

Background: The human public T cell response to a dominant CMV epitope features high clonal diversity.
Results: Structures of two public TCRs bound to this CMV epitope and HLA-A2 reveal different recognition strategies.
Conclusion: These structures show how the same public complementarity-determining region 3α (CDR3α) motif can associate with different variable α regions and pair with different CDR3βs.
Significance: This structural interchangeability generates a clonally diverse public TCR repertoire.
Keywords: complex, herpesvirus, major histocompatibility complex (MHC), T-cell receptor (TCR), x-ray crystallography
Abstract
Cytomegalovirus (CMV) is a ubiquitous and persistent human pathogen that is kept in check by CD8+ cytotoxic T lymphocytes. Individuals expressing the major histocompatibility complex (MHC) class I molecule HLA-A2 produce cytotoxic T lymphocytes bearing T cell receptors (TCRs) that recognize the immunodominant CMV epitope NLVPMVATV (NLV). The NLV-specific T cell repertoire is characterized by a high prevalence of TCRs that are frequently observed in multiple unrelated individuals. These public TCRs feature identical, or nearly identical, complementarity-determining region 3α (CDR3α) and/or CDR3β sequences. The TCRs may express public CDR3α motifs alone, public CDR3β motifs alone, or dual public CDR3αβ motifs. In addition, the same public CDR3α motif may pair with different CDR3β motifs (and the reverse), giving rise to highly diverse NLV-specific TCR repertoires. To investigate the structural underpinnings of this clonal diversity, we determined crystal structures of two public TCRs (C7 and C25) in complex with NLV·HLA-A2. These TCRs utilize completely different CDR3α and CDR3β motifs that, in addition, can associate with multiple variable α and variable β regions in NLV-specific T cell repertoires. The C7·NLV·HLA-A2 and C25·NLV·HLA-A2 complexes exhibit divergent TCR footprints on peptide-MHC such that C25 is more focused on the central portion of the NLV peptide than is C7. These structures combined with molecular modeling show how the public CDR3α motif of C25 may associate with different variable α regions and how the public CDR3α motif of C7 may pair with different CDR3β motifs. This interchangeability of TCR V regions and CDR3 motifs permits multiple structural solutions to binding an identical peptide-MHC ligand and thereby the generation of a clonally diverse public T cell response to CMV.
Introduction
Human cytomegalovirus (CMV)3 is a ubiquitous herpesvirus that infects 60–90% of the world population. Although CMV infections are usually kept in check by the immune system of immunocompetent individuals, they can cause life-threatening diseases in immunocompromised patients and are a major health concern in patients undergoing bone marrow transplantation (1, 2). In addition, congenital CMV infection is the most common cause of infectious complications in newborns, resulting in deafness and other developmental abnormalities (3). CD8+ cytotoxic T lymphocytes (CTLs) play a vital role in controlling CMV infection in humans (4–6). The dominant CTL response is directed against the CMV tegument protein pp65 (6). Individuals expressing the widely distributed major histocompatibility complex (MHC) class I molecule HLA-A*0201 (HLA-A2) produce CMV-specific CTLs bearing T cell receptors (TCRs) that mainly recognize an epitope corresponding to residues 495–503 of pp65 (NLVPMVATV; herein referred to as NLV) (6, 7).
TCRs bind peptide-MHC (pMHC) via their six complementarity-determining region (CDR) loops, three from the variable α (Vα) domain and three from Vβ. The first and second CDRs (CDR1 and CDR2) are encoded within the TCR Vα and Vβ gene segments; CDR3 is formed by DNA recombination involving juxtaposition of Vα and Jα segments for the α chain genes and of Vβ, D, and Jβ segments for the β chain genes. Direct in vivo estimates of TCR diversity in humans have placed the number of unique structures at >2.5 × 107 (8).
Recent advances in high-throughput DNA sequencing (next generation sequencing) have revolutionized the study of human TCR repertoires in response to infection with CMV, human immunodeficiency virus (HIV), and other viruses (9–11). In addition, new strategies have been developed for concurrent characterization of the CDR3 sequences of TCR α and β chains in epitope-specific CD8+ T cells through simultaneous amplification of CDR3α and CDR3β transcripts from single cells (12–14). Because CDR3 loops interact specifically with the MHC-bound antigenic peptide (15), CDR3 sequences serve as unique markers for clonal expansion after T cell activation. Together, high-throughput sequencing and single-cell analysis have greatly improved our understanding of the breadth of virus-specific T cell responses and the degree of overlap of TCR repertoires among individuals. Thus, studies of CD8+ T cell responses to infection with CMV and influenza virus have revealed highly diverse TCR repertoires directed against immunodominant epitopes, such as the CMV NLV peptide, accompanied by antigen-driven selection of high avidity T cell clones that presumably eliminate infected cells more rapidly (12, 13, 16, 17). However, clonal focusing does not appear to result in overall repertoire narrowing, suggesting a strategy to optimize CTL responses while safeguarding TCR structural diversity (18). This diversity assures protection from viral escape (19) and the provision of a wide range of avidities that fulfill requirements for functional heterogeneity (20).
In most T cells responses, the TCR repertoires elicited by a particular antigenic epitope are distinct between individuals (private T cell response). By contrast, certain other epitope-specific TCR repertoires contain TCRs that are frequently observed in multiple unrelated individuals (public T cell response). Indeed, public TCRs have been described in immune responses to a variety of human viruses, including CMV, HIV, and Epstein-Barr virus (EBV) (21). In particular, clonotypic analysis of the CMV NLV-specific T cell repertoire has revealed a high prevalence of public TCRs, as manifested by usage of identical, or nearly identical, CDR3α and/or CDR3β sequences in TCRs from different individuals (12, 13, 16). Seven public CDR3α and six public CDR3β motifs have been identified to date, which collectively account for ∼70% of the total NLV-specific TCR response (12). These TCRs are characterized by usage of public CDR3α motifs alone (∼25%), public CDR3β motifs alone (∼25%), or both public CDR3α and public CDR3β motifs (∼50%) (12). Therefore, although public CDR3α motifs often pair with public CDR3β motifs, other pairings are equally common. Moreover, even among TCRs expressing dual public CDR3α/CDR3β motifs the same public CDR3α motif may pair with different public CDR3β motifs (and vice versa). The association of different Vα (or Vβ) gene segments with the same public CDR3α (or CDR3β) motif increases the diversity of the NLV-specific TCR response even further (12, 13, 16). These findings reveal the multiplicity of solutions that TCRs can employ to bind the same NLV·HLA-A2 ligand. However, the structural principles underlying this remarkable diversity are unclear.
Several structures of public TCRs in complex with pMHC have been reported: TCR JM22 bound to an influenza-derived peptide presented by HLA-A2 (22), TCRs bound to EBV-derived peptides presented by HLA-B8 (23–26), and TCR RA14 bound to CMV NLV presented by HLA-A2 (27). In some cases these studies have identified unusual structural features of the selecting pMHC ligand, such as limited solvent accessibility or bulging of the viral peptide, which may explain the selection of dominant TCRs (22, 23, 26), whereas in other cases such features are not evident (24, 25, 27). Here, we have addressed the structural basis for the surprising diversity of the public TCR response to CMV revealed by recent single-cell clonotypic analyses of NLV-specific T cell repertoires (12, 13). To do so we determined crystal structures of two public TCRs (C7 and C25) in complex with NLV·HLA-A2. One of these TCRs (C7) uses the same CDR3α motif as RA14 but an unrelated CDR3β. The other TCR (C25) uses completely different public CDR3α and CDR3β motifs that, in addition, can associate with multiple Vα and Vβ regions in NLV-specific T cell repertoires. These structures in conjunction with molecular modeling of other TCR·NLV·HLA-A2 complexes provide new insights into how public TCRs expressing highly diverse α/β chain pairs can mediate high affinity recognition of an identical peptide-MHC ligand.
Experimental Procedures
Study Subjects
Two HLA-A2+ healthy male donors (33 and 55 years old) participated in this study. The protocol was approved by the National Institute on Aging Institutional Review Board.
Isolation of CMV NLV-specific CD8+ T Cells
The isolation of CD8+ T cells from peripheral blood was previously described (28). Briefly, peripheral blood mononuclear cells were isolated from leukapheresis cells by Ficoll gradient centrifugation. CD8+ T cells were isolated from peripheral blood mononuclear cells by immunomagnetic separation using a custom-made antibody mixture (28) and BigMag goat anti-mouse IgG beads (Qiagen). CMV NLV-specific CD8+ T cells were expanded in an artificial antigen presenting system as previously described (29, 30). Briefly, CD8+ T cells were stimulated with NLV peptide (NLVPMVATV; BioMer Technology) presented by artificial antigen-presenting cells for 14 days (29). The expanded cells were stained with antigen-presenting cell- and FITC-conjugated NLV DextramerTM (Immudex). FITC and antigen-presenting cell double-positive cells were sorted by flow cytometry.
Identification of Paired TCR Chains on the Single-cell Level
Single CMV NLV-specific CD8+ T cells were sorted into a 384-well plate containing 5 μl of lysis buffer from an Invitrogen CellsDirect One-Step qRT-PCR kit (Life Technologies). The plate was centrifuged for 5 min and then heated at 75 °C for 10 min immediately after sorting. The cDNA was synthesized in a 10-μl reaction system with TCR α and β chain constant region primers, TRA-RT and TRA-BT, and SuperScript III (Life Technologies) at 50 °C for 60 min. After that 5 μl of cDNA products were used for the first round PCR with HiFi Taq (Life Technologies) and TCR α and β chain variable region primers and constant region interior primers. All forward primers included a sequence (UF) at the 5′ end that was used as a primer for subsequent rounds of PCR. One microliter of the first round PCR products was used for the second round PCR with UF and either α or β chain constant region interior primers. The final PCR products were column-purified (Qiagen) and sequenced (GeneWiz). The TCR V, J, and CDR3 were identified using IMGT (International ImMunoGeneTics database) V-Quest tools (31). All primers and PCR program conditions are listed in supplemental Table 1.
