

Background: The role of slow off-rates of processive enzymes acting on recalcitrant polysaccharides is poorly understood.
Results: Chitinase variants with high off-rates and weak product binding were inefficient in degradation of recalcitrant chitin.
Conclusion: Slow off-rates and strong product binding are required for high efficiency and processivity.
Significance: Knowledge of determinants of processivity aids to design better enzymes.
Keywords: cellobiohydrolase, cellulose, chitin, chitinase, processivity, off-rate, product binding
Abstract
Processive glycoside hydrolases are the key components of enzymatic machineries that decompose recalcitrant polysaccharides, such as chitin and cellulose. The intrinsic processivity (PIntr) of cellulases has been shown to be governed by the rate constant of dissociation from polymer chain (koff). However, the reported koff values of cellulases are strongly dependent on the method used for their measurement. Here, we developed a new method for determining koff, based on measuring the exchange rate of the enzyme between a non-labeled and a 14C-labeled polymeric substrate. The method was applied to the study of the processive chitinase ChiA from Serratia marcescens. In parallel, ChiA variants with weaker binding of the N-acetylglucosamine unit either in substrate-binding site −3 (ChiA-W167A) or the product-binding site +1 (ChiA-W275A) were studied. Both ChiA variants showed increased off-rates and lower apparent processivity on α-chitin. The rate of the production of insoluble reducing groups on the reduced α-chitin was an order of magnitude higher than koff, suggesting that the enzyme can initiate several processive runs without leaving the substrate. On crystalline chitin, the general activity of the wild type enzyme was higher, and the difference was magnifying with hydrolysis time. On amorphous chitin, the variants clearly outperformed the wild type. A model is proposed whereby strong interactions with polymer in the substrate-binding sites (low off-rates) and strong binding of the product in the product-binding sites (high pushing potential) are required for the removal of obstacles, like disintegration of chitin microfibrils.
Introduction
Structural polysaccharides such as cellulose (the homopolymer of β-1,4-linked glucose units) and chitin (the homopolymer of β-1,4-linked N-acetylglucosamine (NAG)3 units) are abundant sources of renewable carbon. Their enzymatic depolymerization is an important route to products of high value, which is important for chemical industry. Both cellulose and chitin have evolved to crystalline structures that make them recalcitrant to enzymatic degradation (1). Likewise, there are remarkable structural and functional similarities in enzymatic machineries employed in degradation of cellulose and chitin in nature. Therefore, the lessons learned from chitinase research have turned out to be useful in dissecting the mechanism of cellulose hydrolysis and vice versa (2). The best characterized enzymatic machineries of recalcitrant polysaccharide degradation are the cellulolytic system of the fungus Trichoderma reesei (3) and the chitinolytic system of the bacterium Serratia marcescens (4). The key components of both enzyme systems, the cellobiohydrolase TrCel7A of T. reesei and the chitinase ChiA of S. marcescens, are processive enzymes that move toward the non-reducing end of the polymer and produce disaccharides as the major product. Their processive abilities are governed by multiple interactions with consecutive monomer units along polymer chains and more or less closed architectures of active sites. The active site of ChiA rests in a deep cleft containing four substrate (−4 to −1) and three product (+1 to +3) NAG unit binding sites in the catalytic domain (5–8). Furthermore, the substrate binding region is extended to the carbohydrate binding module (CBM), resulting in a total of 13 substrate-binding sites (6, 9). The long chitin binding cleft is lined with aromatic residues, mostly Trp and Phe (Fig. 1). The hydrophobic interactions form a flexible sheath necessary for substrate recognition and sliding of the polymer chain between successive glycosidic bond cleavages. It has been shown that the replacement of the single Trp in the binding site −3 to Ala (W167A) drastically reduces the processivity of ChiA in the hydrolysis of chitosan, a soluble derivative of chitin (7). The same has been demonstrated with another chitinase of S. marcescens, ChiB (10). The reduced processivity of the ChiA variants was accompanied by better performance on chitosan. In contrast, the activity on crystalline chitin was poor, indicating the importance of processivity in the crystalline substrate degradation (7, 11). The reduction of processivity upon changing Trp to Ala in substrate-binding sites has been demonstrated also for fungal (12) as well as bacterial cellulases (13–16).
FIGURE 1.
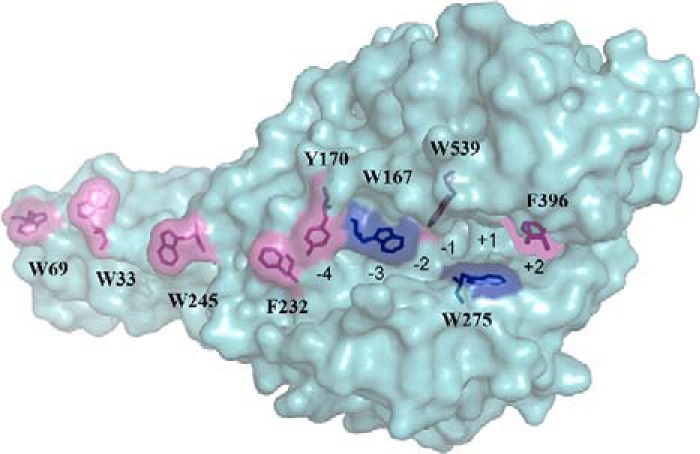
Structure of S. marcescens ChiA. The chitin binding cleft of ChiA is aligned with aromatic residues. The active site rests in the catalytic domain containing four substrate (−4 to −1) and three product (+1 to +3) NAG unit binding sites. The hydrolysis of glycosidic bond takes place between binding sites −1 and +1. The CBM is rigidly connected to the catalytic domain and provides additional NAG unit substrate-binding sites (−13 to −5). Two Trp residues that were replaced with Ala in ChiA variants, ChiA-W167A and ChiA-W275A, studied here are indicated with dark blue color. The rest of the aromatic residues involved in chitin binding are colored pink.
The processive ability of an enzyme is described by its intrinsic processivity (PIntr). Numerically, PIntr represents an average number of catalytic events performed per one productive binding event on an ideal polymer (i.e. on a polymer, where the enzyme dissociation probability is independent of the position of the enzyme on the polymer). Soluble homopolymers with no secondary structure can serve as an example of an ideal polymer. For processive enzymes, PIntr is given by the ratio of the catalytic constant and the dissociation rate constant, PIntr = kcat/koff (17, 18). PIntr has been shown to be related to the free energy of the binding of a polymer chain to the enzymes' active site (19, 20). For cellulases, it has been proposed that PIntr is governed by koff, because non-processive endoglucanases and processive cellobiohydrolases have evolved to similar kcat values but different koff values (17). In the same study it was also concluded that the apparent processivity (Papp, i.e. the experimentally measured processivity on a real polymer) is much lower than PIntr predicted from the kcat/koff ratio, indicating that the true processive ability is seldom realized. Furthermore, numerous studies have pointed out that the overall rate of processive cellulose hydrolysis is limited by the slow dissociation of the cellobiohydrolases (12, 17, 21–26). This raises the question as to why processive cellulases have evolved to such low koff values. Although the importance of koff is well recognized, there are only few experimentally measured koff values available in the literature. Moreover, the koff values are strongly dependent on the method used for measuring. Fluorescence recovery after photobleaching (FRAP) measurements have revealed the koff value in the order of 10−6 s−1 for the dissociation of TrCel7A from the microfibrils of the bacterial microcrystalline cellulose (27). In case the TrCel7A-generated insoluble reducing groups on reduced cellulose under single-hit conditions were fluorescence-labeled, the koff value on the order of 10−3 s−1 was obtained (17). Using the global kinetic modeling of progress curves (12, 26) and single molecule fluorescence imaging (28), koff values for TrCel7A in the order of 10−2 s−1 were obtained. Using high speed atomic force microscopy, the koff values measured for TrCel7A were in the order of 10−1 s−1 (29, 30). For the cellulases of the bacterium Thermobifida fusca, the koff values between 10−2 and 10−3 s−1 have been measured using FRAP (31). To the best of our knowledge, the koff values for chitinases are not available.
Here, we have developed a method for measuring off-rates for enzymes acting on chitin. The method is based on measuring the rate of exchange of a family 18 chitinase between non-labeled and 14C-labeled chitin. We found that the koff value of ChiA depends on both the nature of the substrate and the incubation time with chitin. The ChiA variants ChiA-W167A and ChiA-W275A, formerly showed to have reduced processivity on the soluble substrate chitosan (7), displayed higher off-rates and lower processivity compared with the wild type enzyme on the crystalline chitin substrate. Initial rates analysis demonstrated that both variants were less efficient on highly crystalline chitin but outperformed the wild type enzyme on amorphous chitin. ChiA was also demonstrated to have high probability of endo-mode initiation. The comparison of the rate of the generation of insoluble reducing groups on the reduced chitin by ChiA with the off-rates of total dissociation suggested that the enzyme can initiate a new processive run without leaving the substrate after the completion of the previous processive run.
Experimental Procedures
Materials
Crab chitin (Sigma C7170), chitobiose (Sigma D1523), chitosan, 4-methylumbelliferyl β-diacetylchitobioside (MU-NAG2) hydrate (Sigma M9763), anthranilic acid (AA), sodium cyanoborohydride, sodium borohydride, and bovine serum albumin (BSA) were purchased from Sigma. The scintillation mixture was purchased from Merck. All chemicals were used as purchased.
Enzymes
S. marcescens ChiA and its variants ChiA-W167A and ChiA-W275A were produced and purified as described before (7, 32). Chitobiase was expressed in Escherichia coli and purified by ammonium sulfate precipitation and ion-exchange chromatography as described previously (33).
Chitin Substrates
Crystalline α-chitin was prepared from crab chitin. The chitin was suspended in water and incubated in 0.55 m HCl for 2 h at room temperature with three changes. After three washes with water, the chitin was incubated with 0.3 m NaOH at 70 °C for 3 h with three changes followed by several washes with water. Next, the chitin was washed with ethanol and incubated in acetone for 1 h with two changes at room temperature. Finally, the purified chitin was washed repeatedly with water and ground in a mortar. The purified chitin was N-acetylated with acetic anhydride. For that, chitin was washed three times with methanol and finally resuspended in methanol to give the chitin a concentration of 20 mg ml−1. 1 ml of acetic anhydride was added per 1 g of chitin, and the mixture was incubated overnight at room temperature, with stirring. Next, O-deacetylation was carried out by adding 100 mm KOH in methanol and incubating for 4 h at room temperature, with stirring. After that, the chitin was washed repeatedly with water and 50 mm sodium acetate, pH 6.1. Finally, 0.01% NaN3 was added, and the chitin was stored at 4 °C.
Chitin nanowhiskers (CNWs) and 14C-labeled chitin nanowhiskers (14C-CNWs) were prepared by HCl treatment of crab chitin as described previously (34). The specific radioactivity of the 14C-CNW preparation was 4.18 × 106 dpm mg−1.
