

Abstract
Background
Acute myeloid leukemia (AML) patients with highly active AKT tend to do poorly. Cell cycle arrest and apoptosis are tightly regulated by AKT via phosphorylation of GSK3α and β isoforms which inactivates these kinases. In the current study we examine the prognostic role of AKT mediated GSK3 phosphorylation in AML.
Methods
We analyzed GSK3α/β phosphorylation by reverse phase protein analysis (RPPA) in a cohort of 511 acute myeloid leukemia (AML) patients. Levels of phosphorylated GSK3 were correlated with patient characteristics including survival and with expression of other proteins important in AML cell survival.
Results
High levels of p-GSK3α/β correlated with adverse overall survival and a lower incidence of complete remission duration in patients with intermediate cytogenetics, but not in those with unfavorable cytogenetics. Intermediate cytogenetic patients with FLT3 mutation also fared better respectively when p-GSK3α/β levels were lower. Phosphorylated GSK3α/β expression was compared and contrasted with that of 229 related cell cycle arrest and/or apoptosis proteins. Consistent with p-GSK3α/β as an indicator of AKT activation, RPPA revealed that p-GSK3α/β positively correlated with phosphorylation of AKT, BAD, and P70S6K, and negatively correlated with β-catenin and FOXO3A. PKCδ also positively correlated with p-GSK3α/β expression, suggesting crosstalk between the AKT and PKC signaling pathways in AML cells.
Conclusions
These findings suggest that AKT-mediated phosphorylation of GSK3α/β may be beneficial to AML cell survival, and hence detrimental to the overall survival of AML patients. Intrinsically, p-GSK3α/β may serve as an important adverse prognostic factor for a subset of AML patients.
Keywords: Leukemia, GSK3, AKT, PKC delta, RPPA, Signal transduction
Highlights
-
•
Phospho-GSK3 is prognostic for poor survival in a subset of AML patients.
-
•
Phospho-GSK3 is a biomarker for active AKT in AML.
-
•
AKT is a PKCδ kinase in AML cells.
1. Introduction
Acute myeloid leukemia (AML) remains a highly fatal disease, so a better understanding of the signaling pathways that support leukemia cell growth and survival is necessary in order to develop improved therapies. Strategies designed to target signaling cascades have been suggested as a way to optimize AML therapy [1], [2], [3], [4], [5], [6], [7], [8]. The activation of survival kinases such as AKT, PKC, and ERK has been shown to predict poor clinical outcome for patients with AML [8]. AKT is a key regulator of protein translation, transcription, cell proliferation, and apoptosis, and is emerging as a potentially important target for AML therapy [1], [3], [4], [8].
AKT has been shown to phosphorylate and inactivate GSK3, which is a key regulator of differentiation, metabolism, apoptosis, autophagy, as well as tumorigenesis [9], [10], [11], [12], [13], [14], [15], [16]. Two functional isoforms of GSK3 known as GSK3α and GSK3β are produced from separate genes, but share 97% amino acid homology [9], [10], [11]. GSK3α and GSK3β are unique among kinases in that they are constitutively active unless their activity is blocked by post-translational modification such as phosphorylation [11]. An appraisal of kinase consensus sequences revealed that GSK3 had more possible substrates than any other kinase [12], [13], although many GSK3 substrates require a “priming” phosphorylation event independent of GSK3. The intricate regulation of GSK3 and its substrates likely reflects the importance of modulating GSK3 signaling pathways as these cascades are critical for cellular homeostasis.
A prime example of role of GSK3 role in tumorigenesis involves the regulation of the WNT/β-catenin pathways [11], [16]. GSK3 mediates degradation of β-catenin by phosphorylating the molecule and stabilizing the docking molecule (i.e., axin) required for its destruction [11], [13], [16]. Failure to properly regulate β-catenin can result in aberrant expression of pro-oncogenic molecules such as c-MYC, Cyclin D1, and c-JUN [13]. Thus a more comprehensive, understanding of the AKT–GSK3 signaling axis and it role in leukemogenesis is critically needed.