Cloning, Expression, and Purification of NLV-specific TCRs
The identified α and β chain pairs of CMV NLV-specific TCRs C7 and C25 were amplified from single cell PCR products and cloned into the expression vector pET26b (Novagen). Soluble C7 and C25 were prepared by in vitro folding from inclusion bodies produced in Escherichia coli. The gene encoding residues 1–204 of the C7 α chain (or 1–202 of the C25 α chain) was inserted into pET26b. The gene encoding residues 1–244 of the C7 β chain (or 1–246 of the C25 β chain) was cloned into the same vector. To increase yields and stability of TCR αβ heterodimers, we engineered a CαCys-158–CβCys-171 interchain disulfide in C7 (or CαCys-156–CβCys-173 in C25) (32) using two separate PCR reactions. The first PCR amplified the α or β chain from CDR1 to the mutated cysteine, and the second PCR amplified the remainder of the constant region. To facilitate cloning into pET26b, restriction enzyme sites NdeI/EagI and NdeI/SpeI were introduced in the first PCR of α and β chains, respectively. Restriction sites EagI/XhoI and SpeI/XhoI were introduced in the second PCR of α and β chains, respectively. All primers used for vector cloning are listed in supplemental Table 2.
The mutated TCR α and β chains were expressed separately as inclusion bodies in BL21(DE3) E. coli cells (Agilent Technologies). Bacteria were grown at 37 °C in LB medium to A600 = 0.6–0.8 and induced with 1 mm isopropyl-β-d-thiogalactoside. After incubation for 3 h, the bacteria were harvested by centrifugation and resuspended in 50 mm Tris-HCl (pH 8.0) containing 0.1 m NaCl and 2 mm EDTA; cells were disrupted by sonication. Inclusion bodies were washed extensively with 50 mm Tris-HCl (pH 8.0) and 5% (v/v) Triton X-100. Inclusion bodies were dissolved in 8 m urea, 50 mm Tris-HCl (pH 8.0), 10 mm EDTA, and 10 mm DTT overnight. The mixture was spun at 50,000 × g for 40 min, and the supernatant was retained. For in vitro folding, the TCR α and β chains were mixed in a 1.2:1 molar ratio for 30 min before dilution into ice-cold folding buffer containing 5 m urea, 0.4 m l-arginine-HCl, 100 mm Tris-HCl (pH 8.0), 5 mm EDTA, 3.7 mm cystamine, and 6.6 mm cysteamine to a final protein concentration of 80 mg/liter. The folding mixture was dialyzed against distilled H2O for 48 h at 4 °C, then against 10 mm Tris-HCl (pH 8.0) for 24 h. The mixture was concentrated 20-fold and dialyzed against 25 mm Tris-HCl (pH 8.0). Disulfide-linked TCR C7 or C25 heterodimers were purified using sequential Superdex 200 GL and Mono Q FPLC columns (GE Healthcare).
Production of NLV·HLA-A2
Soluble HLA-A2 loaded with NLV peptide (NLVPMVATV) (GenScript) was prepared by in vitro folding. The HLA-A*0201 heavy chain (residues 1–275) and β2-microglobulin (residues 1–99) were produced separately as inclusion bodies in BL21(DE3) E. coli cells transformed by pET26b containing the corresponding genes. Inclusion bodies, prepared as described above, were dissolved in 8 m urea, 50 mm Tris-HCl (pH 8.0), 10 mm EDTA, and 10 mm DTT. For in vitro folding, the HLA-A*0201 heavy chain, β2-microglobulin and NLV peptide were mixed in a 1:2:10 molar ratio and diluted into a folding solution containing 5 m urea, 0.4 m l-arginine-HCl, 100 mm Tris-HCl (pH 8.0), 5 mm EDTA, 3.7 mm cystamine, and 6.6 mm cysteamine. After 72 h at 4 °C, the folding mixture was concentrated 20-fold and dialyzed against 25 mm Tris-HCl (pH 8.0). Correctly folded NLV·HLA-A2 was purified using sequential HiTrapQ, Superdex 200 GL, and Mono Q columns.
Crystallization and Data Collection
For crystallization, TCRs C7 and C25 were each mixed with NLV·HLA-A2 in a 1:1 molar ratio and concentrated to 10 mg/ml. Crystals of the C7·NLV·HLA-A2 complex grew in 30% (w/v) polyethylene glycol (PEG) 400, 0.1 m Tris-HCl (pH 8.5), and 0.2 m MgCl2. For data collection, crystals were transferred to a cryoprotectant solution of mother liquor containing 35% (w/v) PEG 400 before flash-cooling in a nitrogen stream. The C25·NLV·HLA-A2 complex crystallized in 10–15% (w/v) PEG 3000, 0.1 m imidazole (pH 8.0), and 0.2 m calcium acetate. Crystals were cryoprotected with 30% (v/v) glycerol and flash-cooled. X-ray diffraction data for the C7·NLV·HLA-A2 and C25·NLV·HLA-A2 complexes were collected at beamline 22ID of the Advanced Photon Source, Argonne National Laboratory with a MAR 300 CCD detector. Diffraction data were indexed, integrated, and scaled with the program HKL2000 (33). Data collection statistics are presented in Table 1.
TABLE 1.
Data collection and structure refinement statistics
| C7·NLV·HLA-A2 | C25·NLV·HLA-A2 | |
|---|---|---|
| Data collection | ||
| Space group | C2221 | P212121 |
| Cell dimensions | ||
| a (Å) | 151.8 | 84.3 |
| b (Å) | 366.6 | 124.9 |
| c (Å) | 152.0 | 193.8 |
| α, β, γ (o) | 90, 90, 90 | 90, 90, 90 |
| Resolution range (Å)a | 49.5–3.51 (3.64–3.51) | 40.7–2.10 (2.17–2.10) |
| Unique reflectionsa | 52,979 (4,991) | 119,101 (11,500) |
| Rmerge (%)a,b | 20.3 (89.0) | 8.4 (59.4) |
| Mean I/σ(I)a | 10.7 (2.4) | 22.6 (3.6) |
| Completeness (%)a | 99.5 (95.0) | 99.2 (97.1) |
| Refinement | ||
| Resolution range (Å)a | 49.5-3.51 | 40.7–2.10 |
| Rwork(%)/Rfree(%)a,c | 27.0 (31.0)/35.5 (38.1) | 20.1 (26.2)/25.4 (29.6) |
| No. of protein atoms | 25,218 | 13,250 |
| No. of water molecules | 54 | 814 |
| r.m.s.d. from ideality | ||
| Bond lengths (Å) | 0.009 | 0.016 |
| Bond angles (o) | 1.59 | 1.15 |
| Ramachandran statistics (%) | ||
| Most favored | 87.2 | 96.0 |
| Allowed | 12.2 | 3.9 |
| Disallowed | 0.6 | 0.1 |
aValues in parentheses are statistics for the highest resolution shell.
bRmerge = Σ|j − 〈I〉|/ΣIj, where j is the intensity of an individual reflection, and 〈I〉 is the average intensity of that reflection.
cRwork = Σ‖Fo| − |Fc‖/Σ|Fo|, where Fc is the calculated structure factor. Rfree is as for Rwork but calculated for a randomly selected 5.0% of reflections not included in the refinement.
Structure Determination and Refinement
The structures of the C7·NLV·HLA-A2 and C25·NLV·HLA-A2 complexes were solved by molecular replacement with the program Phaser (34). For the C7·NLV·HLA-A2 complex, a gliadin-specific TCR (PDB accession code 4OZF) (35), and NLV·HLA-A2 (PDB accession code 3GSN) (27) were used as search models with CDRs and the peptide removed, respectively. Three complex molecules in the asymmetric unit were located first; the fourth was found according to non-crystallographic symmetry. Structure refinement was performed using Phenix (36) followed by manual model building with Coot (37) based on 2Fo − Fc and Fo − Fc maps with NLV peptide omitted in the initial refinement. The final Rwork and Rfree values for the C7·NLV·HLA-A2 complex are 27.0% and 35.5%, respectively. Refinement statistics are summarized in Table 1.
For the C25·NLV·HLA-A2 complex, two NLV·HLA-A2 and one C25 were immediately found using NLV·HLA-A2 (PDB accession code 3GSO) (27) and TCR LC13 (PDB accession code 1MI5) (23) as search models. Another C25 TCR was located in a different asymmetric unit. A new search model was generated by deleting that TCR and placing a TCR opposite the located NLV·HLA-A2 molecule. Two C25·NLV·HLA-A2 complexes were then found by Phaser (34). The NLV peptide and CDRs was omitted in the initial refinement. Rigid body and simulated annealing refinement were performed using Phenix (36). Rebuilding and modeling were accomplished manually with Coot (37) according to 2Fo − Fc and Fo − Fc maps. Water molecules were added with a distance cutoff of 3.4 Å. The final Rwork and Rfree values for the C25·NLV·HLA-A2 complex were 20.1% and 25.4%, respectively. Refinement statistics are presented in Table 1. Stereochemical parameters were evaluated by PROCHECK (38).