Amorphous chitin was prepared by the acetylation of chitosan. Chitosan was suspended in water; an equal volume of 20% acetic acid was slowly added, with stirring; and the mixture was diluted five times by slowly adding methanol, with stirring. 1 ml of acetic anhydride was added per 1 g of chitin, with stirring, and the mixture was incubated overnight at room temperature, without stirring. Next, the mixture was diluted five times by slowly adding water and stirring. The acetic acid was neutralized, and the O-deacetylation was carried out by adding NaOH to a final concentration of 50 mm and incubated overnight at room temperature, with stirring. The amorphous chitin was repeatedly washed with water and 50 mm sodium acetate, pH 6.1. Finally, 0.01% NaN3 was added, and the chitin was stored at 4 °C.
Reduced chitin was prepared of α-chitin by NaBH4 treatment. The chitin was washed twice with 0.25 m NaHCO3/Na2CO3, pH 10, and resuspended in the same buffer to give the chitin a concentration of 2 mg ml−1. The mixture was heated to 80 °C, and 5 m sodium borohydride in 0.1 m NaOH was added to give a final concentration of 25 mm sodium borohydride followed by 1 h of incubation. The same amount of 5 m sodium borohydride in 0.1 m NaOH was added four more times with 1 h of incubation at 80 °C after each addition. Next, an equal volume of 0.5 m acetic acid was added, and the mixture was incubated overnight at room temperature, with stirring. The reduced chitin was washed repeatedly with water, and 50 mm sodium acetate, pH 6.1, 0.01% NaN3 was added and stored at 4 °C.
Substrate Exchange Experiment
CNWs (2 mg ml−1) or α-chitin (4 mg ml−1) was incubated with 20 nm ChiA or its variants and 20 nm chitobiase, in 50 mm sodium acetate, pH 6.1, supplemented with BSA (0.2 mg ml−1) at 25 °C, with stirring in the case of α-chitin. After the desired time, an equal volume of 14C-CNWs (2 mg ml−1) in 50 mm sodium acetate, pH 6.1, was added and further incubated at 25 °C. After the selected times, aliquots were withdrawn and stopped by adding NaOH to 0.2 m. 3 g liter−1 CNWs was added; chitin was sedimented by centrifugation (5 min at 104 × g), and the amount of radioactivity in the supernatant was quantified using a liquid scintillation counter. To make the reference curves, 14C-CNWs were mixed with CNWs or α-chitin, and the reaction was started by adding ChiA or its mutants. For t = 0, CNWs or α-chitin and 14C-CNWs were mixed, and NaOH was added before ChiA. The released radioactivity was converted to the concentration of 14C-chitobiose using a previously constructed calibration curve (34).
Chitin Hydrolysis and Binding of Chitinases
α-Chitin (4 mg ml−1) was incubated with 20 nm ChiA or its variants and 20 nm chitobiase in 50 mm sodium acetate, pH 6.1, supplemented with BSA (0.2 mg ml−1) at 25 °C, with stirring. After the selected times, aliquots were withdrawn, filtered through Whatman GF-D filters, and centrifuged for 5 min at 104 × g. The amount of reducing groups in the supernatant was measured using the 3-methyl-2-benzothiazolinone hydrazone hydrochloride (MBTH) method, as described previously (35). The amount of free chitinase was measured by MU-NAG2 hydrolysis assay. For that, 100 μl of supernatant was incubated with 5 μm MU-NAG2 at 25 °C. After the selected times, the reaction was stopped by adding NaOH to the final concentration of 10 mm. The reaction was diluted with 0.1 m ammonium hydroxide, and the concentration of released 4-methylumbelliferone was determined by fluorescence. The excitation and emission wavelengths were set to 360 and 450 nm, respectively. The concentration of free chitinase was calculated from the rate of MU-NAG2 hydrolysis using appropriate reference curves. The MU-NAG2 (5 μm) hydrolyzing activity (v/E0) of ChiAWT, ChiA-W167A, and ChiA-W275A was 1.34 ± 0.03, 0.050 ± 0.005, and 0.0044 ± 0.0003 s−1, respectively. The concentration of chitin-bound chitinase was found as the difference between the concentration of total and free chitinase.
Chitobiose Inhibition of Chitinases
The chitobiose inhibition of chitinase variants ChiA-W167A and ChiA W275A was measured on 14C-CNWs essentially as described previously (34). Inhibition was tested at 14C-CNW concentrations of 0.1, 0.25, and 1.0 mg ml−1. Concentration of chitobiose varied between 0 and 5 mm.
Measuring Initial Rates
α-Chitin, CNWs, or amorphous chitin (0.05–3 mg ml−1) were incubated with 20 nm ChiA or its variants in 50 mm sodium acetate, pH 6.1, supplemented with BSA (0.1 mg ml−1) at 25 °C for 1 min, without stirring. The reaction was stopped by adding NaOH up to 0.2 m. For t = 0, NaOH was added before ChiA. Chitin was sedimented by centrifugation (5 min at 104 × g) after which 500 μl of the supernatant was used for measuring the reducing groups with the MBTH method.
Measuring Apparent Processivity
Reduced chitin (1 mg ml−1) was incubated with 10 nm ChiA or its variants in 50 mm sodium acetate, pH 6.1, supplemented with BSA (0.1 mg ml−1) at 25 °C with stirring. At the desired times, aliquots were withdrawn, and the reaction was stopped by adding 0.2 m NaOH. The chitin was sedimented by centrifugation (2 min at 104 × g), and the amount of soluble reducing groups in the supernatant was measured using the MBTH method. The amount of insoluble reducing groups was determined by fluorescence labeling of the chitin with anthranilic acid (AA). For that, the chitin pellet was washed twice with water, once with 50 mm sodium acetate, pH 6.1, and twice with water. The chitin was resuspended in 200 μl of water, and the AA labeling was carried out in 80% buffered methanol at 80 °C for 2 h as described previously (36). The concentrations of sodium cyanoborohydride and AA were 0.5 m and 50 mm, respectively. The labeled chitin was washed three times with water and three times with 50 mm sodium acetate, pH 6.1. Finally, the chitin was resuspended in 50 mm sodium acetate, pH 6.1, to the final concentration of 0.5 mg ml−1, and the fluorescence of the suspension was measured using excitation and emission wavelengths set to 330 and 425 nm, respectively. Relative fluorescence of 310 intensity units μm−1 determined for AA-labeled NAG in 0.5 mg ml−1 chitin suspension was used for calibration. AA labeling of NAG was done according to the protocol of the preparation of AA/glucose in water (36), and the relative fluorescence of AA-labeled NAG was 450 intensity units μm−1.
Measuring the Probability of Endo-mode Initiation
First, AA-labeled chitin was prepared from α-chitin, using the above described protocol. Prior to enzymatic hydrolysis, the AA-labeled α-chitin was incubated in 0.2 m NaOH for 15 min at room temperature to remove the nonspecific label, followed by three washes with 50 mm sodium acetate, pH 6.1. 1 mg ml−1 AA-labeled α-chitin was incubated with 10 nm ChiA or its variants in 50 mm sodium acetate, pH 6.1, supplemented with BSA (0.1 mg ml−1) at 25 °C, with stirring. At defined times, the reaction was stopped by adding 0.2 m NaOH, and the chitin was pelleted by centrifugation (2 min at 104 × g). The released AA-sugars were determined by measuring the fluorescence in the supernatant using excitation and emission wavelengths set to 330 and 425 nm, respectively. The concentration of the reducing groups in the supernatant was measured using the MBTH method. The probability of the endo-mode initiation was calculated from the concentration of insoluble reducing groups measured in the apparent processivity experiment and the concentration of the released AA-sugars as described previously (17).
Results
Measuring Off-rates of Enzymes in Substrate Exchange Experiments, Theoretical Considerations
In the substrate exchange experiment (SEE), an enzyme (E) was incubated with a polymeric substrate (S) of interest, and after the predefined time, the reaction was supplemented with 14C-labeled polymeric substrate (14C-S). Upon the addition of 14C-S, the release of 14C-labeled product (14C-P) in time was followed. If the concentration of S is saturating for the enzyme, a lag phase in the formation of 14C-P in time was expected, because the enzyme must release from S before it is available for 14C-S. For simplicity, we used a simple two-step reaction mechanism for the formation of 14C-P. According to Scheme 1, the rate of 14C-P formation is proportional to the concentration of enzyme in complex with 14C-S ([E14C-S]).
SCHEME 1.
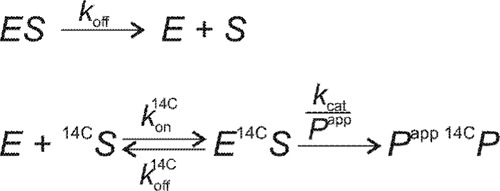
Reaction sequences in SEE. In SEE an enzyme (E) must dissociate from the complex with non-labeled substrate (ES) before it can react with 14C-labeled substrate (14C-S) and release the labeled product (14C-P) to be detected. kon is the association rate constant (liter g−1 s−1); koff is the dissociation rate constant (s−1); kcat is the catalytic constant representing the release of one product molecule (s−1), and Papp is an apparent processivity representing an average number of product molecules released during one processive run. Superscript 14C refers to the parameter for 14C-S.
It can be shown that the time dependence of [E14C-S] is governed by the exponent in the form of (1 − exp(−kofft)). We note that this simple time dependence of [E14C-S] is valid only if koff 14C + kon14C[14C-S] ≫ koff.
To meet these requirements, the concentration of 14C-S must be high enough to ensure that the on-rate (kon14C[14C-S]) is higher than both off-rates (koff14C and koff). In other words, [14C-S] must be saturating for the enzyme. The same is true for S before the addition of 14C-S. This ensures that before the addition of 14C-S, [E] is negligible, so that the dissociation from ES is a prerequisite for the reaction with 14C-S. The analysis of SEE results is somewhat complicated by the absence of true steady state in the hydrolysis of heterogeneous polymeric substrates (37). The progress curves of cellulose hydrolysis seem to follow the fractal-like kinetics with gradual loss of activity in time. Because the complex system does not enable rigorous analytical treatment, empirical equations have been used to describe the progress curves. The simplest equation describing the hydrolysis of cellulose is the two-parameter equation introduced by Kostylev and Wilson (14). According to Equation 1,
In Equation 1, [14C-P] is the product concentration; A is the constant referred to as the enzyme activity, and the constant b is the hydrolysis power term (14, 38). The parameter A is a product of the concentration of productive enzyme-substrate complex and the catalytic constant. It has been shown that in cellulose hydrolysis, A scales with the total enzyme concentration (E0), whereas b is independent on E0 (14). Despite its simplicity, Equation 1 has been shown to describe the progress curves of cellulose hydrolysis over a large range of total conversions of the substrate (14). This encouraged us to use Equation 1 also in the analysis of SEE results. Assuming that at the moment of 14C-S addition the concentration of the free enzyme is negligible, the availability of the enzyme for 14C-S hydrolysis is governed by its release from the non-labeled S. Thus, an apparent A is expected to increase in time analogously to [E14C-S]. Introducing the time dependence into Equation 1 results in Equation 2,
To find numerical values for koff, the following approach was used. First, the reference time course, where S and 14C-S were mixed together before the addition of the enzyme, was fitted to Equation 1 to find the values of parameters A and b. Next, the time course of 14C-P formation in the SEE experiment was fitted to Equation 2. In fitting the SEE results to Equation 2, the value of the parameter b was fixed to the value found from the analysis of the reference time course. This was necessary because of the interdependency between koff and b in Equation 2.The average R values for fitting the reference time courses to Equation 1 and SEE results to Equation 2 were 0.9919 (n = 9) and 0.9951 (n = 19), respectively.
