

Abstract
Background
Neuromyelitis optica (NMO) is an inflammatory disease of the central nervous system that predominantly affects the optic nerves and spinal cord. Although NMO has long been considered a subtype of multiple sclerosis (MS), the effects of interferon-β treatment are different between NMO and MS. Recent findings of NMO-IgG suggest that NMO could be a distinct disease rather than a subtype of MS. However, the underlying molecular mechanism of NMO pathology remains poorly understood.
Methods
OPN in the cerebrospinal fluid and brain of patients with NMO and with MS, as well as of patients with other neurologic disease/idiopathic other neurologic disease was examined using Western blotting, ELISA, immunohistochemistry and Boyden chamber.
Results
Here we show that osteopontin is significantly increased in the cerebrospinal fluid of NMO patients compared with MS patients. Immunohistochemical analyses revealed that osteopontin was markedly elevated in the cerebral white matter of NMO patients and produced by astrocytes, neurons, and oligodendroglia as well as infiltrating macrophages. We also demonstrate that the interaction of the cerebrospinal fluid osteopontin in NMO patients with integrin αvβ3 promoted macrophage chemotaxis by activating phosphoinositide 3-kinase and MEK1/2 signaling pathways.
Conclusion
These results indicate that osteopontin is involved in NMO pathology.
General significance
Thus therapeutic strategies that target osteopontin signaling may be useful to treat NMO.
Abbreviations: Ab, antibody; Abs, antibodies; AQP4, aquaporin-4; CNS, central nervous system; CSF, cerebrospinal fluid; EAE, experimental autoimmune encephalomyelitis; GFAP, glial fibrillary acidic protein; IFN, interferon; mAb, monoclonal antibody; MS, multiple sclerosis; NMO, neuromyelitis optica; OPN, osteopontin; pAb, polyclonal antibody; PI3K, phosphoinositide 3-kinase; RGD, Arg–Gly–Asp; RGE, Arg–Gly–Glu; TRAP, tartrate-resistant form of acid phosphatase
Keywords: Osteopontin, Neuromyelitis optica, Cerebrospinal fluid, Multiple sclerosis, Biomarker, Integrin
Highlights
-
•
Osteopontin is significantly increased in the CSF of NMO patients.
-
•
Osteopontin is markedly elevated in the cerebral white matter of NMO patients.
-
•
NMO osteopontin is produced by astrocytes, neurons, oligodendroglia, and macrophages.
-
•
Interaction of NMO osteopontin with integrin αvβ3 promotes macrophage chemotaxis.
-
•
NMO osteopontin promotes macrophage chemotaxis by activating PI3K and MEK1/2.
1. Introduction
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease of the central nervous system (CNS) that primarily affects the optic nerves and spinal cord leading to paralysis and blindness [28]. NMO has long been considered a subtype of multiple sclerosis (MS), which is characterized by myelin breakdown, oligodendrocyte loss, and axonal damage [8], [12]. However, the attacks of NMO are usually more severe than those of MS, and spinal cord magnetic resonance imaging (MRI) can aid in the diagnosis [28]. The recent discovery of NMO-IgG in the serum of a high percentage of NMO patients (~ 75%) that are usually absent in the conventional form of MS, and the subsequent identification of its target antigen, aquaporin-4 (AQP4), which is the most abundant water channel at the astrocyte endfeet in CNS, provided strong evidence that NMO was distinct from MS [17]. The NMO phenotype can be attributed to NMO-IgG and complement attacking astrocytes in the nerve and spinal cord. In demyelinated active lesions of NMO patients, extensive macrophage infiltration associated with large numbers of perivascular granulocytes and eosinophils and rare T cells was observed [10], [18]. While it remains unclear how these inflammatory or immune cells are involved in NMO pathogenesis, these findings indicate that the most appropriate treatment may differ for NMO and MS. In fact, several case reports have shown the contrasting effects of interferon (IFN)-β treatment; in NMO, IFN-β induces severe relapses and exacerbations, although it is the most commonly prescribed treatment for relapsing remitting MS [1], [21], [26], [27]. It is therefore crucial to distinguish these two disorders to select optimal treatment.
Osteopontin (OPN) is involved in various physiological and pathological events, including mineralization, acute and chronic inflammation, and cancer metastasis [15], [22], [23]. OPN is believed to play an important role in several autoimmune diseases such as MS [6], rheumatoid arthritis [30], systemic lupus erythematosus [13], and asthma [29]. In relapsing remitting MS, increased OPN protein levels in the plasma but not in the cerebrospinal fluid (CSF) are associated with disease activity [24], [25]. It has also been reported that OPN concentrations are increased in CSF during attacks of MS [4]. OPN expression is elevated in the CNS lesions of experimental autoimmune encephalomyelitis (EAE) during the acute phase but not during remission, and OPN plays a critical role in EAE progression [6].
Osteopontin (OPN) is a multiphosphorylated extracellular glycoprotein. The molecular weight of OPN on SDS-PAGE varies from 45 to 75 kDa due to glycosylation and phosphorylation as well as having a highly negative charge resulting from the predominantly acidic amino acid composition [15]. OPN contains an Arg–Gly–Asp (RGD) sequence common to many extracellular matrix proteins, which mediate the association of OPN with multiple integrins such as α5β1, αvβ1, and αvβ3 [14], [15], [23]. Another important receptor for OPN is CD44, and thrombin-cleaved carboxyl-terminal OPN fragments bind to CD44 variants in an RGD-independent manner [3]. The binding of OPN to these cell surface receptors induces cellular signaling pathways such as phosphoinositide 3-kinase (PI3K), MAPK, and JNK to regulate various cell functions, including adhesion, migration, chemotaxis, and proliferation in various types of cells [3], [14], [23].
In this study, we found that OPN was significantly increased in CSF of patients with NMO compared with patients with MS or other diseases. Immunohistochemical analyses using anti-OPN antibody (Ab) showed the aberrant expression of OPN in the cerebral white matter of the NMO patients. Of note, OPN in CSF of NMO patients was found to be a primary protein that promptly induced macrophage chemotaxis.
2. Materials and methods
2.1. Compliance with ethical standards for human subject research
Study approval (number 613) was granted after protocol review by the Ethics Committee of Fukushima Medical University, which is guided by local policy, national law, and the World Medical Association Declaration of Helsinki.