In the current study, we examined the ability of GSK3 total protein expression level, and protein activation state, based on serine 9 and/or serine 21 phosphorylation status, to predict overall survival (OS) and remission duration (RD) in AML patients using reverse phase protein array (RPPA) methodology. As phosphorylation of these sites results in inactivation of the GSK3 kinases, high phosphorylation levels indicate lower activity of these kinase in the tumor cells. Results suggest that AML patients with blast cells with inactivated GSK3, highly phosphorylated, have inferior OS and RD. RPPA revealed correlation of phospho-GSK3 with a number of proteins including phosphorylated AKT, phosphorylated PKCδ, FOXO3A, and β-catenin. These results suggest that the AKT/GSK3 axis is critical in AML, which may be of benefit to the development of therapeutic strategies in AML patients classified with intermediate cytogenetics.
2. Material and methods
2.1. Patient samples
Peripheral blood and bone marrow specimens were collected from 511 patients with newly diagnosed AML evaluated at The University of Texas M.D. Anderson Cancer Center (MDACC) between September 1999 and March 2007. Samples were acquired during routine diagnostic assessments in accordance with the regulations and protocols (Lab 01-473) approved by the Investigational Review Board of MDACC. Informed consent was obtained in accordance with the Declaration of Helsinki. Samples were analyzed under an Institutional Review Board approved laboratory protocol (Lab 05-0654). The patient characteristics and sample preparation are described in Table 1.
Table 1.
Patient characteristics of all AML patients in the study.
| Demographics for GSKp219 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Raw numbers | |||||||||||||
| Treated |
Treated |
||||||||||||
| All cases | Lowest 2/3rd | Highest 3rd | P | Test used | All cases | Lowest 2/3rd | Highest 3rd | P | Test used | ||||
| Number | 511 | 341 | 170 | NA | Chi square | Number | 511 | 341 | 170 | NA | Chi square | ||
| Gender | M | 291 | 203 | 88 | 0.1153 | Gender | M | 56.9% | 59.5% | 51.8% | 0.1153 | ||
| F | 220 | 138 | 82 | F | 43.1% | 40.5% | 48.2% | ||||||
| Age | Min | 15.8 | 16.01 | 15.83 | t-Test | Age | Min | 15.8 | 16.01 | 15.83 | F test | ||
| Max | 87.23 | 87.23 | 86.8 | Max | 87.23 | 87.23 | 86.8 | ||||||
| Median | 65.7 | 65 | 65.21 | 0.76 | Median | 65.7 | 65 | 65.21 | 0.76 | ||||
| FAB | M0 | 29 | 20 | 9 | < 0.00001 | Chi square | FAB | MO | 5.7% | 5.9% | 5.3% | < 0.00001 | Chi square |
| M1 | 55 | 39 | 16 | M1 | 10.8% | 11.4% | 9.4% | ||||||
| M2 | 169 | 127 | 42 | M2 | 33.1% | 37.2% | 24.7% | ||||||
| M4 | 119 | 66 | 53 | M4 | 23.3% | 19.4% | 31.2% | ||||||
| M5 | 50 | 19 | 31 | M5 | 9.8% | 5.6% | 18.2% | ||||||
| M6 | 27 | 21 | 6 | M6 | 5.3% | 6.2% | 3.5% | ||||||
| M7 | 10 | 7 | 3 | M7 | 2.0% | 2.1% | 1.8% | ||||||
| RAEBT | 33 | 29 | 4 | RAEBT | 6.5% | 8.5% | 2.4% | ||||||
| Unknown | 19 | 13 | 6 | Unknown | 3.7% | 3.8% | 3.5% | ||||||
| WHO | Abnormal cytogenetics | 47 | 33 | 14 | 0.1 | Chi square | WHO | Abnormal cytogenetics | 9.2% | 9.7% | 8.2% | 0.1 | Chi square |
| Therapy related | 73 | 49 | 24 | Therapy related | 14.3% | 14.4% | 14.1% | ||||||
| Multilineage dysplasia | 110 | 83 | 27 | Multilineage dysplasia | 21.5% | 24.3% | 15.9% | ||||||
| Not in others | 281 | 176 | 105 | Not in others | 55.0% | 51.6% | 61.8% | ||||||