Substrate Exchange Experiments with CNWs
Recently, we have described the preparation and kinetic measurements with 14C-CNWs (34). 14C-CNWs were used as the labeled substrate throughout this study. In the first trials of measuring the off-rates of ChiA we used non-labeled CNWs as the substrate. The release of 14C-soluble sugars from 14C-CNWs upon hydrolysis by ChiA is shown in Fig. 2. For the reference curve of SEE, the 14C-CNWs and CNWs were mixed together in equal amounts, and the reaction was started by the addition of ChiA. The presence of CNWs resulted in a 2-fold reduction in the release of 14C-sugars, confirming the equivalence of CNWs and 14C-CNWs (data not shown). In the SEE, the CNWs were first incubated with ChiA for 2 h before the addition of 14C-CNWs. The release of 14C in SEE revealed a transient lag phase followed by a near-linear increase of [14C] in time. However, the inspection of the reference curve reveals that there is no steady state as the rate of 14C release gradually decreases with hydrolysis time (Fig. 2). These results suggest that the apparent linearity observed in SEE may be caused by the counterbalancing effects of the release of ChiA from CNWs and the decrease in hydrolysis rates of 14C-CNWs with hydrolysis time. Therefore, the results were analyzed using equations for fractal-like kinetics, Equation 1 for the reference curve, and Equation 2 for SEE experiments. The koff value of 0.012 ± 0.002 s−1 for the dissociation from CNWs was found. Reference curves made using 5 and 10 nm ChiA resulted in the values of parameter A (Equation 1) of 0.15 ± 0.04 and 0.29 ± 0.05, respectively. Corresponding values for the power term b were 0.66 ± 0.06 and 0.63 ± 0.03. These results are in accord with the results of cellulose hydrolysis, demonstrating that A scales with the enzyme concentration, whereas b is independent (14). When the incubation time with CNWs was extended to 24 h, the lag phase in SEE was absent (Fig. 2). This suggests that changes in CNWs have occurred, resulting in either an increased koff value or the presence of a significant amount of free ChiA before the addition of 14C-CNWs. Because the suspension properties of CNWs did not permit the measurement of free ChiA, it was not possible to discriminate between these two possibilities.
FIGURE 2.
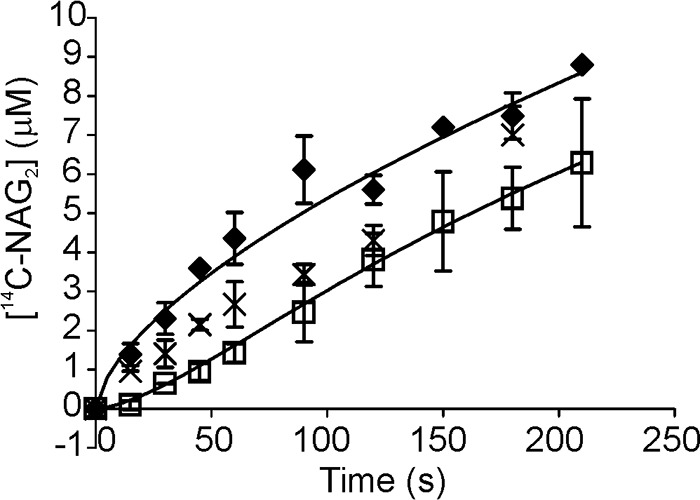
SEE with CNWs. CNWs (2 mg ml−1) were pre-incubated with 20 nm ChiA for 2 h (□) or 24 h (×), after which an equal volume of 14C-CNWs (final concentration 1 mg ml−1) was added and the release of 14C (expressed in NAG2 equivalents) in time was followed. In the control experiments (♦) the mixture of 14C-CNWs (1 mg ml−1) and CNWs (1 mg ml−1) was incubated with 10 nm ChiA. Error bars show S.D. and are from three independent experiments. Solid lines represent the best fit according to Equation 1 (control) or Equation 2 (SEE).
Substrate Exchange Experiments with α-Chitin
The α-chitin used in SEE experiments was the parent substrate used for the preparation of CNWs by heterogeneous acid hydrolysis. Thus, the α-chitin is expected to have lower crystallinity and a higher degree of polymerization compared with CNWs. Besides the wild type enzyme, two ChiA variants, ChiA-W167A and ChiA-W275A, were used in SEE experiments. First, we tested the product NAG2's inhibition of ChiA on 14C-CNWs. There was no difference in inhibition strengths measured at three different 14C-CNW concentrations, 0.1, 0.25, and 1.0 mg ml−1, suggesting non-competitive type of inhibition (34). 23 ± 3 (data from Ref. 34), 40 ± 5, and 86 ± 8% of the activity of ChiAWT, ChiA-W167A, and ChiA-W275A, respectively, were retained in the presence of 5 mm NAG2. To avoid possible complications because of the product inhibition in the long trials, the SEE reactions were supplemented with chitobiase to transform NAG2 to two NAG moieties. Comparing the reference curves made with the mixture of α-chitin and 14C-CNWs with those made with 14C-CNWs only revealed that 14C-CNWs effectively out-competed ChiA from α-chitin (Fig. 3). During the first 5 min of hydrolysis of α-chitin, the activities of both ChiA-W167A and ChiA-W275A were comparable with the activity of ChiAWT. However, with increasing hydrolysis times, ChiAWT clearly outperformed both variants (Fig. 4A). In SEE, the incubation time of 10 min with α-chitin was first tested. The characteristic lag phase was seen with ChiAWT and ChiA-W275A but not with ChiA-W167A (Fig. 3). To test whether the absence of lag phase with ChiA-W167A was caused by high off-rates or a significant amount of free enzyme present before the addition of 14C-CNWs, we measured the binding to α-chitin. The concentration of bound enzyme achieved the plateau value after 5–10 min in cases of all enzymes (Fig. 4B). At the α-chitin concentration of 2 mg ml−1, the concentration of free enzyme was less than 10%, suggesting that the absence of lag phase in SEE with ChiA-W167A was not caused by the presence of a high concentration of free enzyme. Using 2 h of incubation with α-chitin before the addition of 14C-CNWs revealed a lag phase in case of all enzyme variants (Fig. 3). The koff values increased in the following order: ChiAWT < ChiA-W275 < ChiA-W167A (Table 1). The apparent time dependence of koff prompted us to measure the koff values at longer pre-incubation times with α-chitin. In the case of wild type ChiA, the same koff values were measured for the series with pre-incubation times between 24 and 360 h. For ChiAWT, the average koff value for 24–360-h pre-incubation times was within the error limits with the koff value measured using 2 h of pre-incubation time (Table 1). Contrary to ChiAWT, no lag phase was observed with ChiA-W167A and ChiA-W275A in the series with pre-incubation times between 24 and 360 h (Fig. 3). Binding measurements revealed that for all enzymes the concentration of free enzyme measured after 24–360 h of incubation with α-chitin was within a few percent of the total enzyme concentration (data not shown). This suggests that it is the apparent value of koff of ChiA-W167A and ChiA-W275A that has increased with pre-incubation time. The general activity of ChiA-W167A was within the error limits with that of ChiA-W275A. However, both variants were ∼3-fold less active than ChiAWT (Fig. 4C). The extent of α-chitin degradation after 360 h of incubation was 18, 5, and 4.5% for ChiAWT, ChiA-W275A, and ChiA-W167A, respectively.
FIGURE 3.
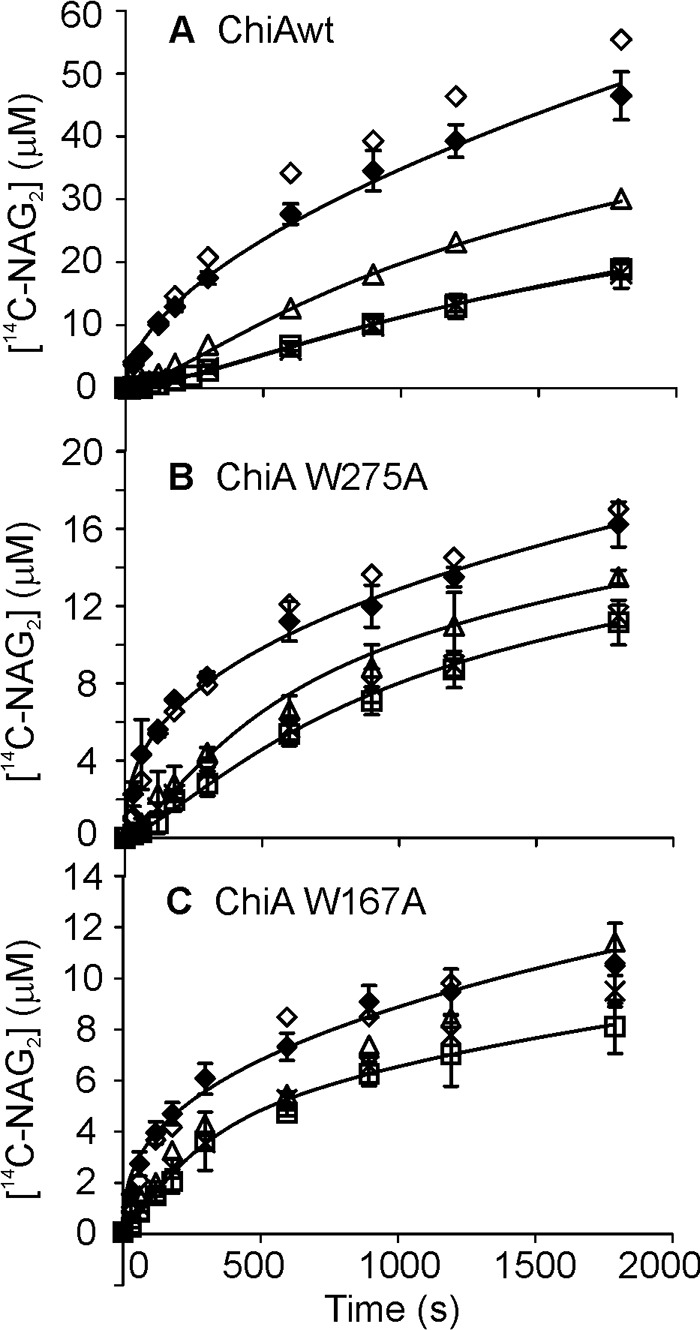
SEE with α-chitin. α-Chitin (4 mg ml−1) was pre-incubated with 20 nm ChiA for 10 min (▵), 2 h (□), or 24–360 h (×), after which an equal volume of 14C-CNWs (final concentration 1 mg ml−1) was added, and the release of 14C (expressed in NAG2 equivalents) in time was followed. In the control experiments (♢), 14C-CNWs (1 mg ml−1) was incubated with 10 nm ChiA or (♦) the mixture of 14C-CNWs (1 mg ml−1) and α-chitin (2 mg ml−1) was incubated with 10 nm ChiA. Error bars show S.D. and are from three independent experiments. Solid lines represent the best fit according to Equation 1 (control) or Equation 2 (SEE). A, wild type ChiA; B, ChiA-W275A; C, ChiA-W167A.
