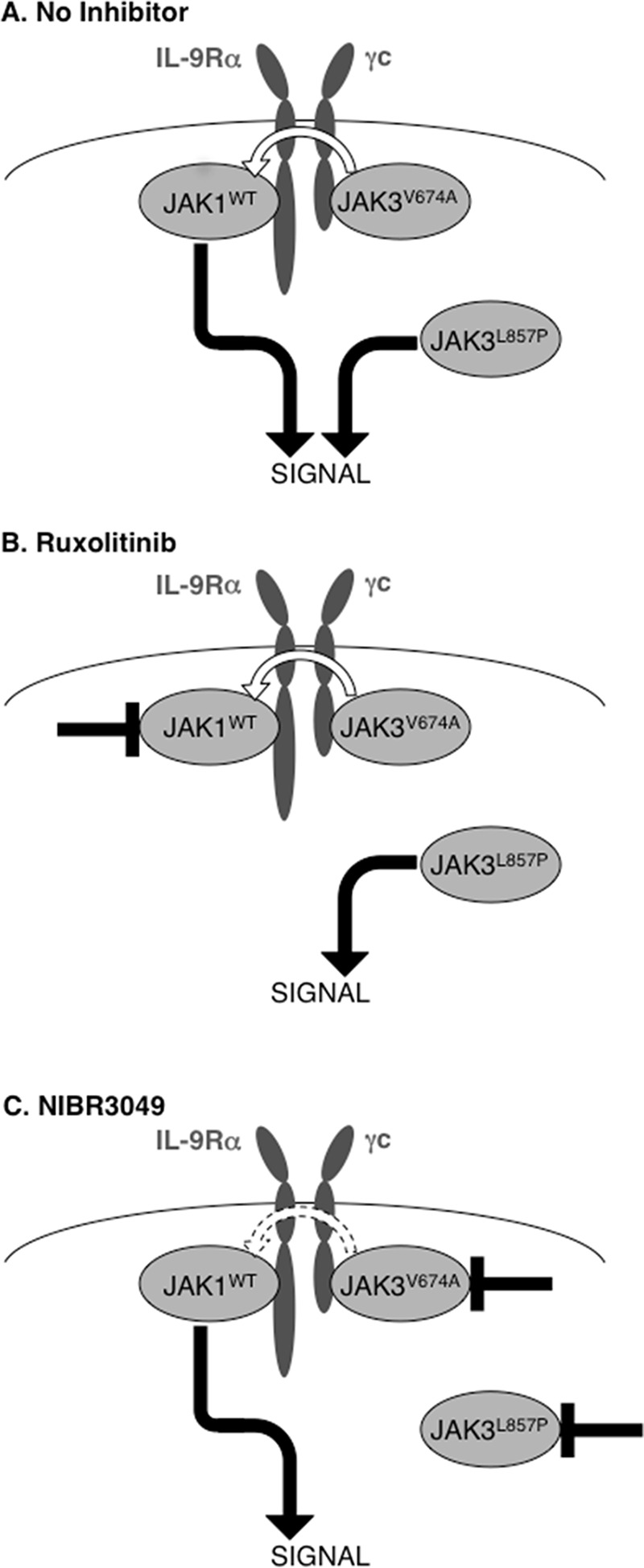
Schematic representation of the activity of JAK3 mutants under JAK inhibitor treatment.
Our findings provide further evidence that although up to 30% of ALL cases might harbor activating mutations of the IL-7 receptor-JAK1-JAK3 pathway, working with JAK inhibitors to treat such hematological neoplasms will not be a simple task. The selection and dosing of the inhibitor must not only be based on the kinase affected but also on the specific mutation observed. We previously showed that activating mutations affecting Phe958 and Pro960 of JAK1 not only promoted autonomous cell proliferation but also conferred resistance to ATP-competitive inhibitors, suggesting that mutations of the same kinase could require different JAK inhibitor dosages (25). This also proved true for mutations of the JAK2 Tyr931 residue (25) and, furthermore, applied to the homologous mutation of the Tyr904 residue of JAK3 (16). Our current study has demonstrated that different JAK inhibitors are required to block different JAK3 mutants, depending on their mechanisms of signal transduction activation. Although JAK1 (L910P) and JAK2 (L884P) mutations that are homologous to L857P also induce cytokine receptor-independent constitutive signaling, their effect on JAK inhibitor sensitivity should be less critical, because many JAK2-associated receptors are homodimeric, and the kinase activity of JAK1 appears essential for γc-associated receptors.