| Cytogenetics | Favorable | 34 | 26 | 8 | 0.36 | Chi square | Cytogenetics | Favorable | 6.7% | 7.6% | 4.7% | 0.36 | Chi square |
| Intermediate | 225 | 170 | 82 | Intermediate | 44.0% | 49.9% | 48.2% | ||||||
| Unfavorable | 252 | 145 | 80 | Unfavorable | 49.3% | 42.5% | 47.1% | ||||||
| FLT3 | ITD | 83 | 49 | 34 | 0.145 | Chi square | FLT3 | ITD | 16.7% | 9.9% | 6.9% | 0.145 | Chi square |
| D835 | 25 | 17 | 8 | 0.92 | D835 | 5.0% | 3.4% | 1.6% | 0.92 | ||||
| Both | 9 | Both | |||||||||||
| NPM1 | 4BP insert | 55 | 179 | 89 | 0.76 | Chi square | NPM1 | 4BP insert | 16.3% | 83.6% | 81.7% | 0.76 | Chi square |
| WT | 283 | 35 | 20 | WT | 83.7% | 16.4% | 18.3% | ||||||
| ND | 173 | 127 | 61 | ND | 33.9% | 37.2% | 35.9% | ||||||
| Zubrod PS | 3 or 4 | 17 | 11 | 6 | Chi square | Zubrod PS | 3 or 4 | 3.3% | 3.2% | 3.5% | Chi square | ||
| AHD | ≥ 2 mo | 204 | 143 | 61 | 0.23 | Chi square | AHD | ≥ 2 mo | 39.9% | 41.9% | 35.9% | 0.23 | Chi square |
| Infection | Yes | 101 | 62 | 39 | 0.35 | Chi square | Infection | Yes | 19.8% | 18.2% | 22.9% | 0.35 | Chi square |
| WBC | Median | 8.8 | 6.9 | 19.25 | 0.02 | F test | WBC | Median | 8.8 | 6.9 | 19.3 | 0.02 | F test |
| Absolute blast count | Median | 1.8 | 1.13 | 3.54 | 0.28 | F test | Absolute blast count | Median | 1.8 | 1.1 | 3.5 | 0.28 | F test |
| Platelet | Median | 56.0 | 55 | 57 | 0.08 | F test | Platelet | Median | 56.0 | 55.0 | 57.0 | 0.08 | F test |
| Hemoglobin | Median | 9.6 | 9.6 | 9.45 | 0.15 | F test | Hemoglobin | Median | 9.6 | 9.6 | 9.5 | 0.15 | F test |
| % marrow blasts | Median | 46.0 | 44 | 57 | 0.0002 | F test | % marrow blasts | Median | 46.0 | 44.0 | 57.0 | 0.0002 | F test |
| % blood blasts | Median | 19.0 | 18 | 20.5 | 0.3 | F test | % blood blasts | Median | 19.0 | 18.0 | 20.5 | 0.3 | F test |
| Response | CR | 231 | 153 | 78 | 0.17 | Chi square | Response | CR | 56.5% | 44.9% | 45.9% | 0.17 | Chi square |
| Resistant | 136 | 101 | 35 | Resistant | 33.3% | 36.7% | 26.1% | ||||||
| Fail | 42 | 21 | 21 | Fail | 10.3% | 7.6% | 15.7% | ||||||
| Relapse | Yes | 151 | 93 | 58 | 0.09 | Chi square | Relapse | Yes | 65.4% | 60.8% | 74.4% | 0.09 | Chi square |
| Alive | Yes | 103 | 76 | 27 | 0.14 | Chi square | Alive | Yes | 20.2% | 22.3% | 15.9% | 0.14 | Chi square |
| Overall survival | Median, weeks | 40.85 | 44.14 | 35.14 | Overall survival | Median, weeks | 40.85 | 49.1 | |||||
| Remission duration | Median, weeks | 51.6 | 50 | 34.5 | Remission duration | Median, weeks | 51.6 | 52 | |||||
2.2. RPPA method
Proteomic profiling was done on samples from patients with AML using RPPA. The method and validation of the technique are fully described in previous publications [7], [19]. Briefly, patient samples were printed in five serial dilutions onto slides along with normalization and expression controls. Slides were probed with strictly validated primary antibodies. Antibodies against total GSK3α/β (Santa Cruz Biological, Catalog # 9331, primary dilution 1:200) and the serine 9 and serine 21 sites on GSK3α/B (Cell Signaling, Catalog #9331, primary antibody dilution 1:200) were used. This same array was probed with 229 other antibodies as listed in Supplemental File 1. An IgG subtype specific secondary antibody (1:15,000 dilution) was used to amplify the signal and finally a stable dye is precipitated. The stained slides were analyzed using the Microvigene software (Vigene Tech, Version 3.0) to produce quantified data.