FIGURE 4.
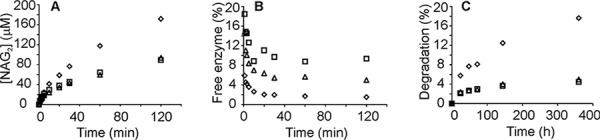
Activity and binding measured with α-chitin. Wild type ChiA (♢), ChiA-W275A (▵), and ChiA-W167A (□) are shown. The concentration of α-chitin was 4 mg ml−1 except in the binding experiments with ChiA-W275A and ChiA-W167A, where it was 2 mg ml−1. The concentration of ChiA was 20 nm. Reactions were supplemented with chitobiase (20 nm), and the release of reducing groups (expressed in NAG2 equivalents) in time was followed. A, hydrolytic activity in the short to medium time scale experiments. B, concentration of free enzyme in the short to medium time scale experiment. C, hydrolytic activity in the long time scale experiments.
TABLE 1.
koff values measured at different pre-incubation times with α-chitin
| Enzyme | A | ba |
koff (10−3 s−1)b |
||
|---|---|---|---|---|---|
| 10 minc | 2 hc | 24–360 hd | |||
| μm s−1 | |||||
| ChiAWT | 0.72 ± 0.06 (43)e | 0.561 ± 0.022 (11)e | 2.8 ± 0.3 (22)e | 1.5 ± 0.5 (31)e | 1.7 ± 0.3 (25)e |
| W275A | 0.87 ± 0.20 (44)e | 0.392 ± 0.027 (17)e | 3.5 ± 1.2 (46)e | 2.2 ± 0.7 (41)e | fastf |
| W167A | 0.61 ± 0.11 (43)e | 0.389 ± 0.030 (17)e | fastf | 6.4 ± 3.7 (49)e | fastf |
a The parameter values of reference curves were found by non-linear regression analysis of data in Fig. 3 (controls, where 14C-CNWs and α-chitin were mixed together before incubation with ChiA) according to Equation 1.
b The koff values were found by non-linear regression analysis of data in Fig. 3 (SEE data, where ChiA was pre-incubated with α-chitin before the addition of 14C-CNWs) according to Equation 2. Because of the interdependence between koff and b, the value of parameter b was fixed to the value found for reference curves (listed in this table) while fitting the data to Equation 2.
c The times of pre-incubation of ChiA with α-chitin before the addition of 14C-CNWs are shown.
d The average of experiments with the times of pre-incubation of ChiA with α-chitin before the addition of 14C-CNWs are as indicated.
e Standard deviations are from three independent measurements. The numbers in parentheses shows the average deviation (in %) of upper and lower 95% confidence intervals from the mean value.
f The dissociation was considered to be fast in the cases, when the lag phase in 14C-P formation in SEE was not detectable.
Initial Rates at Different Chitin Substrates
Progressing difference in the activity of the variants and ChiAWT with hydrolysis time suggests that the variants may be deficient in the hydrolysis of the more recalcitrant portion of α-chitin. This prompted us to measure the initial rates with different chitin substrates. Initial rates were measured after 1 min of hydrolysis. In this time scale, the progress curves were near-linear. We note that the linearity of progress curves does not necessarily imply that the reaction is in true steady state. With all the enzymes and substrates tested, the initial rates measured at different substrate concentration followed Michaelis-Menten saturation kinetics (data not shown). Although originally derived for enzymes acting on soluble substrates, it has been shown that the Michaelis-Menten equation is applicable also for the processive enzymes acting on insoluble substrates (25, 26, 39).
 |
In Equation 3, pvss is the initial (steady state) rate of the processive hydrolysis; S0 is the initial concentration of the substrate, and pVmax and pKm are processive analogs of the maximum velocity and Michaelis constant, respectively (25, 39). To get meaningful estimates for pVmax and pKm, the substrate must be in excess (25). In practice, this condition can be met by using very low enzyme concentrations (E0) (25). Experiments made using 20 and 40 nm ChiA showed that activity was scaled proportionally with enzyme concentration in the case of all substrates and concentrations used (data not shown). This confirms that the substrate is in excess in our study conditions. pVmax and pKm values were found by non-linear regression analysis of pvss versus S0 curves according to Equation 3 and are listed in Table 2. On CNWs, the substrates with highest crystallinity, the pVmax/E0 values of the variants were more than 2-fold lower than that of ChiAWT. On α-chitin, the substrate with medium crystallinity, the pVmax/E0 values of ChiAWT and both variants were the same within the error limits. However, with amorphous chitin both variants displayed more than 5-fold higher pVmax/E0 values than ChiAWT. With all substrates tested, the pKm values of both variants were higher than the corresponding figures for ChiAWT (Table 2).
TABLE 2.
Initial rates based Michaelis-Menten kinetics parameter values on different chitin substrates
| Enzyme |
pVmax/E0 (s−1)a |
pKm (mg ml−1)a |
||||
|---|---|---|---|---|---|---|
| CNWs | α-Chitin | Amorphous chitin | CNWs | α-Chitin | Amorphous chitin | |
| ChiAWT | 11.7 ± 0.9 | 7.6 ± 1.8 | 2.1 ± 0.2 | 0.34 ± 0.07 | 1.5 ± 0.6 | 0.31 ± 0.28 |
| W275A | 4.9 ± 0.7 | 7.5 ± 2.1 | 12.3 ± 0.9 | 0.55 ± 0.20 | 1.8 ± 0.5 | 0.83 ± 0.04 |
| W167A | 5.2 ± 0.8 | 8.9 ± 0.9 | 10.7 ± 1.4 | 0.90 ± 0.52 | 2.3 ± 0.8 | 1.14 ± 0.18 |
a pVmax and pKm values were found by non-linear regression analysis of pvss versus S0 curves according to Equation 3. Standard deviations are from three independent measurements.
Measuring Apparent Processivity with Reduced α-Chitin
Apparent processivity (Papp) is defined as the number of processive catalytic events (Ncatal) divided by the number of the initiations of processive runs (Ninit). It has been shown that if the hydrolysis of reduced cellulose is followed under single-hit conditions, the number of insoluble reducing groups (IRG) generated to the reduced cellulose equals the sum of the reducing-end-exo and endo-mode initiations. However, the total number of enzyme-generated reducing groups (RGtot) equals the Ncatal (17, 18). RGtot is given by the sum of released soluble reducing groups (SRGs) and IRGs. Therefore, for the enzymes that employ reducing-end-exo and/or endo-mode initiation, the slope of the plot in coordinates [RGtot] versus [IRG] equals to Papp (17, 18, 40). Here, we followed the hydrolysis of reduced α-chitin by ChiAWT and its variants ChiA-W275A and ChiA-W167A. The activity on reduced α-chitin was within the error limits with the activity on α-chitin. pVmax/E0 values measured with reduced α-chitin were 7.8 ± 0.4, 7.1 ± 0.3, and 7.0 ± 1.2 s−1 for ChiAWT, ChiA-W275A, and ChiA-W167A, respectively. Corresponding pKm values were 1.1 ± 0.2, 2.1 ± 0.4, and 1.4 ± 0.5 mg ml−1. For parameter values with α-chitin, see Table 2. The number of IRGs was measured by fluorescence labeling of chitinase-treated reduced α-chitin. [RGtot] versus [IRG] plots for ChiA are shown in Fig. 5. We note that the lines in coordinates [RGtot] versus [IRG] became curved upward at longer incubation times (higher IRG concentrations). This is indicative of a deviation from single-hit conditions, because repeated hits on the reducing end of the same chain produce SRGs but not IRGs. However, because the deviation from the linearity was not prominent, considering the error limits, full datasets were used in linear regression analysis. The slope of the linear regression line equals the value of Papp, and the corresponding figures are listed in Table 3. Lower Papp values of variants ChiA-W275A and ChiA-W167A compared with ChiAWT were caused by both the increased rates of the generation of IRGs (Fig. 6A) and the decreased rates of the release of SRGs (Fig. 6B). Within the time interval studied, the time courses of IRG formation were non-linear (Fig. 6A). The apparent rate constant for IRG formation (kIRG) was found from the rate of IRG formation (vIRG) as kIRG = vIRG/E0. With all enzyme variants, the kIRG values were between 0.1 s−1 (measured after 5 min of hydrolysis) and 0.02 s−1 (measured after 1 h of hydrolysis). For ChiAWT, these figures are approximately an order of magnitude higher than koff values measured using SEE experiments (Table 1).
FIGURE 5.
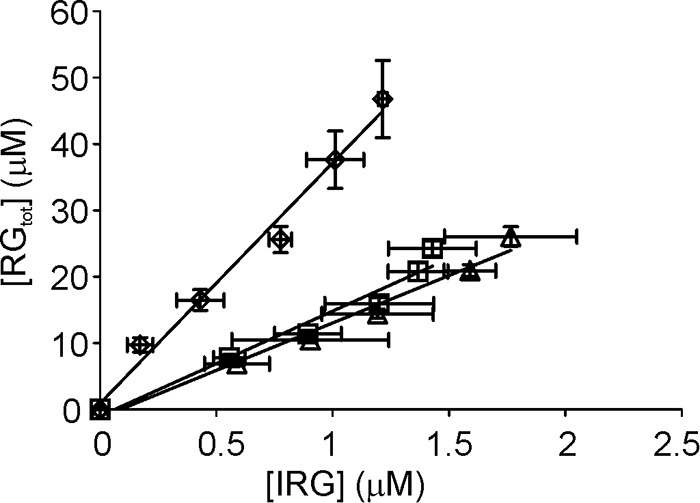
Apparent processivity (Papp) measured with reduced α-chitin. Wild type ChiA (♢), ChiA-W275A (▵), and ChiA-W167A (□) are shown. Reduced α-chitin (1 mg ml−1) was incubated with 10 nm ChiA in 50 mm NaAc, pH 6.1, at 25 °C. SRGs were measured with the MBTH method, and the formation of IRGs was measured with fluorescence labeling. RGtot was found as the sum of IRGs and SRGs. Error bars show S.D. and are from three independent experiments. Solid lines represent the best fit of linear regression and the slope of the line equals the Papp.