Surface Plasmon Resonance Analysis
The interaction of TCRs C7 and C25 with NLV·HLA-A2 was assessed by surface plasmon resonance using a BIAcore T100 biosensor at 25 °C. Biotin-tagged NLV·HLA-A2 (NIH Tetramer Core Facility) was immobilized on a streptavidin-coated BIAcore SA chip (GE Healthcare) at 1500–2000 resonance units followed by blocking the remaining streptavidin sites with 20 μm biotin solution. An additional flow cell was injected only with free biotin to serve as a blank control. For analysis of TCR binding, solutions containing different concentrations of C7 or C25 were flowed sequentially over the chips immobilized with NLV·HLA-A2 and the blank. Injections of TCR were stopped at 30 s after surface plasmon resonance signals reached a plateau. Equilibrium data were fitted with a 1:1 binding model using BIAevaluation 3.1 software to obtain dissociation constants (KD values).
Modeling of TCR·NLV·HLA-A2 Complexes
Initial structural models of the E4.1 (12) and RA11 (16) TCRs were produced using the Lyra web server (39). We used the Modeler program (40) to remodel the CDR3α for both structures using the shared residues from the C25 CDR3α (NNNDMR) as a template. Additionally for RA11, we noted that the Lyra server selected CDR1α and CDR2α loop templates containing proline residues despite the absence of prolines from these CDRs. Given that proline residues can often impact CDR loop conformations (41), we remodeled these loops for the RA11 TCR, along with CDR3α, using CDR1α and CDR2α loops from AS01 (PDB accession code 3O4L) and DMF5 (PDB accession code 3QEU) TCR structures, respectively, as they were homologous in sequence to the RA11 CDRs without containing proline residues.
Docking simulations of E4.1 and RA11 TCR models to NLV·HLA-A2 were performed using a previously developed TCR-pMHC docking algorithm, TCRFlexDock (42). For docking input, TCRs were positioned over the pMHCs as in the original TCRFlexDock study (45° crossing angle, 0° tilt) using the pMHC structure from the C25·NLV·HLA-A2 complex. Approximately 1000 TCR-pMHC models were generated per docking simulation, which employed a Monte Carlo approach to iteratively sample side chains, rigid-body docking orientation, and backbone conformation of peptide and CDR loops (with additional backbone flexibility for CDR3 loops). Models were ranked using ZRANK2 (43).
Protein Data Bank Accession Codes
Coordinates and structure factors for the C7·NLV·HLA-A2 and C25·NLV·HLA-A2 complexes have been deposited under accession codes and PDB accession codes 5D2L and 5D2N, respectively.
Results
Interaction of TCRs C25 and C7 with NLV·HLA-A2
The CMV NLV-specific TCRs C25 and C7 were isolated from CD8+ T cells from the peripheral blood of two HLA-A2+ healthy male donors as described under “Experimental Procedures.” C25 utilizes gene segments TRAV26–2 and TRAJ43 for the α chain, and TRBV7–6, TRBD1, and TRBJ1–4 for the β chain, whereas C7 utilizes TRAV24 and TRAJ49 for the α chain, and TRBV7–2, TRBD2 and TRBJ2–5 for the β chain. We used surface plasmon resonance to measure the binding of TCRs C25 and C7 to HLA-A2 loaded with NLV peptide (Fig. 1, A and C). To characterize the interaction of C25 and C7 with NLV·HLA-A2, we expressed these recombinant proteins by in vitro folding from bacterial inclusion bodies. Biotinylated NLV·HLA-A2 was directionally coupled to a streptavidin-coated biosensor surface, and different concentrations of C25 or C7 were flowed sequentially over the immobilized pMHC ligand. Dissociation constants (KD values) were obtained by fitting equilibrium data to a 1:1 binding model. Both C25 (KD = 4.7 μm) and C7 (KD = 5.1 μm) bound NLV·HLA-A2 with affinities at the high end of the range for TCR-pMHC interactions (Fig. 1B and D), consistent with the affinities of most anti-microbial MHC class I-restricted TCRs characterized to date (44). Notably, C25 and C7 each bind NLV·HLA-A2 ∼5-fold more tightly than does TCR RA14 (KD = 28 μm) (27).
FIGURE 1.

Surface plasmon resonance analysis of the binding of TCRs C25 and C7 to NLV·HLA-A2. A, TCR C25 at concentrations of 83.8, 41.9, 21.0, 10.5, 5.3, 2.7, 1.4, 0.7, 0.4, and 0.2 μm was injected over immobilized NLV·HLA-A2 (1000 resonance units (RU)). B, fitting curve for equilibrium binding that resulted in a KD of 4.7 μm for the C25·NLV·HLA-A2 interaction. C, TCR C7 at concentrations of 106.8, 53.4, 13.4, 6.7, 3.4, 1.7, 0.9, and 0.5 μm was injected over immobilized NLV·HLA-A2 (1000 resonance units). D, fitting curve for equilibrium binding that gave a KD of 5.1 μm for the C7·NLV·HLA-A2 interaction.
Overview of the C25·NLV·HLA-A2 and C7·NLV·HLA-A2 Complexes
To understand how TCRs C25 and C7 recognize NLV·HLA-A2 and to explain the prevalence of the public CDR3 motifs of C24 and C7 in the T cell response to CMV (12, 13. 16), we determined the structures of the C25·NLV·HLA-A2 and C7·NLV·HLA-A2 complexes to 2.1 Å and 3.5 Å resolution, respectively (Table 1; Fig. 2, A and C). The resolution of the C25·NLV·HLA-A2 complex is one of the highest reported for TCR-pMHC class I or II complexes, which seldom exceed 2.5 Å (15). In both the C25·NLV·HLA-A2 and C7·NLV·HLA-A2 structures, the interface between TCR and pMHC was in unambiguous electron density for each of the two (C25·NLV·HLA-A2) or four (C7·NLV·HLA-A2) complex molecules in the asymmetric unit of the crystal (Fig. 2, B and D). The root mean square difference (r.m.s.d.) in α-carbon positions for the TCR VαVβ and MHC α1α2 modules, including the NLV peptide, was 0.40 Å for the two C25·NLV·HLA-A2 complexes. The corresponding r.m.s.d. for the four C7·NLV·HLA-A2 complexes ranged from 0.50 to 0.83 Å. Based on these close similarities, the following descriptions of TCR-pMHC interactions apply to all complex molecules in the asymmetric unit of the C25·NLV·HLA-A2 or C7·NLV·HLA-A2 crystal.
FIGURE 2.

Structure of TCR·NLV·HLA-A2 complexes. A, side view of the C25·NLV·HLA-A2 complex (ribbon diagram). Cyan, TCR α chain; green, TCR β chain; orange, HLA-A2 heavy chain; gray, β2 microglobulin (β2m); magenta, NLV peptide. B, electron density in the interface of the C25·NLV·HLA-A2 complex. Density from the final 2Fo − Fc map at 2.1 Å resolution is contoured at 1σ. C, side view of the C7·NLV·HLA-A2 complex. D, electron density in the interface of the C7·NLV·HLA-A2 complex. Density from the final 2Fo − Fc map at 3.5 Å resolution is contoured at 1σ.
Both C25 and C7 dock symmetrically over NLV·HLA-A2 in a canonical diagonal orientation, with crossing angles of TCR to pMHC (45) of 61° and 29°, respectively. Upon binding NLV·HLA-A2, C25 and C7 bury 89% (272 Å2) and 86% (314 Å2), respectively, of the peptide solvent-accessible surface. These percentages are at the higher end of the range for TCR-pMHC class I complexes, which varies from 60 to 91% in other structures (15). Extensive peptide burial, which is also a salient feature of the RA14·NLV·HLA-A2 complex (27), enables C25 and C7 to maximize readout of the NLV peptide. However, C25 and C7 recognize NLV in distinct ways, as described below.
As depicted by the footprints of C25 and C7 on the pMHC surface (Fig. 3, A and B), both TCRs establish contacts with the N-terminal half of the NLV peptide mainly via the CDR1α and CDR3α loops, whereas the CDR3β loop mostly contacts the C-terminal half. C25 utilizes CDR1α and CDR2α to interact with the HLA-A2 α2 helix, whereas all three Vβ CDRs interact with the HLA-A2 α1 helix, with the majority of contacts (57 of 74 total) mediated by CDR2β (Fig. 3C). In contrast to C25, C7 interacts with HLA-A2 in a more Vα-dominant fashion such that the Vα CDRs contribute 74% of interactions with HLA-A2 (Fig. 3D). Thus, C25 and C7 engage HLA-A2 through different strategies.
FIGURE 3.

Comparison of TCR footprints on NLV·HLA-A2. A, positions of CDR loops of TCRs C25 and RA14 (PDB accession code 3GSN) (27) on NLV·HLA-A2 (top view). CDRs of C25 are shown as numbered red loops. CDRs of RA14 are green. HLA-A2 is gray. The NLV peptide is blue. B, positions of CDR loops of TCRs C7 and RA14 on NLV·HLA-A2. CRDs of C7 are orange. CDRs of RA14 are green. C, footprint of TCR C25 on NLV·HLA-A2. The top of the MHC molecule is depicted as a gray surface. The areas contacted by individual CDR loops are color-coded: green, CDR1α; red, CDR2α; blue, CDR3α; magenta, CDR1β; orange, CDR2β; cyan, CDR3β. D, footprint of TCR C7 on NLV·HLA-A2.