The mechanisms enabling the receptor-independent constitutive activation of JAK3L857P are still subject to speculation. For JAK3, receptor-independent constitutive activation is observed after replacing the leucine with different residues (Fig. 5), except for glutamic acid. JAK3L857Q has been described in ALL patients (13) and is also receptor-independent. The L910Q mutation of JAK1 has already been described as activating in a random mutagenesis screening analysis (26) and introducing the Y107A mutation (homologous to the JAK3 Y100A mutation) in the FERM domain of JAK1L910Q has been found to not completely abolish STAT5 constitutive phosphorylation. The diversity of these residues suggests that the loss of leucine 857 is the key factor inducing constitutive activation. Interestingly, the leucine 857 of JAK3 is not only conserved among all JAK kinases, but also in other tyrosine kinases, such as c-Src, LCK, c-ABL, EGFR, FGF receptor, and PDGF receptor. The relevance of this residue for kinase activity regulation is illustrated by the EGFR exon 19 insertions leading to the L747P mutation described in patients with lung adenocarcinoma or by the L747S mutation of the EGFR, reported in patients acquiring secondary resistance to EGFR inhibitors. It is believed to shift the equilibrium toward the receptor's active conformation (27–29). Other arguments point toward the importance of the JAK αC helix in the regulation of the cytokine receptor dependence. As illustrated in 4B, the Leu857 residue is situated in front of the αC helix of the N-lobe of the JAK3 kinase domain. This alpha helix is conserved among all kinases and constitutes a key mediator of conformational change between the active and inactive states of the kinase (30). Additionally, another mutation located within the αC helix of JAK3, L875H, is present in patients with ALL, exhibiting the same characteristics as L857P, namely constitutive activation and receptor independence.
A further puzzling observation is that when the Y100A mutation was added to the active JAK3L857P mutant, STAT5 activation was seen to increase in HEK293 cells. A preliminary explanation for this could be that Y100A increases L857P-induced constitutive kinase activity. Previous studies have, in fact, suggested that the FERM domain fulfills a regulatory role in kinase activity through interaction between the kinase and FERM domains (2, 31, 32). Moreover, electron microscopy JAK1 imaging revealed this protein as exhibiting a high flexibility, able to shuttle from open to closed states (32). In the compact conformation, the FERM domain comes in close proximity to the kinase domain. The Y100A mutation could change these interactions and increase the kinase activity induced by L857P. Another explanation could reside in the subcellular location of the kinase. JAK3V674A needs to bind to a cytokine receptor to be active, with a kinase activity located close to the plasma membrane. The localization of JAK3L857P/Y100A is unknown, yet cytokine-receptor independent. The constitutive kinase activity could therefore take place in a location where the activation of downstream signaling is more efficient, such as the cytoplasm. Further studies will be needed to address these issues. Because of their particular mode of action, the cytokine receptor-independent active mutants of JAK kinases described here open new perspectives, not only for treating patients with alterations of the JAK-STAT pathway but also for a better understanding of this pathway.
Author Contributions
L. K., J.-C. R., E. L., T. K., and L. S. designed the study. E. L., J.-C. R., and L. K. wrote the paper. E. L., S. D., and O. G. performed the experiments. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgment
We are grateful to Prof. Claude Haan (Life Sciences Research Unit-Signal Transduction Laboratory, University of Luxembourg, Luxembourg) for critical reading of the manuscript.