TABLE 3.
Papp and PEndo measured with α-chitin
a Standard deviations are from at least three independent measurements.
FIGURE 6.
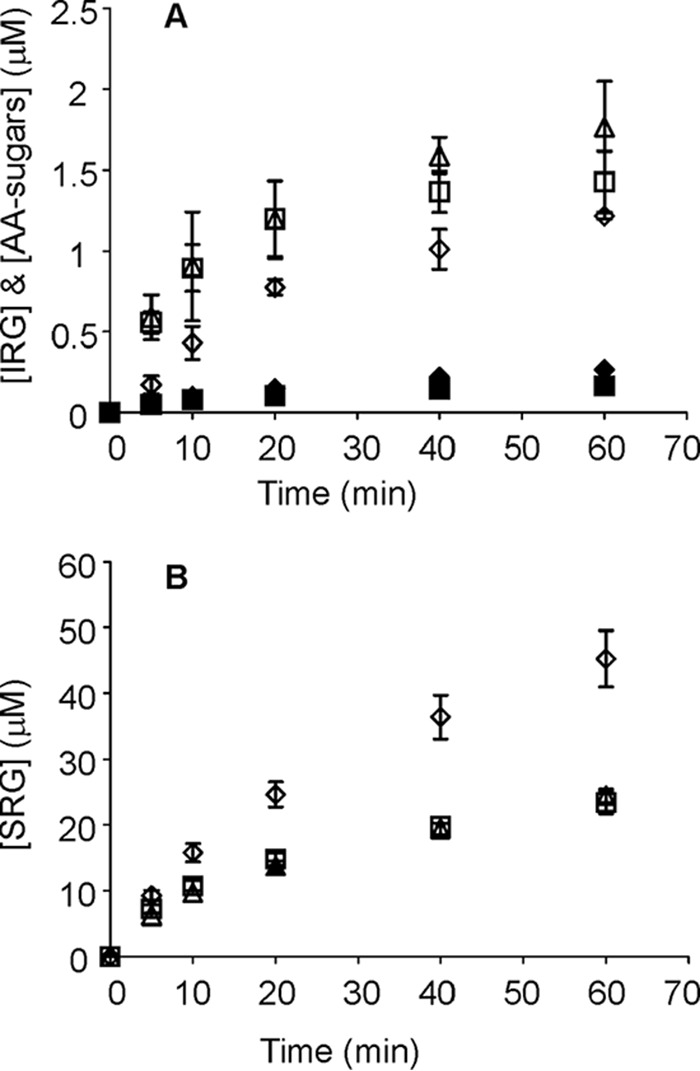
Comparative hydrolysis of reducing end AA-labeled α-chitin (AA-α-chitin) and reduced α-chitin reveals the use of endo-mode initiation. Wild type ChiA (♢), ChiA-W275A (▵), and ChiA-W167A (□) are shown. 1 mg ml−1 substrate was incubated with 10 nm ChiA in 50 mm NaAc, pH 6.1, at 25 °C. A, filled symbols (♦, ▴, and ■) refer to the release of AA-sugars from AA-α-chitin, and the open symbols (♢, ▵, and □) refer to the formation of IRGs on reduced α-chitin. Error bars show S.D. and are from three independent experiments. B, release of SRGs in hydrolysis of both substrates measured with the MBTH method. Error bars show S.D. and are from three independent experiments with AA-α-chitin and three independent experiments with reduced α-chitin taken together.
Measuring the Probability of Endo-mode Initiation
The results of previous studies have suggested that ChiA and an analogous enzyme CHI60 from Serratia sp. can, besides reducing-end-exo initiation, also employ endo-mode initiation (40, 41). Therefore, we estimated the probability of endo-initiation (PEndo) of ChiA by measuring the release of AA-sugars from reducing end AA-labeled α-chitin and comparing it with the number of IRGs generated in the hydrolysis of reduced α-chitin under the same experiment conditions. The release of AA-sugars is representative of the number of initiations from the reducing end ((Ninit)R-exo), whereas the generation of IRGs represents the sum of endo-mode initiations ((Ninit)Endo) and (Ninit)R-exo. Therefore, PEndo = ([IRG] − [AA-sugar])/[IRG] (17). Note that we cannot cope with non-reducing-end exo-initiations, and PEndo represents the use of endo-mode initiation relative to the reducing-end-exo initiations. The AA group in the reducing end of AA-α-chitin did not affect the general activity of ChiA and its variants compared with the activity on reduced α-chitin (Fig. 6B). Thus, the amounts of IRGs and AA-sugars measured at the same hydrolysis times are expected to be comparable. With ChiA and its variants, the amount of IRGs generated to the reduced α-chitin clearly exceeded the amount of AA-sugars released from AA-α-chitin, indicating the predominance of endo-mode initiation (Fig. 6A). The PEndo values for both variants ChiA-W167A and ChiA-W275A were similar to each other but higher than the PEndo value for ChiAWT (Table 3). The higher PEndo of variants was primarily caused by the increased production of IRGs compared with ChiAWT, although ChiA-W167A also released significantly less AA-sugars from AA-α-chitin.
Discussion
Processivity is an important kinetic property of polymer active enzymes (42). Processive enzymes remain attached to the polymer chain after the catalytic event. Provided that processivity is high enough, the complete processing of a polymer chain can be achieved after the initial productive binding. It has been proposed that the main determinant of PIntr of cellulases is koff (17). Although the importance of the koff value of the enzymes processing recalcitrant polysaccharides is widely recognized, the experimental approaches to measure this parameter are scarce.
Here, we developed a new method for measuring the off-rates of a chitinase from the crystalline polysaccharide chitin. We found that the observed off-rates were dependent on both the nature of the chitin substrate as well as the hydrolysis time. After the incubation of ChiAWT with the CNW substrate for 2 h, an apparent koff of 0.012 ± 0.002 s−1 was found. The corresponding value for α-chitin was an order of magnitude lower, 0.0015 ± 0.0005 s−1 (Table 1). Within the series with different incubation times with α-chitin, there was an initial drop in the value of koff of ChiAWT and its variants (Table 1). However, after 2 h of incubation, the koff of ChiAWT achieved a constant value that did not change during further incubation up to 360 h studied. The situation was different with the ChiA variants, ChiA-W275A and ChiA-W167A, dissociation of which was too fast to be measured after the long term (24–360 h) incubation with α-chitin. The fast dissociation of ChiA-W275A and ChiA-W167A was accompanied by inefficiency in α-chitin degradation as evidenced by increasing deficiency of variants compared with ChiAWT with hydrolysis time (Fig. 4, A and C). This indicates that changes in α-chitin structure have occurred during the hydrolysis and that the remaining chitin was more recalcitrant to enzymatic degradation. Inefficiency of ChiA-W275A and ChiA-W167A in degradation of recalcitrant crystalline chitin is also revealed in initial rates based on pVmax /E0 values (Table 2). On the substrate with the highest crystallinity, CNWs, ChiAWT outperformed both variants. In contrast to CNWs, on amorphous chitin both variants clearly outperformed ChiAWT, indicating that strong interaction with the chitin chain comes as a penalty on amorphous substrate. Better performance of ChiA-W275A and ChiA-W167A compared with ChiAWT on partially acetylated chitosan, a soluble chitin derivative, has been demonstrated before (7). At the same time, the activity of ChiA-W275A and ChiA-W167A on crystalline β-chitin was reduced by 50 and 70%, respectively (7). Similar trends have also been observed with processivity-deficient variants of the other S. marcescens chitinase, ChiB (10). Studies of TrCel7A variants with lower processivity also revealed higher activity compared with the wild type enzyme on amorphous cellulose (12, 43). Thus, the negative effect of processivity (slow off-rate) in hydrolysis of amorphous substrates seems to be a general feature among glycoside hydrolases evolved to degrade recalcitrant polysaccharides (7, 43). In the study by Zakariassen et al. (7), the processivity of ChiAWT, ChiA-W275A, and ChiA-W167A was assessed by measuring the product profile of the hydrolysis of soluble chitosan. This method provides a qualitative rather than quantitative measure of processivity (18). It was found that the processivity of ChiA-W167A was reduced dramatically, whereas ChiA-W275A had a less prominent effect on processivity (7). This is in qualitative agreement with the koff values measured here. The larger effect of Trp167 to Ala substitution in reducing koff is expected, because Trp167 interacts with the polymeric part of the substrate, whereas Trp275 interacts with the dimeric product. The strong effect of Trp275 to Ala substitution in relieving chitobiose inhibition (see under “Results”) and increasing the Km value for chitotetraose (44) is also in accord with the role of Trp275 in ligand binding at the product-binding site +1.