2.2. CSF, serum, and plasma samples
CSF samples were obtained at the time of relapse from anti-AQP4 Ab-positive NMO patients (n = 19; all females; mean age, 47.4 ± 14.7 years), and MS patients (n = 19; one male and 18 females; mean age, 34.4 ± 10.8 years). The mean times from symptom onset to lumbar puncture were 5.5 days (range: 0–17 days) for NMO and 13.4 days (range: 2–56 days) for MS. The mean disease durations were 5.2 ± 10.8 years (range: 25 days–46 years) for NMO and 4.6 ± 8.0 years (range: 19 days–36 years) for MS. Fourteen patients with other neurologic disease/idiopathic other neurologic disease (OND/IOND) (1 sacroiliac joint dysfunction; 1 Parkinson's disease; 1 cervical spondylosis; 1 spinocerebellar ataxia type 6; 1 multiple system atrophy-P; 1 sarcoidosis; 1 HTLV-1-associated myelopathy; 1 subterranean clover stunt disease; and 3 NMDAR Ab-negative and 3 NMDAR Ab-positive limbic encephalitis; two males and 12 females: mean age, 47.7 ± 19.1 years) served as disease controls. The mean disease duration was 4.5 ± 6.7 years (range: 26 days–23 years) for the control group. The age at onset was higher in NMO than in MS (p < 0.05) while in control it did not differ significantly in NMO (NMO vs. control) and in MS (MS vs. control). The disease durations did not differ significantly among the three. After obtaining informed consent from the patients, the CSF samples were obtained by lumbar puncture and then centrifuged to remove cells and other insoluble materials. The cell-free CSF supernatant was aliquoted in tubes and stored at − 80 °C until use. Blood samples collected from patients were either heparinized and directly centrifuged at 1000 g for 10 min at room temperature to obtain plasma or allowed to clot at 4 °C overnight before centrifugation, after which serum was transferred and stored at − 80 °C until use.
2.3. Abs and reagents
Rabbit polyclonal Abs (pAbs) were used with the following specificities: against human OPN (O-17; IBL, Cat# 18625), and against Akt (Cell Signaling, Cat# 9272). Mouse monoclonal Abs (mAbs) were used with the following specificities: against OPN (clone 53; Assay Designs, Cat# 905–629 or clone 10A16; IBL, Cat# 10011), against integrin αvβ3 (clone LM609; Millipore, Cat# MAB1976), against phospho-p44/42 MAPK (clone E10; Cell Signaling, Cat# 9106S), against CNPase (clone 11-5B; GeneTex, Cat# GTX72341), and against ERK1 (clone MK12; Cat# 610031), JNK/SAPK1 (clone 37/pan-JNK/SAPK1; Cat# 610627), phospho-JNK/SAPK [pT183/pY185, clone 41/JNK/SAPK (pT183/pY185); Cat# 612540], p38α (clone 27/p38α/SAPK2a; Cat# 612168), and phospho-p38 MAPK [pT180/pY182, clone 30/p38 MAPK (pT180/pY182); Cat# 612280] from BD Transduction laboratories. A rat mAb against CD44 (clone Hermes-1; Cat# MA4400) was from Thermo Scientific. Rabbit mAbs against phospho-Akt1 (pS473, clone EP2109Y; Cat #2118-1), and against glial fibrillary acidic protein (GFAP; clone EPR1034Y; Cat #2301-1) were from Epitomics. The control rat (Cat# sc-2026), and mouse (Cat# sc-2025) IgGs, and a mouse mAb against integrin αvβ3 (clone 23C6; Cat# sc-7312) for FACS analysis were obtained from Santa Cruz Biotechnology, Inc.; Alexa Fluor 488-conjugated goat anti-rabbit IgG (Cat# A11008) and Alexa Fluor 546-conjugated goat anti-mouse IgG (Cat# A11030) were purchased from Life Technologies. The PI3K inhibitor LY294002 (Cat# 70920) was from Cayman Chemical, and the MEK1/2 inhibitor U0126 (Cat# 662005), the JNK inhibitor SP600125 (Cat# 420119), and the p38MAPK inhibitor SB203580 (Cat# 559389) were from Calbiochem. GRGDSP (Cat# SP001) and GRGESP (Cat# SP002) peptides were from Takara Bio Inc., and calf intestine alkaline phosphatase (CIAP; Cat# CAP-101) was purchased from Toyobo. Recombinant OPN was purified from human wild-type OPN overexpressing 293T cells as described previously [14].
2.4. SDS-PAGE and Western blot
SDS-PAGE was performed on 5–20% SuperSep™ Ace gradient gels (Wako, Cat# 194-15021) under reducing conditions. For Western blot analyses, proteins resolved by SDS-PAGE were transferred to nitrocellulose membranes. The blots were probed with each specific Ab. Immunoreactive bands were detected using a SuperSignal West Dura Extended Duration Substrate kit (Thermo Scientific, Cat# 34075). The band intensity was calculated using NIH ImageJ software.
2.5. ELISA assay
OPN levels in CSF samples were measured using a human osteopontin ELISA kit (R&D systems, Cat# DOST00), according to the manufacturer's protocols.
2.6. Dephosphorylation of CSF and serum samples
CSF and serum samples were boiled for 5 min at 95 °C, followed by incubation for 1 h at 37 °C in the presence of 3 units of purified CIAP in 10 mM Tris–HCl (pH 8.0) buffer.
2.7. Immunohistochemistry
Paraffin-embedded sections of human tissues were deparaffinized, rehydrated, and immersed in 0.3% hydrogen peroxidase-containing methanol for 20 min at room temperature to inactivate the intrinsic peroxidase. After microwave treatment for antigen retrieval for 15 min in 10 mM sodium citrate buffer (pH 6.0), the sections were incubated with each mAb at 4 °C overnight. After washing with PBS three times, peroxidase-labeled goat anti-mouse IgG (Fab′) (Histofine simple stain MAX-PO (MULTI) kit; Nichirei Corp., Cat# 424152) was used as a secondary Ab and was applied for 30 min at room temperature. After washing with PBS three times, peroxidase was visualized with 3,3′-diaminobenzidine, using the Histofine DAB substrate kit (Nichirei Corp., Cat# 425011). The slides were then counterstained with Mayer's hematoxylin solution (Wako, Cat# 131-09665). Images were obtained using an optical microscope PROVIS AX-80 equipped with a DP70 camera and DP manager software (Olympus) or a NanoZoomer 2.0-RS (Hamamatsu). For immunofluorescent staining, Alexa Fluor 488-conjugated anti-rabbit IgG and Alexa Fluor 546-conjugated anti-mouse IgG were used as secondary Abs. Coverslips were mounted using Fluorescent mounting medium (Dako). Fluorescence images were obtained using an IX71 fluorescent microscope (use of LUCPlan-FLN 20 ×/0.45 objective) connected to a DP70 camera equipped with DP manager software (Olympus).
2.8. Cell culture
THP-1 is a human monocytic cell line derived from an acute monocytic leukemia patient. It can be differentiated into macrophage-like cells by PMA. THP-1 cell lines were cultured in RPMI 1640 medium (Wako, Cat# 168-23191) supplemented with 10% FBS (Nichirei Corp.) and penicillin and streptomycin (Wako, Cat# 189-02025).