Interaction of TCR C25 with HLA-A2
The C25·NLV·HLA-A2 complex buries a total solvent-accessible surface of 1857 Å2, comparable with that in other TCR-pMHC complexes (15). The buried surface area on Vβ (516 Å2, 60%) is considerably greater than that on Vα (333 Å2, 40%). Such dominance by Vβ is unusual among TCR-pMHC class I complexes, in which Vα and Vβ typically contribute roughly equal buried surfaces, as in RA14·NLV·HLA-A2 (Vα: 52%; Vβ: 48%) (27) and C7·NLV·HLA-A2 (Vα: 55%; Vβ: 45%), or in which Vα dominates (15). Indeed, only three other TCR-pMHC class I complexes displaying a similar degree of Vβ dominance as C25·NLV·HLA-A2 have been reported, involving the HLA-A2-restricted TCR JM22 (67%) (22), the H-2Kb-restricted TCR BM3.3 (63%) (46), and the HLA-E-restricted TCR KK50.4 (61%) (47). Overall, Vβ makes 69 van der Waals contacts and 5 hydrogen bonds with HLA-A2, compared with only 15 van der Waals contacts and 3 hydrogen bonds by Vα. These contacts are mediated by 8 Vβ and 4 Vα residues and involve 15 MHC residues, of which 10 are contacted by RA14 and -7 by C7 (Table 2).
TABLE 2.
Interactions between TCR and MHC molecules in the C25·NLV·HLA-A2, RA14·NLV·HLA-A2, and C7·NLV·HLA-A2 complexes
| Hydrogen bonds | Van der Waals contacts | Hydrogen bonds | Van der Waals contacts | Hydrogen bonds | Van der Waals contacts | |
|---|---|---|---|---|---|---|
| HLA-A2 | C25 | C25 | RA14 | RA14 | C7 | C7 |
| E63H | E63H(Oδ2) N29α(Nδ2) | |||||
| R65H | R65H(O) Q55β(Nϵ2) | Q55β | T94α | |||
| K66H | N29α | K66H(Nζ) N29α(Oδ1) | N29α | |||
| A69H | Q55β | A69H(O) N96α(Nδ2) | G95α | G95α | ||
| N96α | N96α | |||||
| Q72H | Q72H(Oϵ1) N50β(Nδ2) | N50β | Q72H(Oϵ1) N96α(Nδ2) | Y48β | Q72H(Oϵ1) N96α(Nδ2) | N96α |
| Q72H(Nϵ2) Q55β(Oϵ1) | Q55β | Q72H(Nϵ2) Y48β(Oη) Q72H(Nϵ2) D56β(Oϵ1) | V50β | Q72H(Nϵ2) D56β(Oϵ1) | P55β | |
| T73H | P98β | N96α | ||||
| R75H | R75H(Nη1) E52β(Oϵ1) R75H(Nη2) E52β(Oϵ2) | Tyr-51β | I54β | |||
| Glu-52β | ||||||
| V76H | Val-30β | V50β | ||||
| I54β | ||||||
| K146H | T101β | K146H(Nζ) E30β(Oϵ2) | Y96β | T97β | ||
| W147H | T101β | |||||
| A149H | A149H(O) Y101β(Oη) | Y101β | ||||
| A150H | A150H(O) Y31α(OH) | Tyr-31α | Y101β | A150H(O) W100β(Nϵ1) | E101β | |
| T100β | ||||||
| T101β | ||||||
| H151H | H151H(O) Y31α(OH) | Tyr-31α | ||||
| V152H | T100β | W100β | ||||
| E154H | E154H(Oϵ1) T51α(Oγ1) | Leu-50α | L52α | |||
| Q155H | Leu-50α | Y31α | Y31α | |||
| T100β | T51α | T51α | ||||
| I100β | W100β | |||||
| A158H | G28α | L52α | ||||
| Y159H | T29α |
Of the total buried surface on HLA-A2, excluding NLV, CDR1α, CDR2α and CDR3α contribute 18%, 13%, and 6%, respectively, compared with 1%, 37%, and 28%, respectively, for CDR1β, CDR2β and CDR3β. Hence, CDR2β of TCR C25 accounts for more of the binding interface with MHC than any other CDR. The unusually large contribution of CDR2β to the C25-HLA-A2 interface (37%) is highlighted by a comparison with 34 other TCR-pMHC class I structures, in which CDR2β accounts for an average of only 12% of buried surface (15). In particular, CDR2β contributes 16% to the buried surface on HLA-A2 in the RA14·NLV·HLA-A2 complex (27) and 24% in the C7·NLV·HLA-A2 complex. Residues Asn-50β, Glu-52β, and Gln-55β of CDR2β form a dense network of five side-chain–side-chain hydrogen bonds linking C25 to residues Arg-65, Gln-72, and Arg-75 of the HLA-A2 α1 helix (Table 2; Fig. 4A). These polar interactions are reinforced by 56 hydrophobic contacts that further anchor CDR2β to the α1 helix.
FIGURE 4.

Interactions of TCR C25 with HLA-A2 and the NLV peptide. A, interactions between CDR2β (green) of C25 and the HLA-A2 α1 helix (orange). The side chains of contacting residues are drawn in stick representation with carbon atoms in green (CDR2β) or orange (HLA-A2), nitrogen atoms in blue, and oxygen atoms in red. Hydrogen bonds are indicated by red dashed lines. B, interactions between CDR1α (cyan) of C25 and the HLA-A2 α2 helix (orange). C, interactions between C25 and the NLV peptide (magenta). Peptide residues are identified by a one-letter amino acid designation followed by position (P) number. CDR3α (cyan) and CDR3β (green) form a pocket that accommodates the side chain of P5 Met. The sulfur atom of P5 Met is yellow. D, conformational stabilization of CDR3α of C25 by a dense network of eight intraloop hydrogen bonds.
Interestingly, the HLA-B-restricted EBV-specific TCR LC13 (23) utilizes nearly the same Vα/Vβ gene pair (TRAV26–2/TRBV7–8) as C25 (TRAV26–2/TRBV7–6), resulting in the same CDR1α, CDR2α, and CDR1β, and a very similar CDR2β. Moreover, LC13 mediates similar germ-line-encoded interactions with MHC as C25, in agreement with the hypothesis that the canonical diagonal docking orientation of TCR on MHC observed in TCR-pMHC complexes is at least partly the result of co-evolution of TCR and MHC molecules (48, 49). Thus, Tyr-31α, Gln-50β and Glu-52β of LC13 make hydrogen bonds with Arg-151H, Gln-72H, and Arg-75H, respectively, of HLA-B (23). The C25·NLV·HLA-A2 complex contains structurally equivalent hydrogen bonds: C25 Tyr-31α OH-O His-151H HLA-A2, C25 Asn-50β Nδ2-Oϵ1 Gln-72H HLA-A2, and C25 Glu-52β Oϵ2-Nη1 Arg-75H HLA-A2 (Table 2). However, LC13 and C25 have unrelated CDR3 sequences, which explains their different specificities.
TCR C25 contacts the HLA-A2 α2 helix through CDR1α and CDR2α (Table 2; Fig. 3A). In particular, the side chain of CDR1α Tyr-31 binds to a site formed by HLA-A2 α2 residues Ala-150H and His-151H (Fig. 4B), in a manner resembling that observed for other MHC class I-restricted TCRs bearing a CDR1α Tyr/Phe31 motif (49). In sharp contrast to RA14 (27) and C7, whose CDR3α and CDR3β loops interact extensively with HLA-A2, the CDR3 loops of C25 do not engage MHC, except for some minor contacts involving CDR3β (Table 2). Therefore, MHC recognition by C25 is almost exclusively germ-line-encoded.
Peptide Recognition by TCR C25
Excluding several contacts between CDR1α Thr30 and P4 Pro of NLV, all interactions between C25 and the CMV peptide are mediated by the somatically generated CDR3 loops, with CDR3α and CDR3β accounting for 16 and 29 contacts, respectively. Peptide specificity is conferred mainly by shape complementarity, since the C25-NLV interface includes only two hydrogen bonds: C25 Thr-100β Oγ1-O P5 Met and C25 Thr-100β Ν-O P6 Val (Table 3; Fig. 4C). C25 engages nearly all solvent-exposed NLV residues (P4 Pro, P5 Met, P6 Val, P7 Ala, P8 Thr), but the principal focus is on P5 Met, at the center of the MHC-bound peptide, which alone accounts for 50% of all contacts with TCR (Fig. 5A). The CDR3 loops of C25 form a hydrophobic pocket that accommodates the side chain of P5 Met (Fig. 4C). The conformation of CDR3α is stabilized by eight main-chain–side-chain hydrogen bonds within the Asp-91–Asn-92–Asn-93–Asn-94–Asp-95–Met-96 (DNNNDM) motif at the tip of this loop (Fig. 4D), suggesting very restricted CDR3α flexibility. The C25-NLV interface is more dominated by nonpolar interactions than the RA14-NLV or C7-NLV interface, both of which include multiple hydrogen bonds to P8 Thr at the NLV C terminus that are not made by C25 (Table 3, Fig. 5, B and C). Indeed, C25 makes only one van der Waals contact with P8 Thr and, unlike RA14 or C7, does not engage N-terminal NLV residues Asn P1 and Val P3 at all (Fig. 5A). Thus, C25 is more focused on the central portion of NLV, comprising residues P4-P7, than are RA14 and C7.
TABLE 3.