When looking for correlations between the values of parameters of enzyme kinetics and the performance of enzymes in chitin degradation, we first note that there is no direct correlation between koff and enzymes performance. This is because ChiA-W167A has higher off-rates than ChiA-W275A (Table 1) but similar hydrolytic activity on all the substrates and time frames tested (Fig. 4, A and C, and Table 2). Although partially acetylated soluble chitosan is not a homopolymer, it is expected to be more close to an ideal polymer than insoluble chitin. Therefore, the processivity on chitosan is expected to reflect the intrinsic processivity of the enzymes. Unfortunately, the koff values on chitosan are not available. From the results of this study, we can estimate PIntr on α-chitin. Using the pVmax/E0 values listed in Table 2 as a measure of the kcat and koff values measured after 2 h of incubation with α-chitin (Table 1), the PIntr values of 5000 ± 1600, 3500 ± 1000, and 1400 ± 800 can be calculated for ChiAWT, ChiA-W275A, and ChiA-W167A, respectively. It has been shown that PIntr is related to the free energy of the binding of the polymer chain to the enzyme's active site (19, 20). We note that the PIntr value estimated here for ChiA is in the same order with the PIntr value found for TrCel7A from the rate of the production of IRGs on reduced bacterial cellulose (17). The active site of TrCel7A has a tunnel-shaped architecture, whereas the active site of ChiA resides in a deep opened groove. Thus, our data are in line with the general paradigm that the closed active site architecture is not a sole determinant of processivity (42). However, there are at least two reasons why PIntr values reported here must be treated with caution. (i) The pVmax/E0 values were taken as estimates of the kcat values. On CNWs, we recently demonstrated that a significant fraction of ChiA is bound non-productively with the active site free from the chitin chain even at the substrate concentrations saturating for the activity (34). If the same is true for α-chitin, pVmax/E0 underestimates kcat and also PIntr. (ii) The koff values were measured from the rates of the total dissociation of enzymes (Table 1), but in order to be used in calculating PIntr, koff should represent the dissociation of the chitin chain from the active site. If the dissociation of the CBM is slow, the koff for the total dissociation of enzyme may underestimate the koff for the dissociation from the active site, and PIntr is overestimated. The importance of aromatic residues in the CBM of ChiA (Trp33 and Trp69) was clearly demonstrated by Uchiyama et al. (9) as substitution to Ala drastically reduced binding to crystalline β-chitin. At the same time, the substitution of Trp167 or Trp275 to Ala had only moderate effects on binding to β-chitin (45). The rates of the production of IRGs on reduced α-chitin measured here also suggest a significant contribution of the CBM in the total dissociation of the enzyme. For ChiA, the rate constant of the production of IRGs (vIRG/E0) varied between 0.057 ± 0.005 and 0.021 ± 0.003 s−1 as measured after 5 min and 1 h of hydrolysis, respectively. These figures are about an order of magnitude higher than the koff values (Table 1), meaning that the frequency of the initiations of processive runs is much higher than the frequency of the total dissociation of the enzyme from chitin. This is indicative of “lateral processivity,” where, upon the dissociation of the chitin chain from the active site, an enzyme remains attached to the crystal surface and seeks a new site for initiation through lateral diffusion. Lateral diffusion of cellulases, as well as their isolated CBMs, has been demonstrated in several FRAP studies (46–49). To the best of our knowledge, such data are not available for chitinases. Recently, we proposed that the rate-limiting step for ChiA acting on CNWs is the complexation with polymer chain (34). Because the generation of IRGs to the reduced chitin cannot be slower than complexation, the results presented here suggest that the complexation must be at least an order of magnitude faster than full dissociation of the enzyme from α-chitin. Unfortunately, it is not possible to decide which step, complexation with chitin chain or dissociation of chitin chain from the catalytic domain, is rate-limiting for the production of IRGs on the reduced α-chitin.
Similarly to what has been observed with processive cellulases (17, 50–53), and for ChiA acting on soluble chitosan (40), we note that ChiA displayed high probability of endo-mode initiation (Table 3). If endo-mode initiation is included, the possible number of initiation sites is expected to be much higher than one can predict from the degree of polymerization of α-chitin (1230 ± 110 in our study as judged by AA labeling). Increasing the possibility of productive complex formation on a polymer with a low number of reducing ends may be the reason why there seems to be no strict exo-acting enzymes among recalcitrant polysaccharide active enzymes.
Regardless of the mode of initiation, the question of why the processive enzymes have evolved to such low off-rates (high PIntr values) remains. Measuring the processivity of glycoside hydrolases is not straightforward, and the results are dependent on the method used (18, 54). The lengths of the processive runs of TrCel7A measured on different celluloses using different methods for measuring are in the range of 10–70 (17, 23, 26, 29, 43, 55, 56). Clearly these figures are far lower than PIntr values of 4000 and 560 measured on bacterial cellulose and amorphous cellulose, respectively (17). Experimentally measured Papp values for ChiA range between 5 and 30, depending on the substrate and the method used for measuring (40, 57–59). Therefore, the large discrepancy between Papp and PIntr seems to hold also for ChiA. It has been postulated that Papp is limited by the length of the obstacle-free path on the polymer. Although the molecular nature of these obstacles has not been clearly identified, cellulose chains on eroded surface (60–62), non-productively bound enzymes (60, 61), lignin and hemicelluloses (21, 63), amorphous regions on cellulose (23), and aggregates of cellulose microfibrils (21) have been proposed to serve as obstacles. The encounter of an obstacle stops the processive run and leaves the enzyme in the non-productive complex with the polymer chain end in the substrate-binding site −1 (for enzymes moving toward the non-reducing end of the substrate). In the case of processive enzymes, the strong binding in the product-binding sites +1 and +2 creates a cumulative force to ensure directional progression of the enzyme from the non-productive complex (polymer chain end in −1) to the productive complex (polymer chain end in +2) (3, 19, 64, 65). Therefore, it is tempting to speculate that strong binding of the enzyme in the non-productive complex dictated by low koff values helps to hold the enzyme behind obstacles in a stand-by position, whereas the force directed to fill the product-binding sites generates a “pushing potential” that, if strong enough, can remove or displace obstacles (Fig. 7). In an elegant high speed atomic force microscopy study by Igarashi et al. (66), the authors demonstrated that traffic jams formed by multiple halted TrCel7A molecules on parallel cellulose chains were, through collective pushing, able to disintegrate whole cellulose microfibrils. Disintegration of cellulose microfibrils by cellulases has been reported in several other microscopy studies (48, 49, 67–74). Thus, the evolution of processive cellulases and chitinases toward low off-rates may be related to the need of removing obstacles and disintegrating fibril aggregates. Enzymes with high off-rates (weak binding in the substrate sites) or with low “pushing potential” (weak binding in the product sites) are not capable of removing obstacles and are limited to the hydrolysis of the easily accessible part of the substrate only.
FIGURE 7.
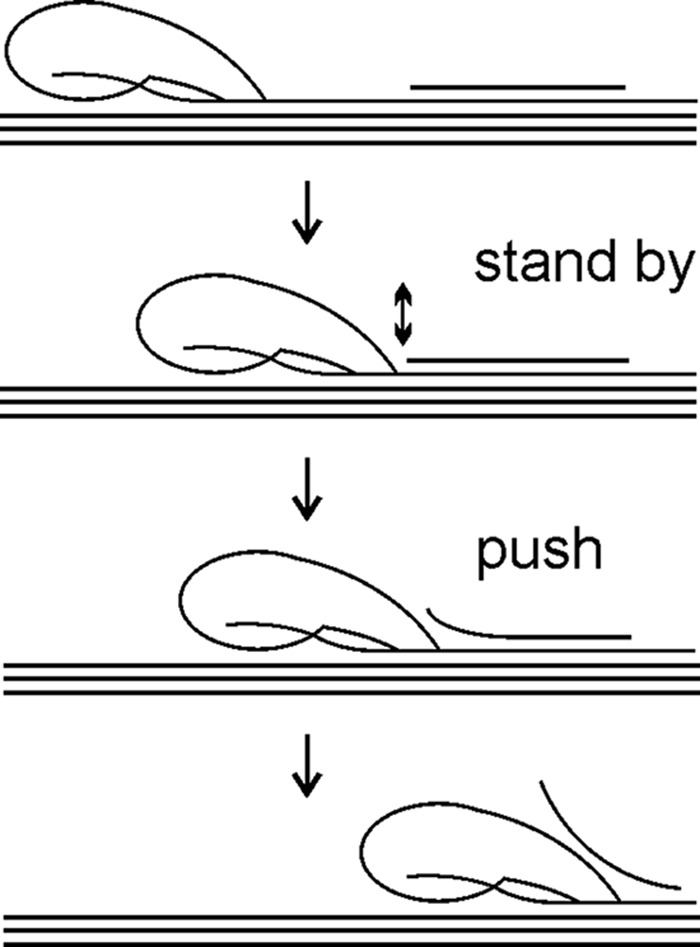
Possible mechanism of degradation of recalcitrant polysaccharide by processive enzyme. An enzyme progresses along polymer chain hydrolyzing it in parallel until it encounters an obstacle (represented by solitary chain on polymer surface). An enzyme halted by the obstacle is bound to the chitin chain with the reducing chain end in the substrate-binding site −1, or +1 if the glycosidic bond is not correctly positioned for hydrolysis. Strong interactions between enzyme and polymer chain in the substrate-binding sites ensure low off-rate and gives to an enzyme a stand by status. At the same time, strong binding of the chain end in the product sites provides the enzyme with pushing potential and forces it to move forward. If obstacle is attached to the polymer surface through non-covalent interactions, a part of it may occasionally “melt” from the polymer surface (represented by two-headed arrow). In such a case, a stand-by enzyme with high pushing potential can move forward and disintegrate the obstacle. Weak binding in substrate-binding sites (like ChiA-W167A) causes the enzyme to dissociate and does not provide sufficient population of stand-by enzymes. Weak binding in product sites (like ChiA-W275A) does not provide the enzyme with strong enough pushing potential to move forward and disintegrate the obstacle. Therefore, an apparent processivity of ChiA-W167A and ChiA-W275A is determined by the average length of obstacle-free bath on polymer. The wild type enzyme can increase the length of the processive runs by pushing away the obstacles.
It must be noted, however, that cellulases are not selected based on their individual activities. It is the performance of the whole synergistic enzyme mixture that is the subject of evolution. In the light of obstacle model, any auxiliary activity that is able to remove obstacles should work synergistically with processive enzymes. Indeed, the results of several studies have pointed to the fact that synergism between processive enzymes and non-processive endo-enzymes (23, 63, 75, 76) or lytic polysaccharide monooxygenases (77, 78) is mediated by the removal of obstacles. Apparently, the molecular nature of the obstacles depends on the source and pre-treatment conditions of the chitin/cellulose substrate. Therefore, the costs and benefits of processivity are expected to depend on the nature of auxiliary activities in combination with the nature of the substrate.
Author Contributions
P. V. and M. S. conceived and coordinated the study and wrote the paper. M. K. and S. K. designed, performed, and analyzed the experiments. P. K. derived the rate equations. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by Norwegian Financial Mechanism Grant EMP171, Danish Agency for Science, Technology, and Innovation, Programme Commission on Sustainable Energy and Environment Grant 2104-07-0028, and Estonian Science Foundation Grant 9227. The authors declare that they have no conflict of interests with the contents of this article.
- NAG
- N-acetylglucosamine
- NAG2
- N,N′-diacetylchitobiose
- AA
- anthranilic acid
- CBM
- carbohydrate binding module
- TrCel7A
- cellobiohydrolase Cel7A from Trichoderma reesei
- ChiA
- family 18 chitinase A from Serratia marcescens
- CNW
- chitin nanowhisker
- FRAP
- fluorescence recovery after photobleaching
- IRG
- insoluble reducing group
- MBTH
- 3-methyl-2-benzothiazolinone hydrazone hydrochloride
- MU-NAG2
- 4-methyl-umbelliferyl-β-d-N,N′-diacetylchitobioside
- RGtot
- the total number of reducing groups
- SEE
- substrate exchange experiment
- SRG
- soluble reducing group.