2.9. Preparation of cell lysates
The cell lysates were prepared in the following manner. The cells were washed two times with cold PBS and then lysed with lysis buffer (1% Triton X-100, 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, and 5 mM EDTA) containing a protease inhibitor cocktail (Nacalai Tesque, Japan, Cat# 07575) and a phosphatase inhibitor cocktail (Roche, Cat# 4906845). After incubation for 10 min on ice, the cell lysates were clarified by centrifugation at 20,400 g for 10 min at 4 °C. The resultant supernatant was used in the following experiments. The protein concentration was determined using a protein assay kit (Nacalai Tesque. Japan, Cat# 29449-44).
2.10. Migration assay
Cell migration was assayed using a 24-well chemotaxis chamber (BD Falcon cell culture companion plates Cat# 353504 and 8.0-μm inserts Cat# 353097; BD Biosciences). THP-1 cells were treated for 48 h with PMA (Wako, Cat# 1544-5) at 50 ng/ml to trigger differentiation of the THP-1 cells into macrophages. Two hundred microliters of 1 × 106 cells/ml PMA-treated THP-1 cells (2 × 105 cells in 0.1% BSA containing serum-free RPMI 1640 medium) were inoculated into each upper well of a chamber, and 800 μl of 0.1% BSA containing serum-free RPMI medium with 120 μl of diluted sample by PBS (total 920 μl) were added to the lower well of a chamber. After 15 h incubation, the cells on the upper side of a membrane were removed with a cotton swab, and the cells on the lower side of a membrane were fixed with 5% glutaraldehyde and stained with 0.5% crystal violet in 20% methanol. Random fields were photographed using a phase contrast microscope, and at least 100 cells were photographed per condition. Then the numbers of migrated cells were counted. For inhibition assays, cell suspensions were incubated with function-blocking Abs, control IgG, or inhibitors for 20 min at room temperature before inoculation.
2.11. Flow cytometric analysis
Cells were detached from a 10-cm dish using trypsin containing 1 mM EDTA. After quenching trypsinization with a medium containing 10% FBS, the cells were washed two times with PBS containing 1 mM EDTA and incubated with primary Ab or control IgG on ice for 30 min. The cells were washed three times with PBS that contained 1 mM EDTA, followed by incubation for 15 min with the appropriate Alexa Fluor-conjugated secondary Ab. After washing three times with PBS that contained 1 mM EDTA, the cells were analyzed by flow cytometry using FACSCalibur and CellQuest software (BD Biosciences).
2.12. Statistical analysis
OPN levels and increased cell signaling were compared between groups using the Kruskal–Wallis test followed by Dunn's post-test, with GraphPad Prism Version 5.0a software (GraphPad Software, Inc.). Comparisons between two groups were made using Student's t-test (two-tailed) and among groups using one-way ANOVA followed by a Bonferroni post-test, with GraphPad Prism Version 5.0a software. A P value of 0.05 was considered the threshold for statistical significance.
3. Results
3.1. Increased OPN levels in NMO CSF
OPN is known to behave as an inflammatory cytokine, a fact considered relevant in MS [6]. To investigate any differences in OPN levels in CSF between NMO and MS, we measured the levels in patients with anti-AQP4 Ab-positive NMO (all at the relapse phase) and with MS (all at the relapse phase), as well as in patients with other diseases (control group). Western blot with 1 μl of CSF and anti-human OPN pAb (clone O-17) revealed that OPN levels were elevated in CSF of the patients with NMO relative to those with MS and with other diseases, including sacroiliac joint dysfunction and Parkinson's disease (Fig. 1A). Similar results arose from Western blot analysis using the same quantity of CSF protein (3 μg) and anti-human OPN pAb (Fig. 1B). To confirm the results of Western blot analyses, we performed ELISA for OPN using CSF samples (Fig. 1C). The patients with NMO showed high OPN levels in their CSF, ranging from 512 to 3079 ng/ml (n = 19). In contrast, the patients with MS (n = 19) and other diseases (n = 14) including sacroiliac joint dysfunction (n = 1), Parkinson's disease (n = 1), cervical spondylosis (n = 1), spinocerebellar ataxia type 6 (n = 1), multiple system atrophy-P (n = 1), sarcoidosis (n = 1), HTLV-1-associated myelopathy (n = 1), subterranean clover stunt disease (n = 1), and NMDAR Ab-negative (n = 3) and -positive (n = 3) limbic encephalitis patients, had low OPN levels, ranging from 94 to 203 ng/ml and from 89 to 210 ng/ml, respectively. Because high OPN levels have been reported in both the serum and plasma in MS [21], [25], we compared these levels in NMO and MS patients. We found no difference in OPN levels between the two groups in the serum (Fig. 1D) or plasma (data not shown). Instead, a higher OPN level in NMO CSF than in MS CSF suggested that OPN in CSF could be a biomarker to discriminate NMO from MS.
Fig. 1.

Increased OPN levels in CSF of NMO patients.
(A) CSF samples (1 μl) from independent patients with diseases as indicated were applied to SDS-PAGE under reducing conditions, followed by Western blot using anti-human OPN pAb. Other diseases (others): sacroiliac joint dysfunction (lane 13) and Parkinson disease (PD) (lane 14). Other procedures are described in Materials and Methods. rOPN, recombinant OPN, for which phosphorylation level is very low, for positive control. The arrowhead indicates the corresponding band in Fig. 1B. (B) The same quantity of protein (3 μg) in the indicated CSF samples from independent patients was loaded to SDS-PAGE under reducing conditions, followed by Western blot using anti-human OPN pAb. Other diseases (others) include sacroiliac joint dysfunction (lane 10), PD (lane 11), cervical spondylosis (lane 12), and spinocerebellar ataxia type 6 (lane 13). The samples in lanes 1–4, 5–9 and 10–11 in Fig. 1B were identical to the samples in lanes 1–4, 7–11, and 13–14 in Fig. 1A, respectively. Vertical line indicates the removal of an intervening lane for presentation purposes. (C) Concentration of OPN in each CSF sample was measured by ELISA. Each point in the scatter plot represents mean OPN concentration of experiments conducted in duplicate. The horizontal lines represent the median of each patient group. Patients with NMO (n = 19) had significantly elevated OPN protein levels compared with those with MS (n = 19) and other diseases (others, n = 14). We used a Kruskal–Wallis test followed by Dunn's post-test to assess differences. The intra assay coefficient of variation (% CV) was lower than 9% (almost always less than 2%). (D) Western blot analysis of OPN in the serum of NMO (lanes 1–3) and MS (lanes 4–7) patients. Each 1 μl of serum samples diluted 1:10 in PBS was run on a 5–20% SDS-PAGE gel under reducing conditions, followed by Western blot using anti-human OPN pAb. Ordinates indicate molecular sizes in kDa of marker proteins. All blots are representative of at least three independent experiments.