Interactions between TCR and NLV peptide in the C25·NLV·HLA-A2, RA14·NLV·HLA-A2, and C7·NLV·HLA-A2 complexes
| Hydrogen bonds | Van der Waals contacts | Hydrogen bonds | Van der Waals contacts | Hydrogen bonds | Van der Waals contacts | |
|---|---|---|---|---|---|---|
| NLV | C25 | C25 | RA14 | RA14 | C7 | C7 |
| N1P | N1P(Nδ2) N29α(Oδ1) | N29α | ||||
| V3P | Y31α | |||||
| P4P | T30α | N29α | P4P(O) G95α(N) | N29α | ||
| T91α | F30α | F30α | ||||
| N992α | Y31α | Y31α | ||||
| G95α | ||||||
| M5P | M5P(O) T100β(Oγ1) | N991α | M5P(Sδ) N96α(N) | Y31α | M5P(Sδ) N96α(N) | Y31α |
| N992α | G95α | I93α | ||||
| N993α | G98β | G95α | ||||
| Gly-99β | G99β | N96α | ||||
| T100β | I100β | W100β | ||||
| N102β | ||||||
| V6P | V6P(O) T100β(N) | G99β | G98β | W100β | ||
| T100β | ||||||
| A7P | T100β | T97β | Q98β | |||
| T101β | G98β | W100β | ||||
| G99β | ||||||
| T8P | T101β | T8P(N) T97β(O) | Q30β | T8P(N) Q98β(Oϵ1) | Q98β | |
| T8P(Oγ1) E30β(O ϵ1) | T97β | T8P(Oγ1) Q98β(Oϵ1) | ||||
| T8P(Oγ1) T97β(O) | ||||||
| T8P(Oγ1) T97β(N) |
FIGURE 5.
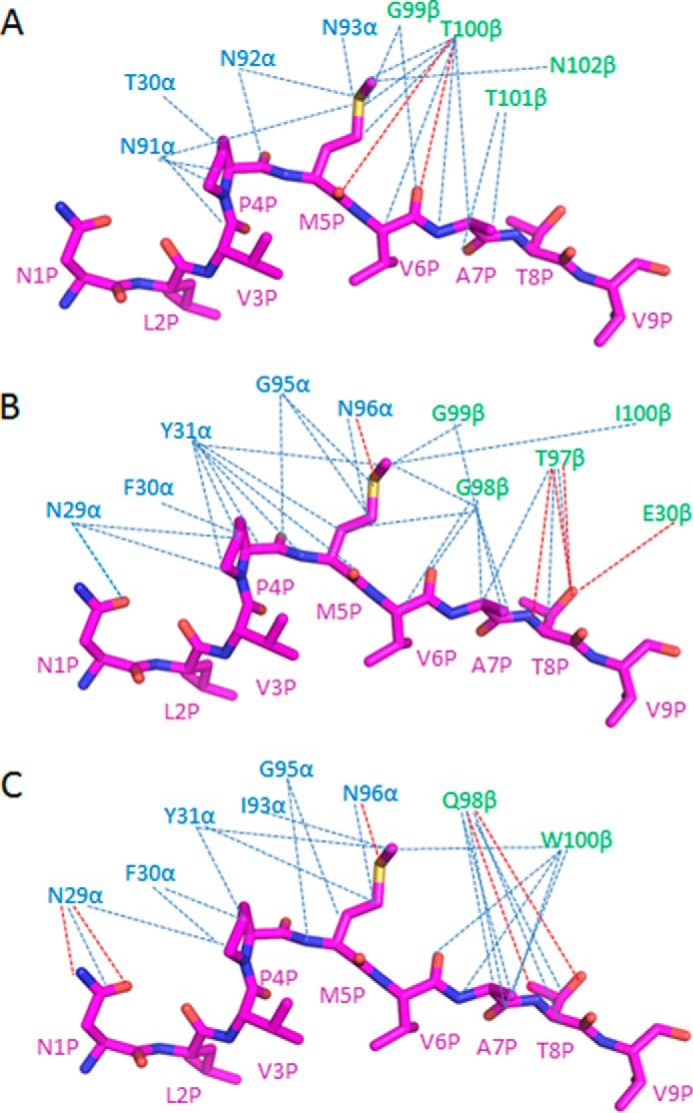
Comparison of interactions between TCRs and the NLV peptide. A, interactions between TCR C25 and NLV. Hydrogen bonds are red dotted lines; van der Waals contacts are blue dotted lines. B, interactions between TCR RA14 (27) and NLV. C, interactions between TCR C7 and NLV.
A recent analysis of human TCRs specific for NLV·HLA-A2 (297 sequences) revealed that the CDR3α sequence used by C25, DNNNDM, is a public CDR3α motif (XNNNDM, where X is variable) that is expressed by 11% of all responses across multiple donors (12). Importantly, this prevalence is second only to the 14% prevalence of the public CDR3α motif used by RA14 and G7 (GNQF). The XNNNDM public CDR3α motif may be associated with several different Vα gene segments, including TRAV26–2 (C25), TRAV18, and TRAV24 (12, 13, 16). Assuming that TCRs expressing the XNNNDM CDR3α motif dock similarly onto the NLV·HLA-A2 ligand, as suggested by the apparent rigidity of CDR3α in the C25·NLV·HLA-A2 structure (Fig. 4D), this diversity of Vα regions is explained, at least in part, by the paucity of contacts between the CDR1α and CDR2α loops of C25 and HLA-A2 and by the conservation of key contacting residues, notably CDR1α Tyr-31 (Fig. 4B), among these Vα segments.
To further examine the structural basis for TRAV18 and TRAV24 TCRs interacting with NLV·HLA-A2 with the public XNNNDM CDR3α motif, we modeled the NLV·HLA-A2 recognition for two previously described TCRs: RA11 (16), which utilizes TRAV18 and TRBV27 germ-line genes, and the CDR3α sequence of AFPYNNNDMR, and E4.1 (12), which utilizes TRAV24 and TRBV27 germ-line genes, and identical CDR3α and CDR3β sequences as RA11. Analysis of top-ranked flexible docking predictions identified E4.1-NLV·HLA-A2 and RA11-NLV·HLA-A2 complex models with shared pMHC docking orientations (Fig. 6A), which is a strong possibility given their shared β chain and CDR3α sequences. With crossing angles (45) of 51° and 50°, respectively, they are approximately halfway between the pMHC crossing angles of the RA14 and C25 TCRs. The modeled RA11 CDR3α loop (Fig. 6B) supports the conserved structure of the XNNNDM motif as well as its position over the NLV peptide with respect to the C25 TCR (2.0 Å backbone r.m.s.d. between NNNDM residues after superposition of pMHC) despite the different CDR3α loop length, TRAV germ-line, and TCR β chain. The RA11-NLV·HLA-A2 model also features a hydrophobic network including residues Tyr-31α, Tyr-100β, and P5 Met that is analogous to a hydrophobic region in the C7·NLV·HLA-A2 complex structure (Fig. 6C); this shows a possible structural basis for the public CDR3β sequence ASSLEGYTEAF and its interaction with NLV·HLA-A2. Although there is a minor shift in the docking position of the TCR α chain in the E4.1 model with respect to the C7 and RA11 TCR complexes with NLV·HLA-A2 (possibly due to the distinct CDR3α and β chain), key features of the TRAV24 germ-line-mediated pMHC interactions, such as Tyr-31α side chain position (Fig. 6D), are generally conserved.
FIGURE 6.

Models of the RA11 and E4.1 TCRs bound to NLV·HLA-A2. A, the RA11 (16) and E4.1 (12) TCR models are superposed with α chains in cyan, β chains in green, peptide in magenta sticks, and MHCs (visible as a single MHC as backbones are identical) in orange. B, the NNNDM CDR3α motif in the RA11-NLV·HLA-A2 model is compared with the corresponding motif from the C25·NLV·HLA-A2 crystal structure (backbone r.m.s.d. 2.0 Å; TCR and peptide from the C25·NLV·HLA-A2 complex are pink). Residues corresponding to the NNNDM motif are labeled and shown as sticks as well as P4 Pro of the NLV peptide. C, comparison of the hydrophobic region near NLV P5 Met between the modeled RA11 complex and C25·NLV·HLA-A2 (gray). Proximal hydrophobic TCR residues are labeled and shown as sticks. D, positioning of the CDR1α loop and Tyr-31α in the E4.1-NLV·HLA-A2 model (cyan) compared with the C25·NLV·HLA-A2 structure (gray) and the RA11-NLV·HLA-A2 model (yellow).
The CDR3β sequence of TCR C25 (SLAPGTTNEKL) is nearly identical to that of RA16 (SLAPGATNEKL) (16) and, therefore, defines a new public CDR3β motif. Hence, C25 belongs to the category of HCMV NLV-specific TCRs, comprising 38% of the total repertoire analyzed to date, which is characterized by usage of both CDR3α and CDR3β public motifs (12). In addition, C25 and RA16 use the same Vβ segment (TRBV7-6).
Interaction of TCR C7 with HLA
A2-The C7·NLV·HLA-A2 complex buries a total solvent-accessible surface of 2103 Å2, significantly more than the C25·NLV·HLA-A2 complex (1857 A2). Unlike the C25·NLV·HLA-A2 complex, in which Vβ is dominant, Vα of C25 contributes more than Vβ to the buried surface: 559 A2 (55%) versus 463 A2 (45%). TCR C7 uses the same Vα region as RA14 (TRAV24) and has a nearly identical CDR3α sequence, ITGNQF, compared with NTGNQF for RA14, a public CDR3α motif (12, 16). However, these two TCRs use unrelated Vβ regions (TRBV7–2 for C7; TRBV6–5 for RA14) and CDR3β sequences (SQTQLWETQ for C7; SPVTGGIYGY for RA14). Because the CDR3β sequence of C7 has not been identified as a public CDR3β motif, C7 belongs to the category of NLV-specific TCRs, comprising 34% of the total repertoire characterized so far, that uses either CDR3α or CDR3β public motifs but not both (12). As expected based on usage of the same Vα region, the overall docking topology of the C7·NLV·HLA-A2 complex is similar to that of the RA14·NLV·HLA-A2 complex (27), with crossing angles of TCR to pMHC (45) of 29° and 39°, respectively (Fig. 3B). However, the detailed interactions with HLA-A2 made by C7 and RA14 differ considerably, even for the shared Vα chain (Table 2).