References
- 1.Himmel M. E., Ding S.-Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., and Foust T. D. (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 2.Eijsink V. G., Vaaje-Kolstad G., Vårum K. M., and Horn S. J. (2008) Toward new enzymes for biofuels: lessons from chitinase research. Trends Biotechnol. 26, 228–235 [DOI] [PubMed] [Google Scholar]
- 3.Payne C. M., Knott B. C., Mayes H. B., Hansson H., Himmel M. E., Sandgren M., Ståhlberg J., and Beckham G. T. (2015) Fungal cellulases. Chem. Rev. 115, 1308–1448 [DOI] [PubMed] [Google Scholar]
- 4.Vaaje-Kolstad G., Horn S. J., Sørlie M., and Eijsink V. G. (2013) The chitinolytic machinery of Serratia marcescens--a model system for enzymatic degradation of recalcitrant polysaccharides. FEBS J. 280, 3028–3049 [DOI] [PubMed] [Google Scholar]
- 5.Papanikolau Y., Prag G., Tavlas G., Vorgias C. E., Oppenheim A. B., and Petratos K. (2001) High resolution structural analyses of mutant chitinase A complexes with substrate provide new insight into the mechanism of catalysis. Biochemistry 40, 11338–11343 [DOI] [PubMed] [Google Scholar]
- 6.Aronson N. N. Jr., Halloran B. A., Alexyev M. F., Amable L., Madura J. D., Pasupulati L., Worth C., and Van Roey P. (2003) Family 18 chitinase-oligosaccharide substrate interaction: subsite preference and anomer selectivity of Serratia marcescens chitinase A. Biochem. J. 376, 87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zakariassen H., Aam B. B., Horn S. J., Vårum K. M., Sørlie M., and Eijsink V. G. (2009) Aromatic residues in the catalytic center of chitinase A from Serratia marcescens affect processivity, enzyme activity, and biomass converting efficiency. J. Biol. Chem. 284, 10610–10617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norberg A. L., Dybvik A. I., Zakariassen H., Mormann M., Peter-Katalinić J., Eijsink V. G., and Sørlie M. (2011) Substrate positioning in chitinase A, a processive chito-biohydrolase from Serratia marcescens. FEBS Lett. 585, 2339–2344 [DOI] [PubMed] [Google Scholar]
- 9.Uchiyama T., Katouno F., Nikaidou N., Nonaka T., Sugiyama J., and Watanabe T. (2001) Roles of exposed aromatic residues in crystalline chitin hydrolysis by chitinase A from Serratia marcescens 2170. J. Biol. Chem. 276, 41343–41349 [DOI] [PubMed] [Google Scholar]
- 10.Horn S. J., Sikorski P., Cederkvist J. B., Vaaje-Kolstad G., Sørlie M., Synstad B., Vriend G., Vårum K. M., and Eijsink V. G. (2006) Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 103, 18089–18094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zakariassen H., Eijsink V. G., and Sørlie M. (2010) Signatures of activation parameters reveal substrate-dependent rate determining steps in polysaccharide turnover by a family 18 chitinase. Carbohydr. Polym. 81, 14–20 [Google Scholar]
- 12.Kari J., Olsen J., Borch K., Cruys-Bagger N., Jensen K., and Westh P. (2014) Kinetics of cellobiohydrolase (Cel7A) variants with lowered substrate affinity. J. Biol. Chem. 289, 32459–32468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kostylev M., Alahuhta M., Chen M., Brunecky R., Himmel M. E., Lunin V. V., Brady J., and Wilson D. B. (2014) Cel48A from Thermobifida fusca: structure and site directed mutagenesis of key residues. Biotechnol. Bioeng. 111, 664–673 [DOI] [PubMed] [Google Scholar]
- 14.Kostylev M., and Wilson D. (2013) Two-parameter kinetic model based on a time-dependent activity coefficient accurately describes enzymatic cellulose digestion. Biochemistry 52, 5656–5664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang S., Irwin D. C., and Wilson D. B. (2000) Site-directed mutation of noncatalytic residues of Thermobifida fusca exocellulase Cel6B. Eur. J. Biochem. 267, 3101–3115 [DOI] [PubMed] [Google Scholar]
- 16.Li Y., Irwin D. C., and Wilson D. B. (2007) Processivity, substrate binding, and mechanism of cellulose hydrolysis by Thermobifida fusca Cel9A. Appl. Environ. Microbiol. 73, 3165–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurasin M., and Väljamäe P. (2011) Processivity of cellobiohydrolases is limited by the substrate. J. Biol. Chem. 286, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horn S. J., Sørlie M., Vårum K. M., Väljamäe P., and Eijsink V. G. (2012) Measuring processivity. Methods Enzymol. 510, 69–95 [DOI] [PubMed] [Google Scholar]
- 19.Payne C. M., Jiang W., Shirts M. R., Himmel M. E., Crowley M. F., and Beckham G. T. (2013) Glycoside hydrolase processivity is directly related to oligosaccharide binding free energy. J. Am. Chem. Soc. 135, 18831–18839 [DOI] [PubMed] [Google Scholar]
- 20.Beckham G. T., Ståhlberg J., Knott B. C., Himmel M. E., Crowley M. F., Sandgren M., Sørlie M., and Payne C. M. (2014) Toward a molecular-level theory of carbohydrate processivity in glycoside hydrolases. Curr. Opin. Biotechnol. 27, 96–106 [DOI] [PubMed] [Google Scholar]
- 21.Jalak J., and Väljamäe P. (2010) Mechanism of initial rapid rate retardation in cellobiohydrolase catalyzed cellulose hydrolysis. Biotechnol. Bioeng. 106, 871–883 [DOI] [PubMed] [Google Scholar]
- 22.Praestgaard E., Elmerdahl J., Murphy L., Nymand S., McFarland K. C., Borch K., and Westh P. (2011) A kinetic model for the burst phase of processive cellulases. FEBS J. 278, 1547–1560 [DOI] [PubMed] [Google Scholar]
- 23.Jalak J., Kurašin M., Teugjas H., and Väljamäe P. (2012) Endo-exo synergism in cellulose hydrolysis revisited. J. Biol. Chem. 287, 28802–28815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruys-Bagger N., Elmerdahl J., Praestgaard E., Tatsumi H., Spodsberg N., Borch K., and Westh P. (2012) Pre-steady state kinetics for the hydrolysis of insoluble cellulose by cellobiohydrolase Cel7A. J. Biol. Chem. 287, 18451–18458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruys-Bagger N., Elmerdahl J., Praestgaard E., Borch K., and Westh P. (2013) A steady-state theory for processive cellulases. FEBS J. 280, 3952–3961 [DOI] [PubMed] [Google Scholar]
- 26.Cruys-Bagger N., Tatsumi H., Ren G. R., Borch K., and Westh P. (2013) Transient kinetics and rate-limiting steps for the processive cellobiohydrolase Cel7A: effects of substrate structure and carbohydrate binding domain. Biochemistry 52, 8938–8948 [DOI] [PubMed] [Google Scholar]
- 27.Luterbacher J. S., Walker L. P., and Moran-Mirabal J. M. (2013) Observing and modeling BMCC degradation by commercial cellulase cocktails with fluorescently labeled Trichoderma reseii Cel7A through confocal microscopy. Biotechnol. Bioeng. 110, 108–117 [DOI] [PubMed] [Google Scholar]
- 28.Jung J., Sethi A., Gaiotto T., Han J. J., Jeoh T., Gnanakaran S., and Goodwin P. M. (2013) Binding and movement of individual Cel7A cellobiohydrolases on crystalline cellulose surfaces revealed by single-molecule fluorescence imaging. J. Biol. Chem. 288, 24164–24172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakamura A., Watanabe H., Ishida T., Uchihashi T., Wada M., Ando T., Igarashi K., and Samejima M. (2014) Trade-off between processivity and hydrolytic velocity of cellobiohydrolases at the surface of crystalline cellulose. J. Am. Chem. Soc. 136, 4584–4592 [DOI] [PubMed] [Google Scholar]
- 30.Shibafuji Y., Nakamura A., Uchihashi T., Sugimoto N., Fukuda S., Watanabe H., Samejima M., Ando T., Noji H., Koivula A., Igarashi K., and Iino R. (2014) Single-molecule imaging analysis of elementary reaction steps of Trichoderma reesei cellobiohydrolase I (Cel7A) hydrolyzing crystalline cellulose Iα and IIII. J. Biol. Chem. 289, 14056–14065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moran-Mirabal J. M., Bolewski J. C., and Walker L. P. (2011) Reversibility and binding kinetics of Thermobifida fusca cellulases studied through fluorescence recovery after photobleaching microscopy. Biophys. Chem. 155, 20–28 [DOI] [PubMed] [Google Scholar]
- 32.Brurberg M. B., Eijsink V. G., and Nes I. F. (1994) Characterization of a chitinase gene (chiA) from Serratia marcescens BJL200 and one-step purification of the gene product. FEMS Microbiol. Lett. 124, 399–404 [DOI] [PubMed] [Google Scholar]
- 33.Tews I., Perrakis A., Oppenheim A., Dauter Z., Wilson K. S., and Vorgias C. E. (1996) Bacterial chitobiase structure provides insight into catalytic mechanism and the basis of Tay-Sachs disease. Nat. Struct. Biol. 3, 638–648 [DOI] [PubMed] [Google Scholar]
- 34.Kuusk S., Sørlie M., and Väljamäe P. (2015) The predominant molecular state of bound enzyme determines the strength and type of product inhibition in the hydrolysis of recalcitrant polysaccharides by processive enzymes. J. Biol. Chem. 290, 11678–11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horn S. J., and Eijsink V. G. (2004) A reliable reducing end assay for chito-oligosaccharides. Carbohydr. Polym. 56, 35–39 [Google Scholar]
- 36.Velleste R., Teugjas H., and Väljamäe P. (2010) Reducing end-specific fluorescence labeled celluloses for cellulase mode of action. Cellulose 17, 125–138 [Google Scholar]
- 37.Bansal P., Hall M., Realff M. J., Lee J. H., and Bommarius A. S. (2009) Modeling cellulase kinetics on lignocellulosic substrates. Biotechnol. Adv. 27, 833–848 [DOI] [PubMed] [Google Scholar]
- 38.Shu Z., Wang Y., An L., and Yao L. (2014) The slowdown of the endoglucanase Trichoderma reesei Cel5A-catalyzed cellulose hydrolysis is related to its initial activity. Biochemistry 53, 7650–7658 [DOI] [PubMed] [Google Scholar]
- 39.Sørensen T. H., Cruys-Bagger N., Windahl M. S., Badino S. F., Borch K., and Westh P. (2015) Temperature effects on kinetic parameters and substrate affinity of Cel7A cellobiohydrolases. J. Biol. Chem. 290, 22193–22202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sikorski P., Sørbotten A., Horn S. J., Eijsink V. G., and Vårum K. M. (2006) Serratia marcescens chitinases with tunnel-shaped substrate-binding grooves show endo activity and different degrees of processivity during enzymatic hydrolysis of chitosan. Biochemistry 45, 9566–9574 [DOI] [PubMed] [Google Scholar]