3.2. Pathological analyses of OPN in NMO
The results in Fig. 1 suggest that the cellular source of OPN in CSF might be independent of that of OPN in the serum because the OPN expression pattern in CSF was not relevant to the pattern in the serum. To confirm the possibility, we investigated the cellular expression patterns of OPN in human NMO brain sections by immunohistochemistry. Immunostaining using mAb specifically recognizing OPN (clone 53) showed that OPN was highly expressed in the white matter of the cerebrum of the NMO patients whereas OPN was weakly expressed in the gray matter (Fig. 2A). In the cerebrum of the patients with MS (Fig. 2B), the OPN signal was quite low compared with that in patients with NMO (Fig. 2C), whereas in the cerebrum of the patients with AD, the OPN signal was almost undetectable (Fig. 2D). In the brain of the patients with NMO, OPN was detected on astrocytes, which are abundant in gray matter and have many fine processes (Fig. 2E; arrows), on neurons, which have a large cell body and large bright nucleus (Fig. 2E; white arrowheads), and on oligodendrocytes, which are abundant in white matter and have several processes (Fig. 2F; arrowheads). OPN was highly expressed on perivascular infiltration of numerous macrophages in the serial cerebral section from NMO patients (Fig. 2G and H). Similar result was obtained using another anti-OPN mAb (clone 10A16) (data not shown). The expression of OPN on astrocytes and on oligodendrocytes was also confirmed by double-immunofluorescent staining for OPN and cell type-specific marker proteins (Fig. 2I and J). These results suggested that OPN expression was highly upregulated in the NMO brain but not in the MS brain, which was consistent with the results in Fig. 1.
Fig. 2.

OPN expression in the cerebral cortex of NMO patients.
(A) High OPN expression level in the white matter of NMO patients. Scale bar: 2.5 mm. OPN expression around the boundary area between white and gray matter in the patient with NMO (B), MS (C), and AD (D). Scale bars: 200 μm. The dashed lines indicate the boundary between white and gray matter. w, white matter; g, gray matter. OPN expression in astrocytes (E; arrows), neurons (E; white arrowheads), and oligodendroglias (F; arrowheads) of the cerebral sections of NMO patients. Scale bars: 50 μm. (G and H) Serial cerebral section from NMO patients demonstrating perivascular infiltration of numerous macrophages expressing OPN. The area in the rectangle in (G) is shown at higher magnification in (H). The arrowheads in (H) indicate macrophages. Scale bars: 200 μm (G), and 50 μm (H). OPN staining was performed using mAb against OPN (clone 53) on paraffin-embedded sections (n = 3) from two patients per disease. Photos in (B–H) are immunoperoxidase-stained with 3,3′-diaminobenzidine and hematoxylin counterstain. (I and J) Representative immunofluorescence staining of cerebral sections of an NMO patient. (I) Staining with OPN (green) and GFAP (red: astrocytes). (J) Staining with OPN (green) and CNPase (red: oligodendroglias). Nuclei were stained blue with Hoechst 33342. Scale bars: 50 μm.
OPN is a multiphosphorylated extracellular glycoprotein and the molecular weight could be affected by phosphorylation [14]. The extent of OPN phosphorylation is variable, and it depends on the cellular source of OPN [15]. To further study the possibility that the cellular source of OPN in CSF is different from that in the serum, we investigated the OPN phosphorylation level in the CSF and serum of NMO patient using alkaline phosphatase, CIAP (Fig. 3). Since dephosphorylated OPN migrated faster than the phosphorylated OPN on SDS-PAGE gel [14], we can determine phosphorylation levels of OPN by checking the band mobility before and after CIAP treatment. After dephosphorylation with CIAP treatment, OPN was checked by Western blot using anti-human OPN pAb (O-17). The 70–75 kDa bands of CSF OPN without CIAP treatment completely disappeared after CIAP treatment and bands around 55 kDa appeared after CIAP treatment, indicative of phosphorylated and nonphosphorylated forms of CSF OPN, respectively. On the other hand, it was difficult to detect any changes in serum OPN band mobility. These results indicated that OPN in the CSF was more phosphorylated than OPN in the serum, and that OPN in CSF and the serum might be produced by different cellular sources.
Fig. 3.
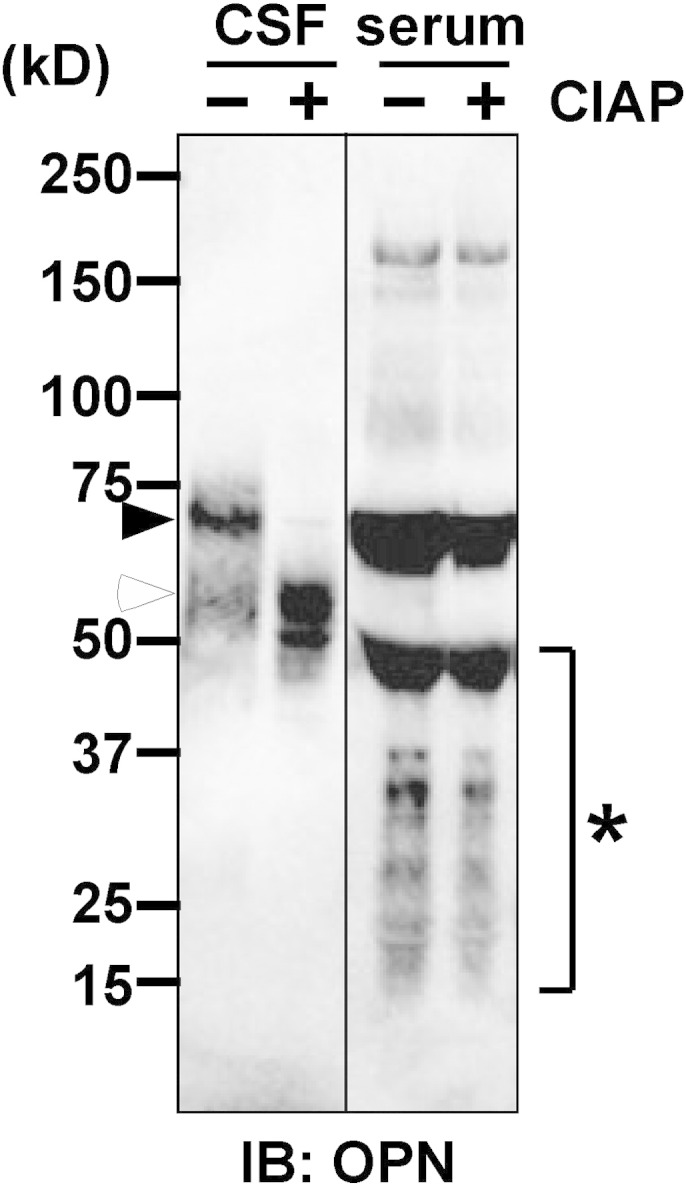
Dephosphorylation analysis of OPN in the CSF and serum of NMO patient.
