

Abstract
Camurati–Engelmann’s disease (CED) is a rare disorder worldwide with just over 200 cases reported. No case of CED has been reported in Afghanistan till date. Most patients of CED (also known as progressive diaphyseal dysplasia and oeteopathica hyperostotica multiplex infantalis) present with extremity pain, muscle weakness, and waddling gait. It tends to be bilateral and symmetrical and can affect any bone but has greater affinity for long bones e.g., humerus, femur, tibia, ulna, and radius. Other common sites include skull and pelvis. Symptomatology relating to cranial nerve impingement is secondary to amorphous increase in the density of skull bones resulting in stenosis of various foramina/spaces within skull.
Case presentation
A 9-year-old boy presented to his clinician with a history of gradual loss of hearing and vision in both eyes over the preceding 7 months. He was also complaining of pain localized to his calf and thigh muscles. There was no history of joint swelling. He had been previously well with no relevant medical history.
Clinical examination revealed the child to be pale, lethargic, and having a waddling gait.
Based on the clinical history, his clinician referred him for magnetic resonance imaging of the brain to exclude any space occupying lesion which may be the cause of his hearing and visual loss.
His magnetic resonance imaging revealed marked hyperostosis of the skull vault bones and the base of skull which is appearing hypointense on T2-weighted images (Fig. 1).
Fig. 1.
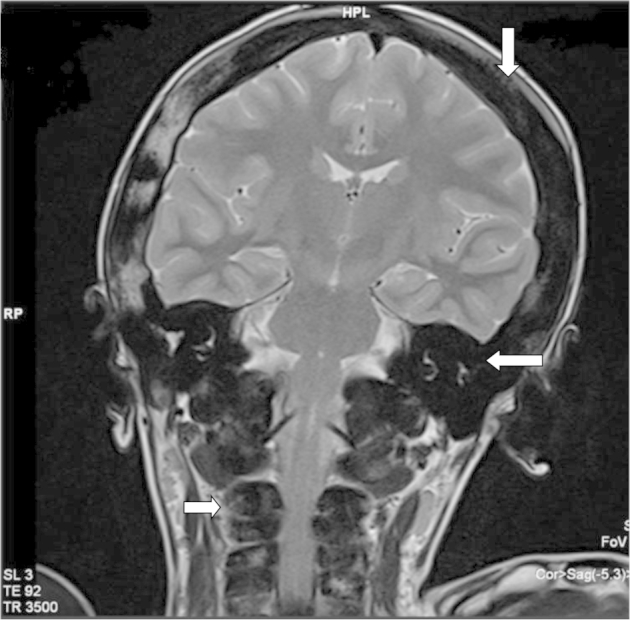
T2-weighted coronal image of the brain revealed marked hyperostosis of the entire calvarium (arrow), temporal bone (arrow), and visualized cervical vertebra (arrow).
Marked narrowing of bilateral optic nerve canal due to hyperostosis. The optic nerves can be seen compressed in the region of bilateral optic canals (Fig. 2).
Fig. 2.
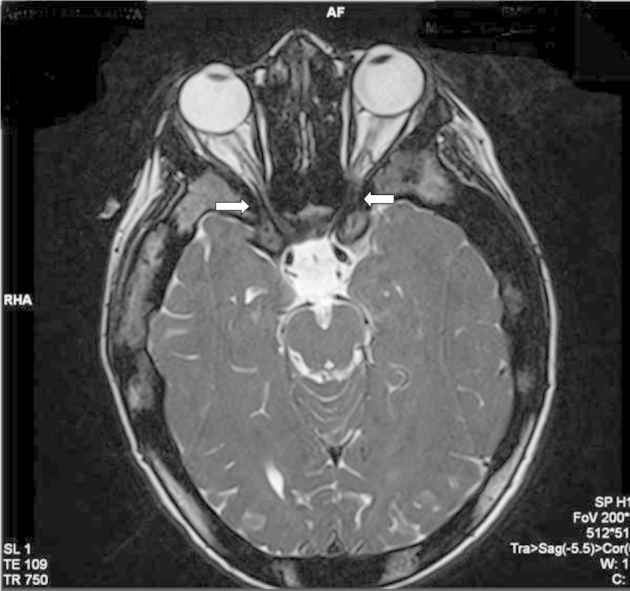
T2-weighted axial image of the brain showing marked narrowing of bilateral optic canals (arrows) due to excessive hyperostosis.
Sclerosis is also seen in the region of petrous part of temporal bone which may be the reason for hearing loss.