- 41.Kuttiyawong K., Nakapong S., and Pichyangkura R. (2008) The dual exo/endo-type mode and the effect of ionic strength on the mode of catalysis of chitinase 60 (CHI60) from Serratia sp. TU09 and its mutants. Carbohydr. Res. 343, 2754–2762 [DOI] [PubMed] [Google Scholar]
- 42.Breyer W. A., and Matthews B. W. (2001) A structural basis for processivity. Protein Sci. 10, 1699–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Ossowski I., Ståhlberg J., Koivula A., Piens K., Becker D., Boer H., Harle R., Harris M., Divne C., Mahdi S., Zhao Y., Driguez H., Claeyssens M., Sinnott M. L., and Teeri T. T. (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A. A comparison with Phanerochaete chrysosporium Cel7D. J. Mol. Biol. 333, 817–829 [DOI] [PubMed] [Google Scholar]
- 44.Hamre A. G., Schaupp D., Eijsink V. G., and Sørlie M. (2015) The directionality of processive enzymes acting on recalcitrant polysaccharides is reflected in the kinetic signatures of oligomer degradation. FEBS Lett. 589, 1807–1812 [DOI] [PubMed] [Google Scholar]
- 45.Zakariassen H., Klemetsen L., Sakuda S., Vaaje-Kolstad G., Vårum K. M., Sørlie M., and Eijsink V. G. (2010) Effect of enzyme processivity on the efficacy of a competitive chitinase inhibitor. Carbohydr. Polym. 82, 779–785 [Google Scholar]
- 46.Jervis E. J., Haynes C. A., and Kilburn D. G. (1997) Surface diffusion of cellulases and their isolated binding domains on cellulose. J. Biol. Chem. 272, 24016–24023 [DOI] [PubMed] [Google Scholar]
- 47.Moran-Mirabal J. M., Bolewski J. C., and Walker L. P. (2013) Thermobifida fusca cellulases exhibit limited surface diffusion on bacterial micro-crystalline cellulose. Biotechnol. Bioeng. 110, 47–56 [DOI] [PubMed] [Google Scholar]
- 48.Luterbacher J. S., Moran-Mirabal J. M., Burkholder E. W., and Walker L. P. (2015) Modeling enzymatic hydrolysis of lignocellulosic substrates using confocal fluorescence microscopy I: filter paper cellulose. Biotechnol. Bioeng. 112, 21–31 [DOI] [PubMed] [Google Scholar]
- 49.Luterbacher J. S., Moran-Mirabal J. M., Burkholder E. W., and Walker L. P. (2015) Modeling enzymatic hydrolysis of lignocellulosic substrates using fluorescent confocal microscopy II: pretreated biomass. Biotechnol. Bioeng. 112, 32–42 [DOI] [PubMed] [Google Scholar]
- 50.Ståhlberg J., Johansson G., and Pettersson G. (1993) Trichoderma reesei has no true exo-cellulase: all intact and truncated cellulases produce new reducing end groups on cellulose. Biochim. Biophys. Acta 1157, 107–113 [DOI] [PubMed] [Google Scholar]
- 51.Reverbel-Leroy C., Pages S., Belaich A., Belaich J.-P., and Tardif C. (1997) The processive endocellulase CelF, a major component of the Clostridium cellulolyticum cellulosome: purification and characterization of the recombinant form. J. Bacteriol. 179, 46–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sakon J., Irwin D., Wilson D. B., and Karplus P. A. (1997) Structure and mechanism of endo/exocellulase E4 from Thermomonospora fusca. Nat. Struct. Biol. 4, 810–818 [DOI] [PubMed] [Google Scholar]
- 53.Boisset C., Fraschini C., Schülein M., Henrissat B., and Chanzy H. (2000) Imaging the enzymatic digestion of bacterial cellulose ribbons reveals the endo character of the cellobiohydrolase Cel6A from Humicola insolens and its mode of synergy with cellobiohydrolase Cel7A. Appl. Environ. Microbiol. 66, 1444–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilson D. B., and Kostylev M. (2012) Cellulase processivity. Methods Mol. Biol. 908, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Medve J., Karlsson J., Lee D., and Tjerneld F. (1998) Hydrolysis of microcrystalline cellulose by cellobiohydrolase I and endoglucanase II from Trichoderma reesei: adsorption, sugar production pattern, and synergism of the enzymes. Biotechnol. Bioeng. 59, 621–634 [PubMed] [Google Scholar]
- 56.Kipper K., Väljamäe P., and Johansson G. (2005) Processive action of cellobiohydrolase Cel7A from Trichoderma reesei is revealed as “burst” kinetics on fluorescent polymeric model substrates. Biochem. J. 385, 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horn S. J., Sørbotten A., Synstad B., Sikorski P., Sørlie M., Vårum K. M., and Eijsink V. G. (2006) Endo/exo mechanism and processivity of family 18 chitinases produced by Serratia marcescens. FEBS J. 273, 491–503 [DOI] [PubMed] [Google Scholar]
- 58.Igarashi K., Uchihashi T., Uchiyama T., Sugimoto H., Wada M., Suzuki K., Sakuda S., Ando T., Watanabe T., and Samejima M. (2014) Two-way traffic of glycoside hydrolase family 18 processive chitinases on crystalline chitin. Nat. Commun. 5, 3975. [DOI] [PubMed] [Google Scholar]
- 59.Hamre A. G., Lorentzen S. B., Väljamäe P., and Sørlie M. (2014) Enzyme processivity changes with the extent of recalcitrant polysaccharide degradation. FEBS Lett. 588, 4620–4624 [DOI] [PubMed] [Google Scholar]
- 60.Väljamäe P., Sild V., Pettersson G., and Johansson G. (1998) The initial kinetics of hydrolysis by cellobiohydrolases I and II is consistent with a cellulose surface-erosion model. Eur. J. Biochem. 253, 469–475 [DOI] [PubMed] [Google Scholar]
- 61.Shang B. Z., Chang R., and Chu J.-W. (2013) Systems-level modeling with molecular resolution elucidates the rate-limiting mechanisms of cellulose decomposition by cellobiohydrolases. J. Biol. Chem. 288, 29081–29089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shang B. Z., and Chu J.-W. (2014) Kinetic modeling at single-molecule resolution elucidates the mechanisms of cellulase synergy. ACS Catal. 4, 2216–2225 [Google Scholar]
- 63.Eriksson T., Karlsson J., and Tjerneld F. (2002) A model explaining declining rate in hydrolysis of lignocellulose substrates with cellobiohydrolase I (Cel7A) and endoglucanase I (Cel7B) of Trichoderma reesei. Appl. Biochem. Biotechnol. 101, 41–60 [DOI] [PubMed] [Google Scholar]
- 64.Knott B. C., Crowley M. F., Himmel M. E., Ståhlberg J., and Beckham G. T. (2014) Carbohydrate-protein interactions that drive processive polysaccharide translocation in enzymes revealed from a computational study of cellobiohydrolase processivity. J. Am. Chem. Soc. 136, 8810–8819 [DOI] [PubMed] [Google Scholar]
- 65.Colussi F., Sørensen T. H., Alasepp K., Kari J., Cruys-Bagger N., Windahl M. S., Olsen J. P., Borch K., and Westh P. (2015) Probing substrate interactions in the active tunnel of a catalytically deficient cellobiohydrolase (Cel7). J. Biol. Chem. 290, 2444–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Igarashi K., Uchihashi T., Koivula A., Wada M., Kimura S., Okamoto T., Penttilä M., Ando T., and Samejima M. (2011) Traffic jams reduce hydrolytic efficiency of cellulase on cellulose surface. Science 333, 1279–1282 [DOI] [PubMed] [Google Scholar]
- 67.White A. R., and Brown R. M. (1981) Enzymatic hydrolysis of cellulose: visual characterization of the process. Proc. Natl. Acad. Sci. U.S.A. 78, 1047–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chanzy H., Henrissat B., Vuong R., and Schülein M. (1983) The action of 1,4-β-d-glucan cellobiohydrolase on Valonia cellulose microcrystals: an electron microscopic study. FEBS Lett. 153, 113–118 [Google Scholar]
- 69.Walker L. P., Wilson D. B., and Irwin D. C. (1990) Measuring fragmentation of cellulose by Thermomonospora fusca cellulase. Enzyme Microb. Technol. 12, 378–386 [Google Scholar]
- 70.Walker L. P., Wilson D. B., Irvin D. C., McQuire C., and Price M. (1992) Fragmentation of cellulose by the major Thermomonospora fusca cellulases, Trichoderma reesei CBHI, and their mixtures. Biotechnol. Bioeng. 40, 1019–1026 [DOI] [PubMed] [Google Scholar]
- 71.Woodward J., Affholter K. A., Noles K. K., Troy N. T., and Gaslightwala S. F. (1992) Does cellobiohydrolase II core protein from Trichoderma reesei disperse cellulose macrofibrils? Enzyme Microb. Technol. 14, 625–630 [Google Scholar]
- 72.Imai T., Boisset C., Samejima M., Igarashi K., and Sugiyama J. (1998) Unidirectional processive action of cellobiohydrolase Cel7A on Valonia cellulose microcrystals. FEBS Lett. 432, 113–116 [DOI] [PubMed] [Google Scholar]
- 73.Santa-Maria M., and Jeoh T. (2010) Molecular-scale investigations of cellulose microstructure during enzymatic hydrolysis. Biomacromolecules 11, 2000–2007 [DOI] [PubMed] [Google Scholar]
- 74.Jeoh T., Santa-Maria M. C., and O'Dell P. J. (2013) Assessing cellulose microfibrillar structure changes due to cellulase action. Carbohydr. Polym. 97, 581–586 [DOI] [PubMed] [Google Scholar]
- 75.Kostylev M., and Wilson D. (2014) A distinct model of synergism between a processive endocellulase (TfCel9A) and an exocellulase (TfCel48A) from Thermobifida fusca. Appl. Environ. Microbiol. 80, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Väljamäe P., Sild V., Nutt A., Pettersson G., and Johansson G. (1999) Acid hydrolysis of bacterial cellulose reveals different modes of synergistic action between cellobiohydrolase I and endoglucanase I. Eur. J. Biochem. 266, 327–334 [DOI] [PubMed] [Google Scholar]
- 77.Eibinger M., Ganner T., Bubner P., Rošker S., Kracher D., Haltrich D., Ludwig R., Plank H., and Nidetzky B. (2014) Cellulose surface degradation by a lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J. Biol. Chem. 289, 35929–35938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakagawa Y. S., Kudo M., Loose J. S., Ishikawa T., Totani K., Eijsink V. G., and Vaaje-Kolstad G. (2015) A small lytic polysaccharide monooxygenase from Streptomyces griseus targeting α- and β-chitin. FEBS J. 282, 1065–1079 [DOI] [PubMed] [Google Scholar]