This work was supported in part by Grant IAP-P7/43 from the Belgian Program on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming (to S. N. C.); Actions de Recherche Concertées of the Communauté Française de Belgique Grant ARC10/15-027 (to S. N. C.); the Fondation contre le Cancer, Belgium (to S. N. C.); the Foundation Salus Sanguinis, Belgium; the Opération Télévie, Belgium; Associazione Italiana per la Ricerca sul Cancro Grants IG2009-8803 and IG2012-3360 (to M. T.); the Fund for Medical Scientific Research, Belgium (to S. N. C.); and the Atlantic Philanthropies New York (to S. N. C.). The authors declare that they have no conflicts of interest with the contents of this article.
M. Tartaglia, unpublished data.
J. Cools, unpublished data.
- γc
- common γ chain
- ALL
- acute lymphoblastic leukemia
- IL-9R
- IL-9 receptor
- LHRE
- lactogenic hormone response element
- SCID
- severe combined immunodeficiency syndrome
- T-ALL
- T cell acute lymphoblastic leukemia
- EGFR
- EGF receptor.
References
- 1.Haan C., Rolvering C., Raulf F., Kapp M., Drückes P., Thoma G., Behrmann I., and Zerwes H. G. (2011) Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem. Biol. 18, 314–323 [DOI] [PubMed] [Google Scholar]
- 2.Saharinen P., and Silvennoinen O. (2002) The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J. Biol. Chem. 277, 47954–47963 [DOI] [PubMed] [Google Scholar]
- 3.Haan C., Is'harc H., Hermanns H. M., Schmitz-Van De Leur H., Kerr I. M., Heinrich P. C., Grötzinger J., and Behrmann I. (2001) Mapping of a region within the N terminus of Jak1 involved in cytokine receptor interaction. J. Biol. Chem. 276, 37451–37458 [DOI] [PubMed] [Google Scholar]
- 4.Tang W., Huo H., Zhu J., Ji H., Zou W., Xu L., Sun L., Zheng Z., Theze J., and Liu X. (2001) Critical sites for the interaction between IL-2Rγ and JAK3 and the following signaling. Biochem. Biophys. Res. Commun. 283, 598–605 [DOI] [PubMed] [Google Scholar]
- 5.Cacalano N. A., Migone T. S., Bazan F., Hanson E. P., Chen M., Candotti F., O'Shea J. J., and Johnston J. A. (1999) Autosomal SCID caused by a point mutation in the N-terminus of Jak3: mapping of the Jak3-receptor interaction domain. EMBO J. 18, 1549–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bains T., Heinrich M. C., Loriaux M. M., Beadling C., Nelson D., Warrick A., Neff T. L., Tyner J. W., Dunlap J., Corless C. L., and Fan G. (2012) Newly described activating JAK3 mutations in T-cell acute lymphoblastic leukemia. Leukemia 26, 2144–2146 [DOI] [PubMed] [Google Scholar]
- 7.Walters D. K., Mercher T., Gu T. L., O'Hare T., Tyner J. W., Loriaux M., Goss V. L., Lee K. A., Eide C. A., Wong M. J., Stoffregen E. P., McGreevey L., Nardone J., Moore S. A., Crispino J., Boggon T. J., Heinrich M. C., Deininger M. W., Polakiewicz R. D., Gilliland D. G., and Druker B. J. (2006) Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell 10, 65–75 [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi H., Okuno Y., Muramatsu H., Yoshida K., Shiraishi Y., Takahashi M., Kon A., Sanada M., Chiba K., Tanaka H., Makishima H., Wang X., Xu Y., Doisaki S., Hama A., Nakanishi K., Takahashi Y., Yoshida N., Maciejewski J. P., Miyano S., Ogawa S., and Kojima S. (2013) Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat. Genet. 45, 937–941 [DOI] [PubMed] [Google Scholar]
- 9.Kudlacz E., Conklyn M., Andresen C., Whitney-Pickett C., and Changelian P. (2008) The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur. J. Pharmacol 582, 154–161 [DOI] [PubMed] [Google Scholar]