The C7-HLA-A2 interaction involves all six CDRs except CDR1β (Table 2). Like RA14 (27), C7 employs CDR1α, CDR3α, and CDR2β to recognize the HLA-A2 α1 helix, with Vα contributing many more contacts than Vβ, as well as three out of four hydrogen bonds: C7 Asn-29α Nδ2-Oδ2 Glu-63H HLA-A2, C7 Asn-29α Oδ1-Nζ Lys-66H HLA-A2, and C7 Asn-96 Nδ2-Oϵ1 Gln-72H HLA-A2 (Fig. 7A). Although the first two of these hydrogen bonds are absent from the RA14·NLV·HLA-A2 structure (Table 2), both C7 and RA14 interact extensively with the HLA-A2 α1 helix through CDR3α Gly-95 and Asn-96, which constitute the core of the XTGNQF public CDR3α motif (12). However, the specific interactions made by these two residues differ in the C7·NLV·HLA-A2 and RA14·NLV·HLA-A2 complexes (Table 2) due to differences in CDR3α loop conformation, as described later. C7 engages the HLA-A2 α2 helix through CDR1α, CDR2α, and CDR3β. A side-chain–main-chain hydrogen bond (C7 Trp-100β Nϵ1-O Ala-150H HLA-A2), not present in the RA14·NLV·HLA-A2 complex (Table 2), provides additional stabilization (Fig. 7B).
FIGURE 7.

Interactions of TCR C7 with HLA-A2 and the NLV peptide. A, interactions of CDR1α, CDR3α, and CDR2β with the HLA-A2 α1 helix. The side chains of contacting residues are drawn in stick representation with carbon atoms in cyan (CDR1α and CDR3α), green (CDR2β), or orange (HLA-A2), nitrogen atoms in blue, and oxygen atoms in red. Hydrogen bonds are indicated by red dashed lines. B, interactions of CDR1α, CDR2α, and CDR3β with the HLA-A2 α2 helix. The side chains of contacting residues are drawn with carbon atoms in cyan (CDR1α and CDR2α), green (CDR3β), or orange (HLA-A2). C, interactions of CDR1α, CDR3α, and CDR3β with the NLV peptide. The side chains of contacting residues are drawn with carbon atoms in cyan (CDR1α and CDR3α), green (CDR3β), or magenta (NLV). D, close-up of interactions between C7 and P5 Met.
Peptide Recognition by TCR C7
TCR C7 binds the NLV peptide through CDR1α, CDR3α, and CDR3β via five hydrogen bonds (Table 3). Like RA14, C7 engages nearly all solvent-exposed NLV residues (P1 Asn, P4 Pro, P5 Met, P6 Val, P7 Ala, P8 Thr), thereby burying 314 Å2 of surface at the C7-NLV interface and enabling maximum readout of the peptide sequence (Fig. 7C). Unlike C25, C7 interacts extensively with both N- and C-terminal residues of NLV, especially P8 Thr (Fig. 5, A and C). P4 Pro is wedged between the side chains of CDR1α Asn-29 and Tyr-31, with which it establishes multiple hydrophobic contacts (Fig. 7C). The side chain of P5 Met alone accounts for 36% of all contacts with C7, mainly through CDR1α and CDR3α (Fig. 7D). In addition to extensive hydrophobic interactions with CDR1α Tyr-31, CDR3α Asn-96, and CDR3β Trp-100, P5 Met forms a hydrogen bond through its sulfur atom with the main-chain nitrogen of CDR3α Asn-96. This hydrogen bond is conserved in the RA14·NLV·HLA-A2 complex (Table 3). Four additional hydrogen bonds reinforce the C7-NLV interaction: C7 Asn-29α Oδ1-Nδ2 P1 Asn, C7 Gly-95α N-O P4 Pro, C7 Gln-98β Oϵ1-N P8 Thr, and C7 Gln-98β Oϵ1-Oγ1 P8 Thr. Therefore, although P5 Met appears to be the most critical peptide residue for TCR recognition, P1 Asn, P4 Pro, and P8 Thr also have important roles.
Influence of CDR3β on CDR3α Loop Conformation in TCR C7
Because TCRs C7 and RA14 employ identical Vα chains (except for a single amino acid difference, CDR3α Ile/Asn-93) to bind identical pMHC ligands, one might have expected the Vα CDR loops to have the same, or at least very similar, conformations in the C7·NLV·HLA-A2 and RA14·NLV·HLA-A2 structures. Indeed, CDR1α and CDR2α display nearly the same conformation in the two complexes: r.m.s.d. in α-carbon positions of 1.0 Å and 1.3 Å for residues SSNFY of CDR1α and TLNGD of CDR2α, respectively (Fig. 3B). By contrast, CDR3α adopts different conformations in TCRs C7 and RA14, with an r.m.s.d. in α-carbon positions of 2.3 Å for residues TGNQ. As a result, CDR3α engages pMHC through a somewhat different set of contacts in the C7·NLV·HLA-A2 and RA14·NLV·HLA-A2 complexes (Tables 2 and 3).
The different conformations of CDR3α observed in C7 and RA14 are attributable to the CDR3β loops of these TCRs, which differ in both sequence and length (SQTQLWETQ for C7; SPVTGGIYGY for RA14). These structural differences in CDR3β are transmitted to CDR3α via interactions between these loops in the TCR binding site. In RA14, the tip of CDR3β points toward CDR3α, with which it makes several van der Waals contacts and a main-chain–side-chain hydrogen bond (CDR3β Gly-98 N-Oδ1 Asn-96 CDR3α) (Fig. 8). These interactions, which are absent in C7 due to an unrelated CDR3β structure, effectively draw CDR3α toward CDR3β in RA14, resulting in displacement of 2.8 Å in the α-carbon position of CDR3α Asn-96 relative to its position in C7.
FIGURE 8.
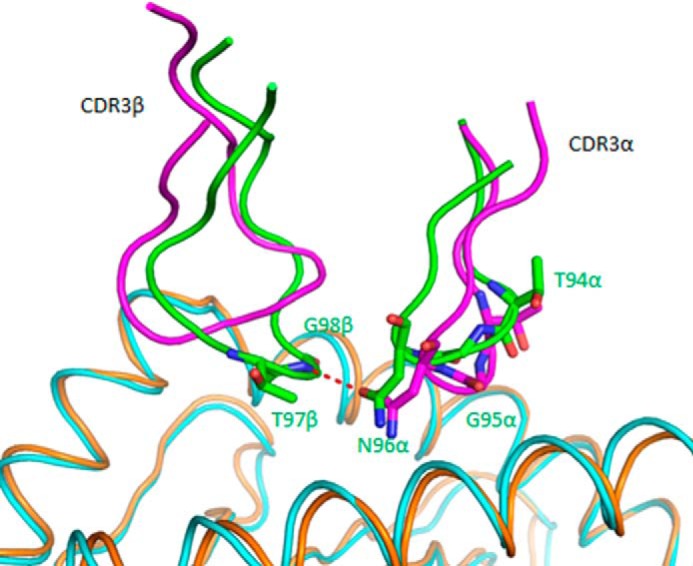
Influence of CDR3β on the conformation of CDR3α in TCR C7. Conformation of CDR3α and CDR3β loops in superposed C7·NLV·HLA-A2 and RA14·NLV·HLA-A2 complexes (pink, TCR C7; cyan, C7-bound NLV·HLA-A2; green, TCR RA14; orange, RA14-bound NLV·HLA-A2). In RA14, but not in C7, the tip of CDR3β points toward CDR3α, resulting in interactions that draw CDR3α close to CDR3β in RA14. These interactions are absent in C7, whose CDR3β sequence (SQTQLWETQ) is unrelated to that of RA14 (SPVTGGIYGY).
Discussion
Previous structural studies of TCR recognition of immunodominant viral epitopes presented by MHC class I molecules have focused mainly on EBV (23–26). Most notably, a comparison of three public TCRs in complex with a bulged EBV peptide presented by HLA-B8 revealed two distinct binding modes: one in which the TCR straddles the bulged peptide but makes few contacts with MHC and one in which the TCR is positioned toward the N-terminal end of the HLA-B8 peptide binding groove, thereby bypassing the bulged peptide (26). By contrast to EBV, knowledge of TCR recognition of CMV has so far been limited to the RA14·NLV·HLA-A2 complex (27). Structural information on how different TCRs are able to bind the same pMHC ligand is particularly relevant in light of a growing appreciation for the surprising diversity of public TCR responses to certain viral epitopes revealed by powerful new technologies for T cell repertoire analysis (9–14, 21).
In the case of the CMV NLV-specific TCR response, seven public CDR3α and seven public CDR3β motifs have so far been identified (12, 13, 16), including one additional public CDR3β motif reported here. Although public CDR3α motifs often pair with public CDR3β motifs, pairings between public and private CDR3α/CDR3β motifs occur with equal frequency. Furthermore, even among NLV-specific TCRs expressing dual public CDR3α/CDR3β motifs, the same public CDR3α motif may pair with different public CDR3β motifs (and the reverse). Importantly, this striking flexibility of CDR3α/CDR3β pairing is not unique to NLV-specific TCRs, as it has now also been documented among TCRs specific for the influenza NP366 epitope (50), which had previously been thought to elicit a narrow TCR repertoire comprising only a few clonotypes (51).