Denatured 6 μl of CSF and 1 μl of serum samples from the same NMO patient were treated with (+) or without (−) CIAP. After incubation for 1 h at 37 °C, samples were applied to SDS-PAGE under reducing conditions, followed by Western blot using anti-human OPN pAb. Black arrowheads indicate the band of phosphorylated OPN. White arrowhead indicates the band of dephosphorylated OPN. Asterisk indicates the cleaved or degraded forms of OPN. Note that OPN in the CSF was reduced in size after CIAP treatment but OPN in the serum maintained almost identical size with and without CIAP treatment, suggesting that CSF contains the more phosphorylated form of OPN than serum. Ordinates indicate molecular sizes in kDa of marker proteins. Blots are representative of at least three independent experiments.
3.3. OPN in NMO CSF promotes macrophage chemotaxis
OPN plays a role as a chemotactic cytokine for macrophages, dendritic cells and neutrophils in the inflammatory region [3], [9], and extensive macrophage infiltration was observed in demyelinated active lesions of NMO patients [10], [18]. In the present study, high OPN expression was observed in the NMO CSF (Fig. 1) and brain (Fig. 2). To investigate whether OPN in NMO CSF might promote macrophage infiltration, we performed a chemotaxis assay using human macrophage-like cells, THP-1 cell lines, and the CSF samples from NMO patients. Compared to the control PBS, 10 μl of NMO CSF including 25 ng OPN significantly increased the migration of THP-1 cells whereas 10 μl of MS CSF including 1.2 ng OPN slightly induced the chemotactic migration of THP-1 cells (Fig. 4A). NMO CSF, but not MS CSF, caused the chemotactic migration of THP-1 cells in a dose-dependent manner (Fig. 4B). Surprisingly, even when the OPN concentration in the MS CSF-containing chamber was higher than that in the NMO CSF-containing chamber, MS CSF-induced chemotaxis was still weaker than NMO CSF-induced chemotaxis (Fig. 4B, NMO 2.3 ng/ml vs MS 2.7, 4.1, 8.2, 16.3 ng/ml). To investigate whether OPN in NMO CSF directly affected such chemotactic migration of THP-1 cells, we examined the inhibitory effect of anti-OPN mAb (clone 53) that blocks OPN function in chemotactic activity. The neutralizing mAb of OPN markedly abrogated NMO CSF-induced chemotaxis but had no inhibitory effect on MS CSF-induced chemotaxis (Fig. 4C). These results indicated that OPN in NMO CSF induced macrophage chemotaxis, whereas OPN in MS CSF might have no ability to induce it.
Fig. 4.

In vitro chemotaxis assay using NMO and MS CSF.
In vitro chemotaxis assay using CSF of NMO (OPN concentration, 859 ng/ml) or MS patients (OPN concentration, 125 ng/ml). The volume of CSF samples was adjusted to be 120 μl with PBS. The 120 μl of sample or control PBS was added to 800 μl of 0.1% BSA containing serum-free medium in the lower well of the chamber. The migrated cells on the lower side of a membrane were photographed using a phase contrast microscope (A), and they were counted (B). Numerical values on the longitudinal axis indicate the final concentration of OPN in the lower chamber (ng/ml). We used a one-way ANOVA followed by a Bonferroni post-test to assess differences. **p < 0.01 vs. 2.3 ng/ml NMO, ***p < 0.001 vs. 2.3 ng/ml NMO. (C) Cells were pretreated with a control mouse IgG (IgG) or mouse anti-OPN mAb (OPN mAb) and plated on the upper chamber in the presence of NMO CSF (OPN concentration in the lower chamber: 9.3 ng/ml) or MS CSF (OPN concentration in the lower chamber: 4.1 ng/ml) in the lower chamber. The relative number of migrated cells to the lower surface of the membrane in the presence of mouse IgG was taken as 100%. We used a two-tailed Student's t-test to assess differences. Data are represented as the mean ± SD from three independent experiments with at least triplicate data points per experiment. Other experimental conditions are described in the Materials and methods section.
OPN often uses integrin αvβ3 and CD44 as a cell surface receptor, and the association of OPN with them stimulates cellular signaling, which induces various cellular activities such as cell adhesion and migration. FACS analysis revealed that after differentiation by PMA, macrophage-like THP-1 cells expressed both integrin αvβ3 and CD44 on their cell surfaces (Fig. 5A). To clarify which receptors were mainly used in NMO CSF-induced chemotaxis, which was primarily attributed to OPN, we performed inhibitory assays using mAbs against integrin αvβ3 (clone LM609) and CD44 (clone Hermes-1). NMO CSF-induced chemotaxis was decreased by 50% in the presence of the neutralizing mAb against integrin αvβ3 (Fig. 5B). RGD sequence-containing peptide, GRGDSP, is a competitor of RGD-recognizing integrin including integrin αvβ3. Similar results were obtained when the GRGDSP peptide, but not the control Arg–Gly–Glu (RGE) sequence-containing peptide, GRGESP, was used instead of the neutralizing mAb against integrin αvβ3 in a chemotaxis inhibition assay (Fig. 5C). In contrast, the neutralizing mAb against CD44 had no effect on NMO CSF-induced chemotaxis (Fig. 5D) although the mAb blocked the chemotaxis of MDA-MB231 cells to about 30% (Fig. S1). These results suggested that OPN in NMO CSF mainly used integrin αvβ3, but not CD44, as a receptor to cause chemotactic activity. However, the results that neither integrin αvβ3 and CD44 mAbs nor the RGD peptide suppressed MS CSF-induced chemotaxis (Fig. 5B–D, MS) were in accordance with the result that OPN function-blocking mAb had no inhibitory effect on MS CSF-induced chemotaxis (Fig. 4C).
Fig. 5.

OPN in NMO CSF promotes macrophage motility through integrin αvβ3.
(A) Expression levels of integrin αvβ3 and CD44 on the surface of THP-1 cells were examined using FACS analysis. Prior to analysis, the cells were incubated with either the indicated Abs or control IgG, followed by incubation with Alexa Fluor-conjugated secondary Abs, as described in Materials and Methods. Inhibitory effect of 20 μg/ml integrin αvβ3 mAb (clone LM609, B), 100 μM GRGDSP peptide (RGD, C), and 20 μg/ml CD44 mAb (clone Hermes-1, D) on NMO or MS CSF-induced cell motility. The relative number of migrated cells to the lower surface of the membrane in the presence of 20 μg/ml control IgG (IgG, B and D) or 100 μM control GRGESP peptide (RGE, C) was taken as 100%. We used a two-tailed Student's t-test to assess differences. Data are means ± SDs from two or three independent experiments with triplicate data points per experiment. Other experimental conditions are described in the Materials and methods section.