Sclerosis of the visualized upper cervical vertebrae also seen.
Findings were suggestive of Camurati–Engelmann's disease (CED). After discussion with the primary physician radiograph of left femur was performed which revealed diffuse diaphyseal hyperostosis (Fig. 3).
Fig. 3.

Radiograph of right femur anterioposterior view revealed diffuse diaphyseal hyperostosis.
Discussion
Camurati Engelmann's disease (CED) also known as progressive diaphyseal dysplasia (PDD) or oeteopathica hyperostotica multiplex infantalis is a rare disorder caused by domain specific mutations in transforming growth factor (TGF)-β1 gene on chromosome 19q13.1 [1], [2], [3]. Its pattern of inheritance is autosomal dominant with variable penetrance [4]. It tends to be bilateral and symmetrical and can affect any bone but has greater affinity for long bones e.g., humerus, femur, tibia, ulna and radius. Other common sites include skull and pelvis [5], [6].
Medical treatment of bony abnormalities with glucocorticoids (prednisolone) has been tried with success in patients of CED [7], [8] but failure of treatment with bisphosphonates has also been reported [9].
Moumoulidis et al. [10] described otological manifestation in CED as a result of narrowing of internal auditory canals caused by bony encroachment on nerves and vessels. This results in retrocochlear hearing loss, meanwhile, the narrowing of tympanic cavities and Eustachian tube may precipitate middle ear infections and/or adhesions of osciles to the tympanic walls [11].
The incidence of hearing impairment in the patients of CED is estimated to be about 18% [12]. Cochlear implantation for auditory rehabilitation in patients of CED has been described by Friedland et al. [11] and Tibesar [13]. Surgical decompression of internal auditory meatus to relieve vertigo in case of CED has also been successfully done [14].
Visual manifestation of CED can be considered as late sequelae of disease process. Wright M et al. [15] reported that visual symptoms may take some time to develop.
Initial symptom may only be of exophthalmos or headache due to raised intracranial pressure, and a decrease in visual acuity may be the presentation later in the disease process [15]. It may well be the chief presenting symptoms as with this case.
Visual symptoms are not limited to CED alone, other conditions of primary bone disorders such as craniometaphyseal dysplasia, osteopetrosis, and aneurysmal bone cyst, all of which have been reported to cause progressive stenosis of optic canals [16], [17], [18]. The etiology of visual symptoms in some case have been presumed to have occurred solely due to compressive optic neuropathy[19], [20]; however, symptoms can also be because of papilloedema secondary to raised intracranial pressure [15].
In our case, visual symptoms were because of marked narrowing of bilateral optic nerve canal due to hyperostosis. The optic nerves can be seen compressed in the region of bilateral optic canals (Fig 2).
Treatment rationale will be different as papilloedema has been treated with diuretics (acetazolamide) [15] and surgical decompression reported as the best management in cases of compressive optic neuropathy. However, in some cases and overlap of both mechanisms may be encountered. It is suggested that some type of cranial vault expansion procedure, possibly combined with an orbital decompression may be beneficial as well [15].
Here it should be kept in mind that, surgery may not be curative and reconstitution of surgically resected bone may be encountered [19], [21].