A comparison of the C7·NLV·HLA-A2 and RA14·NLV·HLA-A2 (27) structures illustrates how the same public CDR3α motif (XTGNQF) can pair with two unrelated CDR3β motifs, one private (SQTQLWETQ for C7) and the other public (SPVTGGIYGY for RA14) yet still maintain high affinity recognition of NLV·HLA-A2 (KD = 5.1 μm for C7; 28 μm for RA14). We have shown that CDR3α adopts different conformations in C7 and RA14 to accommodate large structural differences in CDR3β, which abuts CDR3α in the TCR binding site. Nevertheless, CDR3α maintains key interactions with the HLA-A2 α1 helix and P5 Met of NLV via Gly-95α and Asn-96α, which form the core of the XTGNQF CDR3α motif. As a result, the common Vα domains of C7 and RA14 are able to dock in almost the same way on NLV·HLA-A2 despite their association with totally different Vβ domains in the TCR heterodimer. Similar considerations likely apply to the accommodation of various other CDR3β sequences that have been found to pair with the XTGNQF CDR3α motif (12, 13, 16). More generally, our study shows how the malleability of protein-protein interfaces permits preservation of function (in this case, pMHC specificity) through accommodation of structural changes in the binding partners.
TCR C25 employs a different solution to binding NLV·HLA-A2 than C7, yet one giving an essentially identical KD: 4.7 μm for C25 versus 5.1 μm for C7. These relatively high affinities support an antigen-driven selection process for both public TCRs. However, unlike C7, whose Vα domain contributes more buried surface to the interface with pMHC than Vβ (55 and 45%, respectively), the opposite is true for C25 (Vα, 40%; Vβ, 60%). In addition, the C25·NLV·HLA-A2 and C7·NLV·HLA-A2 complexes are characterized by crossing angles of 61° and 29°, respectively, resulting in divergent TCR footprints on pMHC. The more acute crossing angle of C7 enables this TCR to contact both N and C termini of the NLV peptide, whereas C25 is decidedly more focused on the peptide center, primarily P5 Met.
The public CDR3α motif of TCR C25 (XNNNDM) has been shown to pair with multiple public and private CDR3β motifs that vary in both sequence and length, including SISDLAKNIQ, QLQGHTEA, SVSDVANTEA, SLEGYTEA, and SLAPGATNEKL (12, 13, 16). In the C25·NLV·HLA-A2 complex, CDR3α is rigidified by eight intraloop hydrogen bonds, making it improbable that this loop can alter its conformation substantially to accommodate different CDR3β structures, as observed for CDR3α of C7. Instead, CDR3β loops must likely adapt to CDR3α in NLV-specific TCRs bearing the XNNNDM CDR3α motif. It is intriguing that this motif occurs in the context of varying CDR3α lengths (e.g. DNNNDM for C25 and PYNNNDM for RA11 and E4.1), though our modeling of the RA11-NLV·HLA-A2 and E4.1-NLV·HLA-A2 complexes demonstrates that, in principle, the structure and pMHC interactions of this motif can be conserved.
CMV was recently shown to boost the immune response of young, healthy individuals to influenza (52). Similarly, mice infected with CMV were found to be resistant to infection with the bacterial pathogens Listeria monocytogenes and Yersinia pestis (53) These and related observations have led to the hypothesis that the ubiquity of CMV infection in human and many other species reflects a mutualistic symbiosis that confers benefits on the host (54). Although the mechanisms underlying CMV-mediated cross-protection are unclear, one possibility is that TCRs specific for NLV or other CMV epitopes cross-react with epitopes from other pathogens (52). Indeed, a degree of cross-reactivity of CD8+ T cell epitopes between CMV and influenza has been reported (55). The known promiscuity of TCRs (56), whereby a single receptor can recognize many different peptides, coupled with the structural diversity of CMV NLV-specific TCRs described here, further support the idea of cross-reactivity as a possible mechanism to help explain CMV-mediated heterologous immunity to influenza and other microbial pathogens.
Author Contributions
X. Y. and M. G. determined the crystal structures. G. C. isolated TCR genes. B. G. P. carried out molecular modeling. J. L. assisted with x-ray data collection. X. Y., M. G., G. C., B. G. P., N. W. and R. A. M. analyzed data and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank the National Institute on Aging clinic core for collecting apheresis pack of normal donors. We also thank Peter D. Sun for generous assistance with x-ray data collection. The x-ray SER-CAT beamlines at the Advanced Photon Source were supported by the United States Department of Energy, Basic Energy Sciences, Office of Science, under Contract W-31-109-Eng-38.
This work was supported by National Institutes of Health (NIH) Grant AI036900 (to R. A. M.) and also by the Intramural Research Program NIH (NIA). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables 1 and 2.
The atomic coordinates and structure factors (codes 5D2L and 5D2N) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CMV
- cytomegalovirus
- CTL
- cytotoxic T lymphocyte
- TCR
- T cell receptor
- NLV
- NLVPMVATV epitope of CMV
- pMHC
- peptide MHC
- CDR
- complementarity-determining region
- Vα
- variable α
- Vβ
- variable β
- EBV
- Epstein-Barr virus
- r.m.s.d.
- root mean square difference.
References
- 1.Sissons J. G., Bain M., and Wills M. R. (2002) Latency and reactivation of human cytomegalovirus. J. Infect. 44, 73–77 [DOI] [PubMed] [Google Scholar]
- 2.Gandhi M. K., and Khanna R. (2004) Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect. Dis. 4, 725–738 [DOI] [PubMed] [Google Scholar]
- 3.Dollard S. C., Grosse S. D., and Ross D. S. (2007) New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 17, 355–363 [DOI] [PubMed] [Google Scholar]
- 4.Quinnan G. V. Jr., Burns W. H., Kirmani N., Rook A. H., Manischewitz J., Jackson L., Santos G. W., and Saral R. (1984) HLA-restricted cytotoxic T lymphocytes are an early immune response and important defense mechanism in cytomegalovirus infections. Rev. Infect. Dis. 6, 156–163 [DOI] [PubMed] [Google Scholar]
- 5.Borysiewicz L. K., Hickling J. K., Graham S., Sinclair J., Cranage M. P., Smith G. L., and Sissons J. G. (1988) Human cytomegalovirus-specific cytotoxic T cells. Relative frequency of stage-specific CTL recognizing the 72-kDa immediate early protein and glycoprotein B expressed by recombinant vaccinia viruses. J. Exp. Med. 168, 919–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wills M. R., Carmichael A. J., Mynard K., Jin X., Weekes M. P., Plachter B., and Sissons J. G. (1996) The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol. 70, 7569–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peggs K., Verfuerth S., Pizzey A., Ainsworth J., Moss P., and Mackinnon S. (2002) Characterization of human cytomegalovirus peptide-specific CD8+ T-cell repertoire diversity following in vitro restimulation by antigen-pulsed dendritic cells. Blood 99, 213–223 [DOI] [PubMed] [Google Scholar]
- 8.Arstila T. P., Casrouge A., Baron V., Even J., Kanellopoulos J., and Kourilsky P. (1999) A direct estimate of the human αβ T cell receptor diversity. Science 286, 958–961 [DOI] [PubMed] [Google Scholar]
- 9.Klarenbeek P. L., Remmerswaal E. B., ten Berge I. J., Doorenspleet M. E., van Schaik B. D., Esveldt R. E., Koch S. D., ten Brinke A., van Kampen A. H., Bemelman F. J., Tak P. P., Baas F., de Vries N., and van Lier R. A. (2012) Deep sequencing of antiviral T-cell responses to HCMV and EBV in humans reveals a stable repertoire that is maintained for many years. PLoS Pathog. 8, e1002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suessmuth Y., Mukherjee R., Watkins B., Koura D. T., Finstermeier K., Desmarais C., Stempora L., Horan J. T., Langston A., Qayed M., Khoury H. J., Grizzle A., Cheeseman J. A., Conger J. A., Robertson J., Garrett A., Kirk A. D., Waller E. K., Blazar B. R., Mehta A. K., Robins H. S., and Kean L. S. (2015) CMV reactivation drives posttransplant T-cell reconstitution and results in defects in the underlying TCRβ repertoire. Blood 125, 3835–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miconnet I. (2012) Probing the T-cell receptor repertoire with deep sequencing. Curr. Opin. HIV AIDS 7, 64–70 [DOI] [PubMed] [Google Scholar]
- 12.Wang G. C., Dash P., McCullers J. A., Doherty P. C., and Thomas P. G. (2012) T cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl. Med. 4, 128ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen T. H., Rowntree L. C., Pellicci D. G., Bird N. L., Handel A., Kjer-Nielsen L., Kedzierska K., Kotsimbos T. C., and Mifsud N. A. (2014) Recognition of distinct cross-reactive virus-specific CD8+ T cells reveals a unique TCR signature in a clinical setting. J. Immunol. 192, 5039–5049 [DOI] [PubMed] [Google Scholar]
- 14.Smith C., Gras S., Brennan R. M., Bird N. L., Valkenburg S. A., Twist K. A., Burrows J. M., Miles J. J., Chambers D., Bell S., Campbell S., Kedzierska K., Burrows S. R., Rossjohn J., and Khanna R. (2014) Molecular imprint of exposure to naturally occurring genetic variants of human cytomegalovirus on the T cell repertoire. Sci. Rep. 4, 3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossjohn J., Gras S., Miles J. J., Turner S. J., Godfrey D. I., and McCluskey J. (2015) T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 [DOI] [PubMed] [Google Scholar]