Fig. S1.
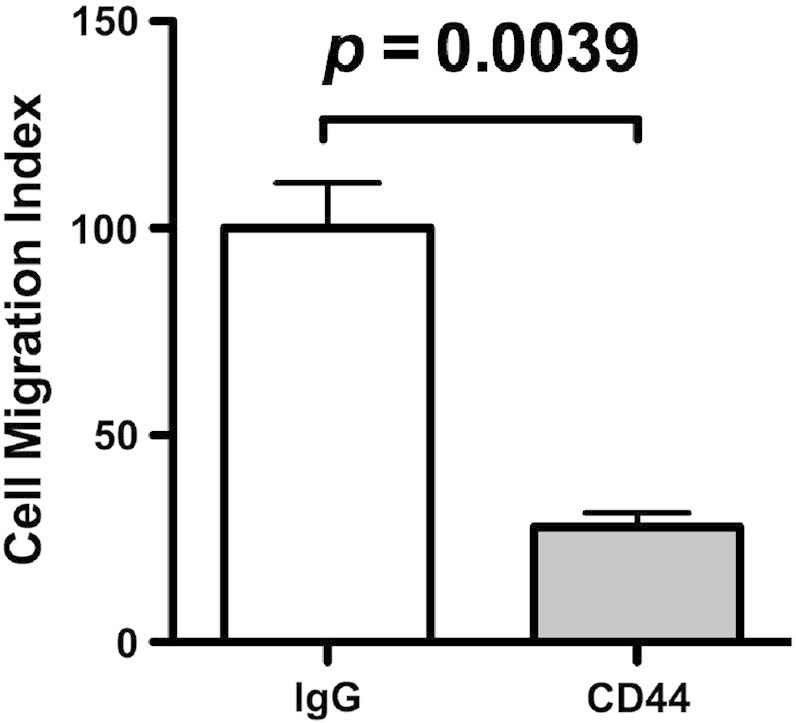
Inhibitory effect of anti-CD44 mAb on MDA-MB231 cell motility.
Two hundred microliters of 1 × 105 cells/ml human breast cancer cell line MDA-MB231 cells (2 × 104 cells in serum-free DMEM) were incubated with 10 μg/ml anti-CD44 mAb (clone Hermes-1) or control IgG. After 20 min incubation at room temperature, the cells were inoculated into each upper well of a chamber, and 750 μl of 10% serum containing DMEM medium was added to the lower well of a chamber. After 18 h incubation, the fixed and stained cells on the lower side of a membrane were photographed using a phase contrast microscope, and the numbers of migrated cells were counted. The relative number of migrated cells to the lower surface of the membrane in the presence of control IgG was taken as 100%. Data are represented as the mean ± SD from three independent experiments.
3.4. Analysis of OPN-activated signaling pathways
To analyze the signaling pathways that OPN in NMO CSF used to induce chemotaxis, we examined the effect of various signaling inhibitors on NMO CSF-induced chemotaxis (Fig. 6A). Compared to the solvent (DMSO) as a control, the chemotactic ability of NMO CSF was markedly reduced by the treatment of cells with the PI3K inhibitor LY294002 or the MEK1/2 inhibitor U0126. In contrast, the JNK inhibitor SP600125 and the p38MAPK inhibitor SB203580 had almost no inhibitory effect on NMO CSF-induced chemotaxis. To confirm the results obtained using these inhibitors, we investigated the phosphorylation state of Akt (downstream of PI3K), MEK1/2, JNK, and p38MAPK in THP-1 cells in the absence (None) or presence of NMO and MS CSF (Fig. 6B). NMO CSF, but not MS CSF, significantly promoted Akt and ERK1/2 phosphorylation but not JNK and p38MAPK phosphorylation compared to the control (Fig. 6B and C), and the Akt and ERK1/2 signaling activations by NMO CSF were reversed with functional blocking mAb against OPN (Fig. 6D), suggesting that OPN in NMO CSF induced the activation of PI3K and MEK1/2, but not of JNK and p38MAPK, signaling pathways. These results indicated that NMO CSF-induced chemotaxis, which was mainly attributed to OPN activity, was mediated by PI3K and MEK1/2, but not by JNK and p38MAPK, signaling cascades.
Fig. 6.

Cell signaling pathways for NMO CSF-induced cell motility.
(A) Effects of various cellular signaling inhibitors on NMO CSF-induced cell motility. THP-1 cells were pretreated with PBS, DMSO, and the indicated inhibitors for 20 min at room temperature and plated on the upper chamber in the presence of NMO CSF in the lower chamber: PI3K inhibitor LY294002 (25 μM), MEK1/2 inhibitor U0126 (20 μM), JNK inhibitor SP600125 (100 μM), and p38MAPK inhibitor SB203580 (20 μM). DMSO was used as a solvent. Data are means ± SDs from two independent experiments with triplicate data points per experiment (one-way ANOVA, Bonferroni post-test). ***p < 0.001 vs. DMSO. NS, not significant. The data were derived by subtracting the numbers of migrated cells in the control (PBS) from those in each condition. (B) PMA-treated THP-1 cells in serum-free medium were cultured in the presence of PBS (None) and CSF of NMO patients (NMO) and of MS patients (MS). After 1 h incubation, cell lysates were collected and immunoblotted against the indicated Abs. All blots are representative of at least three independent experiments. (C) Results of the densitometric analysis are shown as the integrated density of the ratio of phosphorylated protein to total protein bands in Fig. 6B (Kruskal–Wallis test, Dunn's post-test.), which was 1.0 for PBS-treated cells (None). *p < 0.05. **p < 0.01. NS, not significant. (D) Effect of functional blocking mAb against OPN (clone 53) on the increased cell signaling by NMO CSF. PBS and NMO CSF were pretreated with either control mouse IgG or functional blocking mAb against OPN (NMO/Ab) for 15 min at room temperature. Other experimental conditions are described in Fig. 6B. Results of the densitometric analysis are shown as the integrated density of the ratio of phosphorylated protein to total protein bands. *p < 0.05. **p < 0.01. ***p < 0.001.
4. Discussion
For selecting appropriate treatment, it is crucial to distinguish the two disorders, NMO and MS. In this study, we identified OPN as a possible biomarker for NMO. Although AQP4 Ab is a specific biomarker for NMO, an AQP4 Ab assay is required for a high sensitivity cell culture system. Therefore, the AQP4 Ab assay can only be performed in a limited number of institutions. In contrast, OPN can be effectively detected using simple techniques such as Western blot and ELISA, which are a major advantage over the existing methods to detect AQP4 Abs. Thus, the OPN assays could support NMO diagnosis using the AQP4 Ab assay.