In conclusion, the report describes the first case ever of CED to be reported from Afghanistan and its presenting symptomatology.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
References
- 1.Kinoshita A., Saito T., Tomita H., Makita Y., Yoshida K., Ghadami M. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000;26(1):19–20. doi: 10.1038/79128. [DOI] [PubMed] [Google Scholar]
- 2.Simsek S., Janssens K., Kwee M.L., Van Hul W., Veenstra J., Netelenbos J.C. Camurati–Engelmann disease (progressive diaphyseal dysplasia) in a Moroccan family. Osteoporos Int. 2005;16(9):1167–1170. doi: 10.1007/s00198-005-1896-2. [DOI] [PubMed] [Google Scholar]
- 3.Wu S., Liang S., Yan Y., Wang Y., Li F., Deng Y. A novel mutation of TGFβ1 in a Chinese family with Camurati–Engelmann disease. Bone. 2007;40(6):1630–1634. doi: 10.1016/j.bone.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 4.Hundley J.D., Wilson F.C. Progressive diaphyseal dysplasia review of the literature and report of seven cases in one family. J Bone Joint Surg Am. 1973;55(3):461–474. [PubMed] [Google Scholar]
- 5.Camurati M. Di un raro caso di osteite simmetrica ereditaria degli arti inferiori. Chir Organi Mov. 1922;6:662–665. [Google Scholar]
- 6.Engelmann G. Ein Fall von Osteopathia hyperostotica (sclerotisans) multiplex infantilis. Fortschr Roentgenstr. 1929;39:1101–1106. [Google Scholar]
- 7.Heymans O., Gebhart M., Alexiou J., Sokolow Y. Camurati-Engelmann disease. Effects of corticosteroids. Acta clinica Belgica. 1998;53(3):189–192. [PubMed] [Google Scholar]
- 8.Inaoka T., Shuke N., Sato J., Ishikawa Y., Takahashi K., Aburano T. Scintigraphic evaluation of pamidronate and corticosteroid therapy in a patient with progressive diaphyseal dysplasia (Camurati-Engelmann disease) Clin Nucl Med. 2001;26(8):680–682. doi: 10.1097/00003072-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Castro G.R., Appenzeller S., Marques-Neto J.F., Bértolo M.B., Samara A.M., Coimbra I. Camurati-Engelmann disease: failure of response to bisphosphonates: report of two cases. Clin Rheumatol. 2005;24(4):398–401. doi: 10.1007/s10067-004-1056-7. [DOI] [PubMed] [Google Scholar]
- 10.Moumoulidis I., De R., Ramsden R., Moffat D. Unusual otological manifestations in Camurati-Engelmann's disease. J Laryngol Otol. 2006;120(10):892–895. doi: 10.1017/S0022215106001551. [DOI] [PubMed] [Google Scholar]
- 11.Friedland D.R., Wackym P.A., Rhee J.S., Finn M.S. Cochlear implantation for auditory rehabilitation in Camurati-Engelmann disease. Ann Otol Rhinol Laryngol. 2000;109(2):160–162. doi: 10.1177/000348940010900209. [DOI] [PubMed] [Google Scholar]
- 12.Higashi K., Matsuki C. Hearing impairment in Engelmann disease. Am J Otol. 1996;17(1):26–29. [PubMed] [Google Scholar]
- 13.Tibesar R.J., Brissett A.E., Shallop J.K., Driscoll C.L. Internal auditory canal decompression and cochlear implantation in Camurati-Engelmann disease. Otolaryngol Head Neck Surg. 2004;131(6):1004–1006. doi: 10.1016/j.otohns.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 14.Hellier W.P.L., Brookes G.B. Vestibular nerve dysfunction and decompression in Engelmann's disease. J Laryngol Otol. 1996;110(5):462–465. doi: 10.1017/s0022215100133985. [DOI] [PubMed] [Google Scholar]
- 15.Wright M., Miller N.R., McFadzean R.M., Riordan-Eva P., Lee A.G., Sanders M.D. Papilloedema, a complication of progressive diaphyseal dysplasia: a series of three case reports. Br J Ophthalmol. 1998;82(9):1042–1048. doi: 10.1136/bjo.82.9.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puliafito C.A., Wray S.H., Murray J.E., Boger W.P., 3rd Optic atrophy and visual loss in craniometaphyseal dysplasia. Am J Ophthalmol. 1981;92(5):696–701. doi: 10.1016/s0002-9394(14)74664-1. [DOI] [PubMed] [Google Scholar]
- 17.Aasved H. Osteopetrosis from the ophthalmological point of view. A report of two cases. Acta Ophthalmol. 1969;48(4):771–778. [PubMed] [Google Scholar]
- 18.Calderon M., Brady H.R. Fibrous dysplasia of bone with bilateral optic foramina involvement. Am J Ophthalmol. 1969;68(3):513–515. doi: 10.1016/0002-9394(69)90725-9. [DOI] [PubMed] [Google Scholar]
- 19.Yen J.K., Bourke R.S., Popp A.J., Wirth C.R. Camurati-Engelmann disease (progressive hereditary craniodiaphyseal dysplasia). Case report. J Neurosurg. 1978;48(1):138–142. doi: 10.3171/jns.1978.48.1.0138. [DOI] [PubMed] [Google Scholar]
- 20.Walsh F.B., Hoyt W.F. 3rd ed. Williams & Wilkins; Baltimore: 1969. Clinical neuro-ophthalmology. [Google Scholar]
- 21.Krohel G.B., Wirth C.R. Engelmann's disease. Am J Ophthalmol. 1977;84(4):520–525. doi: 10.1016/0002-9394(77)90445-7. [DOI] [PubMed] [Google Scholar]