- 16.Trautmann L., Rimbert M., Echasserieau K., Saulquin X., Neveu B., Dechanet J., Cerundolo V., and Bonneville M. (2005) Selection of T cell clones expressing high-affinity TCRs within cytomegalovirus-specific CD8 T cell responses. J. Immunol. 175, 6123–6132 [DOI] [PubMed] [Google Scholar]
- 17.Cukalac T., Chadderton J., Handel A., Doherty P. C., Turner S. J., Thomas P. G., and La Gruta N. L. (2014) Reproducible selection of high avidity CD8+ T-cell clones following secondary acute virus infection. Proc. Natl. Acad. Sci. U.S.A. 111, 1485–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Gruta N. L., and Thomas P. G. (2013) Interrogating the relationship between naïve and immune antiviral T cell repertoires. Curr. Opin. Virol. 3, 447–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyer-Olson D., Shoukry N. H., Brady K. W., Kim H., Olson D. P., Hartman K., Shintani A. K., Walker C. M., and Kalams S. A. (2004) Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape. J. Exp. Med. 200, 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faroudi M., Utzny C., Salio M., Cerundolo V., Guiraud M., Müller S., and Valitutti S. (2003) Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc. Natl. Acad. Sci. U.S.A. 100, 14145–14150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Ye C., Ji G., and Han J. (2012) Determinants of public T cell responses. Cell Res. 22, 33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart-Jones G. B., McMichael A. J., Bell J. I., Stuart D. I., and Jones E. Y. (2003) A structural basis for immunodominant human T cell receptor recognition. Nat. Immunol. 4, 657–663 [DOI] [PubMed] [Google Scholar]
- 23.Kjer-Nielsen L., Clements C. S., Purcell A. W., Brooks A. G., Whisstock J. C., Burrows S. R., McCluskey J., and Rossjohn J. (2003) A structure basis for the selection of dominant αβ T cell receptors in antiviral immunity. Immunity 18, 53–64 [DOI] [PubMed] [Google Scholar]
- 24.Miles J. J., Bulek A. M., Cole D. K., Gostick E., Schauenburg A. J., Dolton G., Venturi V., Davenport M. P., Tan M. P., Burrows S. R., Wooldridge L., Price D. A., Rizkallah P. J., and Sewell A. K. (2010) Genetic and structural basis for selection of a ubiquitous T cell receptor deployed in Epstein-Barr virus infection. PLoS Pathog. 6, e1001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gras S., Wilmann P. G., Chen Z., Halim H., Liu Y. C., Kjer-Nielsen L., Purcell A. W., Burrows S. R., McCluskey J., and Rossjohn J. (2012) A structural basis for varied αβ TCR usage against an immunodominant EBV antigen restricted to a HLA-B8 molecule. J. Immunol. 188, 311–321 [DOI] [PubMed] [Google Scholar]
- 26.Liu Y. C., Miles J. J., Neller M. A., Gostick E., Price D. A., Purcell A. W., McCluskey J., Burrows S. R., Rossjohn J., and Gras S. (2013) Highly divergent T-cell receptor binding modes underlie specific recognition of a bulged viral peptide bound to a human leukocyte antigen class I molecule. J. Biol. Chem. 288, 15442–15454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gras S., Saulquin X., Reiser J. B., Debeaupuis E., Echasserieau K., Kissenpfennig A., Legoux F., Chouquet A., Le Gorrec M., Machillot P., Neveu B., Thielens N., Malissen B., Bonneville M., and Housset D. (2009) Structural bases for the affinity-driven selection of a public TCR against a dominant human cytomegalovirus epitope. J. Immunol. 183, 430–437 [DOI] [PubMed] [Google Scholar]
- 28.Araki Y., Wang Z., Zang C., Wood W. H. 3rd, Schones D., Cui K., Roh T. Y., Lhotsky B., Wersto R. P., Peng W., Becker K. G., Zhao K., and Weng N. P. (2009) Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 30, 912–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oelke M., Maus M. V., Didiano D., June C. H., Mackensen A., and Schneck J. P. (2003) Ex vivo induction and expansion of antigen-specific cytotoxic T cells by HLA-Ig-coated artificial antigen-presenting cells. Nat. Med. 9, 619–624 [DOI] [PubMed] [Google Scholar]
- 30.Najarro K., Nguyen H., Chen G., Xu M., Alcorta S., Yao X., Zukley L., Metter E. J., Truong T., Lin Y., Li H., Oelke M., Xu X., Ling S. M., Longo D. L., Schneck J., Leng S., Ferrucci L., and Weng N. P. (2015) Telomere length as an indicator of the robustness of B- and T-cell response to influenza in older adults. J. Infect. Dis. 212, 1261–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefranc M. P., Giudicelli V., Ginestoux C., Bodmer J., Müller W., Bontrop R., Lemaitre M., Malik A., Barbié V., and Chaume D. (1999) IMGT, the International ImMunoGeneTics database. Nucleic Acids Res. 27, 209–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulter J. M., Glick M., Todorov P. T., Baston E., Sami M., Rizkallah P., and Jakobsen B. K. (2003) Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 16, 707–711 [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 34.Storoni L. C., McCoy A. J., and Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 [DOI] [PubMed] [Google Scholar]
- 35.Petersen J., Montserrat V., Mujico J. R., Loh K. L., Beringer D. X., van Lummel M., Thompson A., Mearin M. L., Schweizer J., Kooy-Winkelaar Y., van Bergen J., Drijfhout J. W., Kan W. T., La Gruta N. L., Anderson R. P., Reid H. H., Koning F., and Rossjohn J. (2014) T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat. Struct. Mol. Biol. 21, 480–488 [DOI] [PubMed] [Google Scholar]
- 36.Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laskowski R. A., MacArthur M. W., Moss D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereo chemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 39.Klausen M. S., Anderson M. V., Jespersen M. C., Nielsen M., and Marcatili P. (2015) LYRA, a webserver for lymphocyte receptor structural modeling. Nucleic Acids Res. 43, W349–W355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webb B., and Sali A. (2014) Protein structure modeling with MODELLER. Methods Mol. Biol. 1137, 1–15 [DOI] [PubMed] [Google Scholar]
- 41.North B., Lehmann A., and Dunbrack R. L. Jr. (2011) A new clustering of antibody CDR loop conformations. J. Mol. Biol. 406, 228–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pierce B. G., and Weng Z. (2013) A flexible docking approach for prediction of T cell receptor-peptide-MHC complexes. Protein Sci. 22, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce B., and Weng Z. (2008) A combination of rescoring and refinement significantly improves protein docking performance. Proteins 72, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin Y., Li Y., and Mariuzza R. A. (2012) Structural basis for self-recognition by autoimmune T-cell receptors. Immunol. Rev. 250, 32–48 [DOI] [PubMed] [Google Scholar]
- 45.Rudolph M. G., Stanfield R. L., and Wilson I. A. (2006) How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24, 419–466 [DOI] [PubMed] [Google Scholar]
- 46.Reiser J.-B., Darnault C., Guimezanes A., Grégoire C., Mosser T., Schmitt-Verhulst A. M., Fontecilla-Camps J. C., Mazza G., Malissen B., and Housset D. (2000) Crystal structure of a T cell receptor bound to an allogeneic MHC molecule. Nat. Immunol. 1, 291–297 [DOI] [PubMed] [Google Scholar]
- 47.Hoare H. L., Sullivan L. C., Pietra G., Clements C. S., Lee E. J., Ely L. K., Beddoe T., Falco M., Kjer-Nielsen L., Reid H. H., McCluskey J., Moretta L., Rossjohn J., and Brooks A. G. (2006) Structural basis for a major histocompatibility complex class Ib-restricted T cell response. Nat. Immunol. 7, 256–264 [DOI] [PubMed] [Google Scholar]
- 48.Feng D., Bond C. J., Ely L. K., Maynard J., and Garcia K. C. (2007) Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction “codon.” Nat. Immunol. 8, 975–983 [DOI] [PubMed] [Google Scholar]
- 49.Marrack P., Scott-Browne J. P., Dai S., Gapin L., and Kappler J. W. (2008) Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu. Rev. Immunol. 26, 171–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cukalac T., Kan W. T., Dash P., Guan J., Quinn K. M., Gras S., Thomas P. G., and La Gruta N. L. (2015) Paired TCRαβ analysis of virus-specific CD8+ T cells exposes diversity in a previously defined “narrow” repertoire. Immunol. Cell Biol. 10.1038/icb.2015.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kedzierska K., Turner S. J., and Doherty P. C. (2004) Conserved T cell receptor usage in primary and recall responses to an immunodominant influenza virus nucleoprotein epitope. Proc. Natl. Acad. Sci. U.S.A. 101, 4942–4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furman D., Jojic V., Sharma S., Shen-Orr S. S., Angel C. J., Onengut-Gumuscu S., Kidd B. A., Maecker H. T., Concannon P., Dekker C. L., Thomas P. G., and Davis M. M. (2015) Cytomegalovirus infection enhances the immune response to influenza. Sci. Transl. Med. 7, 281ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barton E. S., White D. W, Cathelyn J. S., Brett-McClellan K. A., Engle M., Diamond M. S., Miller V. L., and Virgin H. W. (2007) Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447, 326–329 [DOI] [PubMed] [Google Scholar]
- 54.Roossinck M. J. (2011) The good viruses: viral mutualistic symbioses. Nat. Rev. Microbiol. 9, 99–108 [DOI] [PubMed] [Google Scholar]
- 55.Welsh R. M., Che J. W., Brehm M. A., and Selin L. K. (2010) Heterologous immunity between viruses. Immunol. Rev. 235, 244–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wooldridge L., Ekeruche-Makinde J., van den Berg H. A., Skowera A., Miles J. J., Tan M. P., Dolton G., Clement M., Llewellyn-Lacey S., Price D. A., Peakman M., and Sewell A. K. (2012) A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 287, 1168–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.