- 10.Kudlacz E., Perry B., Sawyer P., Conklyn M., McCurdy S., Brissette W., Flanagan, and Changelian P. (2004) The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am. J. Transplant 4, 51–57 [DOI] [PubMed] [Google Scholar]
- 11.Karaman M. W., Herrgard S., Treiber D. K., Gallant P., Atteridge C. E., Campbell B. T., Chan K. W., Ciceri P., Davis M. I., Edeen P. T., Faraoni R., Floyd M., Hunt J. P., Lockhart D. J., Milanov Z. V., Morrison M. J., Pallares G., Patel H. K., Pritchard S., Wodicka L. M., and Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 [DOI] [PubMed] [Google Scholar]
- 12.Arana Yi C., Tam C. S., and Verstovsek S. (2015) Efficacy and safety of ruxolitinib in the treatment of patients with myelofibrosis. Future Oncol. 11, 719–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Degryse S., de Bock C. E., Cox L., Demeyer S., Gielen O., Mentens N., Jacobs K., Geerdens E., Gianfelici V., Hulselmans G., Fiers M., Aerts S., Meijerink J. P., Tousseyn T., and Cools J. (2014) JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood 124, 3092–3100 [DOI] [PubMed] [Google Scholar]
- 14.Gerland K., Bataillé-Simoneau N., Baslé M., Fourcin M., Gascan H., and Mercier L. (2000) Activation of the Jak/Stat signal transduction pathway in GH-treated rat osteoblast-like cells in culture. Mol. Cell. Endocrinol. 168, 1–9 [DOI] [PubMed] [Google Scholar]
- 15.Dumoutier L., Louahed J., and Renauld J. C. (2000) Cloning and characterization of IL-10-related T cell-derived inducible factor (IL-TIF), a novel cytokine structurally related to IL-10 and inducible by IL-9. J. Immunol. 164, 1814–1819 [DOI] [PubMed] [Google Scholar]
- 16.Springuel L., Hornakova T., Losdyck E., Lambert F., Leroy E., Constantinescu S. N., Flex E., Tartaglia M., Knoops L., and Renauld J. C. (2014) Cooperating JAK1 and JAK3 mutants increase resistance to JAK inhibitors. Blood 124, 3924–3931 [DOI] [PubMed] [Google Scholar]
- 17.Thoma G., Nuninger F., Falchetto R., Hermes E., Tavares G. A., Vangrevelinghe E., and Zerwes H. G. (2011) Identification of a potent Janus kinase 3 inhibitor with high selectivity within the Janus kinase family. J. Med. Chem. 54, 284–288 [DOI] [PubMed] [Google Scholar]
- 18.Yamashita Y., Yuan J., Suetake I., Suzuki H., Ishikawa Y., Choi Y. L., Ueno T., Soda M., Hamada T., Haruta H., Takada S., Miyazaki Y., Kiyoi H., Ito E., Naoe T., Tomonaga M., Toyota M., Tajima S., Iwama A., and Mano H. (2010) Array-based genomic resequencing of human leukemia. Oncogene 29, 3723–3731 [DOI] [PubMed] [Google Scholar]
- 19.Riera L., Lasorsa E., Bonello L., Sismondi F., Tondat F., Di Bello C., Di Celle P. F., Chiarle R., Godio L., Pich A., Facchetti F., Ponzoni M., Marmont F., Zanon C., Bardelli A., and Inghirami G. (2011) Description of a novel Janus kinase 3 P132A mutation in acute megakaryoblastic leukemia and demonstration of previously reported Janus kinase 3 mutations in normal subjects. Leukemia Lymphoma 52, 1742–1750 [DOI] [PubMed] [Google Scholar]
- 20.Lu X., Levine R., Tong W., Wernig G., Pikman Y., Zarnegar S., Gilliland D. G., and Lodish H. (2005) Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc. Natl. Acad. Sci. U.S.A. 102, 18962–18967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hornakova T., Staerk J., Royer Y., Flex E., Tartaglia M., Constantinescu S. N., Knoops L., and Renauld J. C. (2009) Acute lymphoblastic leukemia-associated JAK1 mutants activate the Janus kinase/STAT pathway via interleukin-9 receptor alpha homodimers. J. Biol. Chem. 284, 