The pathological mechanism underlying NMO onset remains poorly understood. Recent reports indicate that NMO-IgG may not be a primary factor for NMO onset because of a report about a patient with NMO who had NMO-IgG years before being symptomatic [20]. This suggests that NMO-IgG by itself cannot induce inflammation; instead, disease onset of NMO and associated inflammatory lesions are required before astrocytes are attacked by NMO-IgG, the anti-AQP4 Ab. The present study showed that in vivo, OPN in NMO CSF was upregulated, and in an in vitro assay, it promoted macrophage motility, which was associated with inflammatory disease. In addition, remarkable vascular inflammatory infiltration of macrophages accompanied by astrocyte loss was observed in the brains of NMO patients (data not shown). Because OPN is known to work as an inflammatory cytokine in some inflammatory diseases and cancer as well as autoimmune disease, OPN may participate in NMO disease onset and in NMO disease progression or both. This will be further addressed in future studies using animal models.
NMO CSF contained a significant amount of OPN and strongly promoted THP-1 motility in an OPN dose-dependent manner. In Fig. 4C, however, neutralizing mAb against OPN did not completely abolish NMO CSF-induced chemotaxis. In contrast, MS CSF induced weak cell motility in an OPN-independent manner. Therefore, not only OPN but also cytokines and other proteins may participate in NMO and MS CSF-induced chemotaxis.
IL-17-producing CD4+ T helper (Th17) cells have recently been identified as a new effector T cell subset involved in the pathogenesis of autoimmune diseases, including rheumatoid arthritis and MS. OPN induces IL-17 production by CD4+ T cells through the integrin αvβ3 [19]. Treatment using anti-integrin αvβ3 Ab and anti-OPN Ab reduced the clinical severity of EAE by reducing IL-17 production [19]. Recent studies using the EAE mouse model indicated that the IFN-β treatment significantly attenuated the progression of EAE symptoms in Th1-induced EAE but exacerbated the disease in Th17-induced EAE [2]. Levels of IL-17, which is secreted from Th17 cells, were significantly elevated in CSF of NMO patients compared with MS patients [11]. In addition, OPN is also secreted from Th17 cells and appears to play a critical role in Th17 differentiation [7]. Thus, elevated OPN levels in NMO might promote Th differentiation to Th17. Considering that the IFN-β treatment induces severe relapses and exacerbations in NMO, the immune response in NMO might mainly be mediated by Th17 cells rather than Th1 cells.
Dephosphorylation with CIAP decreased the molecular size of OPN in NMO CSF but had no effect on the size of OPN in NMO serum, suggesting that OPN in CSF was a more phosphorylated form than that in the serum (Fig. 3). The interaction of OPN with cell surface receptors is determined by its phosphorylation state. Therefore, OPN in CSF could be associated with cell surface receptors distinct from OPN in the serum. The discovery of differences in OPN phosphorylation levels between CSF and the serum among NMO patients might indicate that OPN in the serum does not penetrate CNS blood–brain barrier. The present study demonstrated that OPN in NMO CNS was produced by astrocytes, neurons, oligodendroglias, and macrophages (Figs. 2E–J). Therefore, OPN in CSF of NMO patients could be derived from neural cells and infiltrating immune cells in CNS of the patients but not from their serum.
Recent studies have reported that the genetic deficiency of a tartrate-resistant form of acid phosphatase (TRAP), which is expressed in bone and in immune cells and can be actively secreted, is associated with skeletal dysplasia and cerebral calcifications as well as autoimmunity [5], [16]. Because OPN is known to be a dephosphorylated protein by TRAP, TRAP might regulate OPN functions by controlling its phosphorylation status. In this study, OPN in NMO, but not MS, CSF dose dependently induced cell motility by activating signaling pathways through PI3K and MEK1/2 but not JNK and p38MAPK. Although speculative, the idea that specific phosphorylation sites in the OPN molecules could induce the activation of PI3K and MEK1/2 signaling pathways might explain the different active states between the two disorders. Further studies such as the identification of the phosphorylation sites in OPN from NMO and MS CSF are required to address the question why OPN in MS CSF could not efficiently promote THP-1 cell motility in the chemotaxis assay.
In conclusion, our findings provide a rationale to target OPN as a biomarker and a potential therapeutic approach in NMO diseases.
The following are the supplementary data related to this article.
Conflict of interest
The authors declare no competing financial interests.
Transparency Document
Transparency document.
Acknowledgments
This study was supported by research grants from the Japan Intractable Diseases Research Foundation (Grant number SO23016) to Yoshinobu Kariya; the Ministry of Health, Labor, and Welfare of Japan (Grant number 2006-Nanchi-Ippan-017), Japan Science and Technology Agency (Grant numbers AS221Z00232F, AS231Z01053, 241FT0255, and 149), the Ministry of Education, Culture, Sports, Science, and Technology of Japan [a Grant-in-Aid for Scientific Research on Innovative Areas, Grant number 23110002 (Deciphering sugar chain-based signals regulating integrative neuronal functions)] and a Grant-in-Aid for Scientific Research (Grant number 23590367) to Yasuhiro Hashimoto.
The authors thank Mr. Tomoki Saito, Ms. Mayumi Kanno and Ms. Kyoka Hoshi for their technical assistance. We also would like to thank Dr. Kenneth E. Nollet for his critical reading of the manuscript and comments and Enago for the English language review.
Footnotes
The Transparency document associated with this article can be found, in the online version.
Contributor Information
Yoshinobu Kariya, Email: kariya@fmu.ac.jp.
Yasuhiro Hashimoto, Email: yasuc@fmu.ac.jp.