6773–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malka Y., Hornakova T., Royer Y., Knoops L., Renauld J. C., Constantinescu S. N., and Henis Y. I. (2008) Ligand-independent homomeric and heteromeric complexes between interleukin-2 or -9 receptor subunits and the γ chain. J. Biol. Chem. 283, 33569–33577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wernig G., Gonneville J. R., Crowley B. J., Rodrigues M. S., Reddy M. M., Hudon H. E., Walz C., Reiter A., Podar K., Royer Y., Constantinescu S. N., Tomasson M. H., Griffin J. D., Gilliland D. G., and Sattler M. (2008) The Jak2V617F oncogene associated with myeloproliferative diseases requires a functional FERM domain for transformation and for expression of the Myc and Pim proto-oncogenes. Blood 111, 3751–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bellanger D., Jacquemin V., Chopin M., Pierron G., Bernard O. A., Ghysdael J., and Stern M. H. (2014) Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia 28, 417–419 [DOI] [PubMed] [Google Scholar]
- 25.Hornakova T., Springuel L., Devreux J., Dusa A., Constantinescu S. N., Knoops L., and Renauld J. C. (2011) Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematologica 96, 845–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon G. M., Lambert Q. T., Daniel K. G., and Reuther G. W. (2010) Transforming JAK1 mutations exhibit differential signalling, FERM domain requirements and growth responses to interferon-γ. Biochem. J. 432, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He M., Capelletti M., Nafa K., Yun C. H., Arcila M. E., Miller V. A., Ginsberg M. S., Zhao B., Kris M. G., Eck M. J., Jänne P. A., Ladanyi M., and Oxnard G. R. (2012) EGFR exon 19 insertions: a new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin. Cancer Res. 18, 1790–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaguchi F., Kugawa S., Tateno H., Kokubu F., and Fukuchi K. (2012) Analysis of EGFR, KRAS and P53 mutations in lung cancer using cells in the curette lavage fluid obtained by bronchoscopy. Lung Cancer 78, 201–206 [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi F., Fukuchi K., Yamazaki Y., Takayasu H., Tazawa S., Tateno H., Kato E., Wakabayashi A., Fujimori M., Iwasaki T., Hayashi M., Tsuchiya Y., Yamashita J., Takeda N., and Kokubu F. (2014) Acquired resistance L747S mutation in an epidermal growth factor receptor-tyrosine kinase inhibitor-naive patient: A report of three cases. Oncol. Lett. 7, 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huse M., and Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y. J., Chen M., Cusack N. A., Kimmel L. H., Magnuson K. S., Boyd J. G., Lin W., Roberts J. L., Lengi A., Buckley R. H., Geahlen R. L., Candotti F., Gadina M., Changelian P. S., and O'Shea J. J. (2001) Unexpected effects of FERM domain mutations on catalytic activity of Jak3: structural implication for Janus kinases. Mol. Cell 8, 959–969 [DOI] [PubMed] [Google Scholar]
- 32.Lupardus P. J., Skiniotis G., Rice A. J., Thomas C., Fischer S., Walz T., and Garcia K. C. (2011) Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL-6Ralpha cytokine receptor complex, and the receptor-Jak1 holocomplex. Structure 19, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lupardus P. J., Ultsch M., Wallweber H., Bir Kohli P., Johnson A. R., and Eigenbrot C. (2014) Structure of the pseudokinase-kinase domains from protein kinase TYK2 reveals a mechanism for Janus kinase (JAK) autoinhibition. Proc. Natl. Acad. Sci. U.S.A. 111, 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]