References
- 1.Axtell R.C., Raman C., Steinman L. Interferon-beta exacerbates Th17-mediated inflammatory disease. Trends Immunol. 2011;32:272–277. doi: 10.1016/j.it.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Axtell R.C., de Jong B.A., Boniface K., van der Voort L.F., Bhat R., De Sarno P., Naves R., Han M., Zhong F., Castellanos J.G., Mair R., Christakos A., Kolkowitz I., Katz L., Killestein J., Polman C.H., de Waal Malefyt R., Steinman L., Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat. Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellahcene A., Castronovo V., Ogbureke K.U., Fisher L.W., Fedarko N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat. Rev. Cancer. 2008;8:212–226. doi: 10.1038/nrc2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bornsen L., Khademi M., Olsson T., Sorensen P.S., Sellebjerg F. Osteopontin concentrations are increased in cerebrospinal fluid during attacks of multiple sclerosis. Mult. Scler. 2011;17:32–42. doi: 10.1177/1352458510382247. (Epub 2010 Oct 2014) [DOI] [PubMed] [Google Scholar]
- 5.Briggs T.A. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat. Genet. 2011;43:127–131. doi: 10.1038/ng.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chabas D., Baranzini S.E., Mitchell D., Bernard C.C., Rittling S.R., Denhardt D.T., Sobel R.A., Lock C., Karpuj M., Pedotti R., Heller R., Oksenberg J.R., Steinman L. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–1735. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 7.Chen G., Zhang X., Li R., Fang L., Niu X., Zheng Y., He D., Xu R., Zhang J.Z. Role of osteopontin in synovial Th17 differentiation in rheumatoid arthritis. Arthritis Rheum. 2010;62:2900–2908. doi: 10.1002/art.27603. [DOI] [PubMed] [Google Scholar]
- 8.Compston A., Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 9.Giachelli C.M., Lombardi D., Johnson R.J., Murry C.E., Almeida M. Evidence for a role of osteopontin in macrophage infiltration in response to pathological stimuli in vivo. Am. J. Pathol. 1998;152:353–358. [PMC free article] [PubMed] [Google Scholar]
- 10.Hengstman G.J., Wesseling P., Frenken C.W., Jongen P.J. Neuromyelitis optica with clinical and histopathological involvement of the brain. Mult. Scler. 2007;13:679–682. doi: 10.1177/1352458506070145. [DOI] [PubMed] [Google Scholar]
- 11.Ishizu T., Osoegawa M., Mei F.J., Kikuchi H., Tanaka M., Takakura Y., Minohara M., Murai H., Mihara F., Taniwaki T., Kira J. Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain. 2005;128:988–1002. doi: 10.1093/brain/awh453. [DOI] [PubMed] [Google Scholar]
- 12.Jarius S., Paul F., Franciotta D., Waters P., Zipp F., Hohlfeld R., Vincent A., Wildemann B. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat. Clin. Pract. Neurol. 2008;4:202–214. doi: 10.1038/ncpneuro0764. [DOI] [PubMed] [Google Scholar]
- 13.Kaleta B. Role of osteopontin in systemic lupus erythematosus. Arch. Immunol. Ther. Exp. 2014;11:11. doi: 10.1007/s00005-014-0294-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kariya Y., Kanno M., Matsumoto-Morita K., Konno M., Yamaguchi Y., Hashimoto Y. Osteopontin O-glycosylation contributes to its phosphorylation and cell-adhesion properties. Biochem. J. 2014;463:93–102. doi: 10.1042/BJ20140060. [DOI] [PubMed] [Google Scholar]
- 15.Kazanecki C.C., Uzwiak D.J., Denhardt D.T. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J. Cell. Biochem. 2007;102:912–924. doi: 10.1002/jcb.21558. [DOI] [PubMed] [Google Scholar]
- 16.Lausch E. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat. Genet. 2011;43:132–137. doi: 10.1038/ng.749. [DOI] [PubMed] [Google Scholar]
- 17.Lennon V.A., Wingerchuk D.M., Kryzer T.J., Pittock S.J., Lucchinetti C.F., Fujihara K., Nakashima I., Weinshenker B.G. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 18.Lucchinetti C.F., Mandler R.N., McGavern D., Bruck W., Gleich G., Ransohoff R.M., Trebst C., Weinshenker B., Wingerchuk D., Parisi J.E., Lassmann H. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. 2002;125:1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murugaiyan G., Mittal A., Weiner H.L. Increased osteopontin expression in dendritic cells amplifies IL-17 production by CD4 + T cells in experimental autoimmune encephalomyelitis and in multiple sclerosis. J. Immunol. 2008;181:7480–7488. doi: 10.4049/jimmunol.181.11.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishiyama S., Ito T., Misu T., Takahashi T., Kikuchi A., Suzuki N., Jin K., Aoki M., Fujihara K., Itoyama Y. A case of NMO seropositive for aquaporin-4 antibody more than 10 years before onset. Neurology. 2009;72:1960–1961. doi: 10.1212/WNL.0b013e3181a82621. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu Y., Yokoyama K., Misu T., Takahashi T., Fujihara K., Kikuchi S., Itoyama Y., Iwata M. Development of extensive brain lesions following interferon beta therapy in relapsing neuromyelitis optica and longitudinally extensive myelitis. J. Neurol. 2008;255:305–307. doi: 10.1007/s00415-007-0730-5. [DOI] [PubMed] [Google Scholar]
- 22.Singh R.P., Patarca R., Schwartz J., Singh P., Cantor H. Definition of a specific interaction between the early T lymphocyte activation 1 (Eta-1) protein and murine macrophages in vitro and its effect upon macrophages in vivo. J. Exp. Med. 1990;171:1931–1942. doi: 10.1084/jem.171.6.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sodek J., Ganss B., McKee M.D. Osteopontin. Crit. Rev. Oral Biol. Med. 2000;11:279–303. doi: 10.1177/10454411000110030101. [DOI] [PubMed] [Google Scholar]
- 24.Vogt M.H., Lopatinskaya L., Smits M., Polman C.H., Nagelkerken L. Elevated osteopontin levels in active relapsing-remitting multiple sclerosis. Ann. Neurol. 2003;53:819–822. doi: 10.1002/ana.10606. [DOI] [PubMed] [Google Scholar]
- 25.Vogt M.H., ten Kate J., Drent R.J., Polman C.H., Hupperts R. Increased osteopontin plasma levels in multiple sclerosis patients correlate with bone-specific markers. Mult. Scler. 2010;16:443–449. doi: 10.1177/1352458509359723. [DOI] [PubMed] [Google Scholar]
- 26.Wang A.G., Lin Y.C., Wang S.J., Tsai C.P., Yen M.Y. Early relapse in multiple sclerosis-associated optic neuritis following the use of interferon beta-1a in Chinese patients. Jpn. J. Ophthalmol. 2006;50:537–542. doi: 10.1007/s10384-006-0359-4. [DOI] [PubMed] [Google Scholar]
- 27.Warabi Y., Matsumoto Y., Hayashi H. Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J. Neurol. Sci. 2007;252:57–61. doi: 10.1016/j.jns.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Wingerchuk D.M., Lennon V.A., Lucchinetti C.F., Pittock S.J., Weinshenker B.G. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–815. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 29.Xanthou G., Alissafi T., Semitekolou M., Simoes D.C., Economidou E., Gaga M., Lambrecht B.N., Lloyd C.M., Panoutsakopoulou V. Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat. Med. 2007;13:570–578. doi: 10.1038/nm1580. (Epub 2007 Apr 2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yumoto K., Ishijima M., Rittling S.R., Tsuji K., Tsuchiya Y., Kon S., Nifuji A., Uede T., Denhardt D.T., Noda M. Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4556–4561. doi: 10.1073/pnas.052523599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.