2.3. PKCδ and FOXO3A knockdown cells
FOXO3A was knocked down in OCI-AML3 cells by lentiviral transduction using a gene-specific shRNAmir transfer vector (Clone V3LHS_375386 mature target sequence CGGAACGTGATGCTTCGCA, which targets 1862–1880 on RefSeq NM_001455.3 Clone V3LHS_641765 mature target sequence CGGCACAACCTGTCACTGC, which targets 974–992 on RefSeq NM_001455.3). PKCδ was knocked down in THP-1 and HL60 cells by lentiviral transduction using a gene-specific shRNA transfer vector (Clone TRCN0000010202 mature target sequence GCAAGACAACAGTGGGACCTA, which targets 1334–1354 on RefSeq NM_006254.3) (Open Biosystems, Huntsville, AL). Lentivirus was prepared by cotransfection of HEK293T cells (ATCC) with an equimolar mix of transfer vector and packaging plasmids (psPAX2 and pMD2.G from Addgene, Cambridge, MA) using JetPrime transfection reagent as directed by the manufacturer (Polyplus, Illkirch, France). Fresh lentiviral supernatants were passed through 0.45 μm pore surfactant-free cellulose acetate membranes and then used at once to infect OCI-AML3, THP-1, and HL60 cells by incubation overnight at 37 °C under 5% CO2. OCI-AML3 cells were the kind gift from Mark Minden (Ontario Cancer Institute; Toronto, Canada). THP-1 and HL60 cells were purchased from ATCC (Manassas, VA). Cells were subject to selection with puromycin (Invivogen, San Diego CA) starting at 1 μg/mL. In parallel, all cells were transduced using lentivirus delivering a non-specific control (Open Biosystems). Knockdown was verified by Western blot analysis and by real time PCR.
2.4. Western blot analysis
Cells were treated with various doses of MK-2206 (Selleck Chemicals) for 6 h or 5 μM GSK3 Inhibitor XVI (Calbiochem) for 6 h. Cells were sonicated in lysis buffer (62.5 mM Tris (pH 8.0), 2% SDS, 10% glycerol, 100 μM AEBSF, 80 nM aprotinin, 5 μM bestatin, 1.5 μM E-64, 2 μM leupeptin, 1 μM pepstatin, 500 μM sodium orthovanadate, 500 μM glycerol phosphate, 500 μM sodium pyrophosphate and 50 μM DTT) and protein (5 × 105 cell equivalents) was subjected to electrophoresis using 10–14% acrylamide/0.1% SDS gels. Proteins were transferred to a nitrocellulose membrane and Western blotting analysis was performed with antibodies against phospho-PKCδ (Cell Signaling, Beverly, MA), PKCδ (Santa Cruz Biotechnology), phospho-GSK3α/β (serine 9/21; Cell Signaling), GSK3α/β (Cell Signaling), MYC (Cell Signaling), Foxo3A (Cell Signaling), and Tubulin (Sigma). Signals were detected by using the Odyssey Infrared Imaging System and quantitated by Odyssey software version 3.0 (both LI-COR Biosciences, Lincoln, NE, USA). Tubulin was used as a loading control.
2.5. Statistical analyses
For RPPA, super curve algorithms were used to generate a single value from the five serial dilutions [20]. Loading control and topographical normalization procedures accounted for protein concentration and background staining variations [21]. Analysis using unbiased clustering perturbation bootstrap clustering, and principle component analysis was then done as fully described in a previous publication [7]. The cohort was divided into sixths based on the range of expression of all 511 samples. Based on a similarity of outcomes these were collapsed into two groups with expression in the upper third vs. the lower two thirds for phospho-GSK3α/β. Comparison of the protein levels between paired samples was done by performing paired t test. Association between protein expression levels and categorical clinical variables were assessed in R using standard t-tests, linear regression, or mixed effects linear models. Association between continuous variable and protein levels were assessed by using the Pearson and Spearman correlation and linear regression. Bonferroni corrections were done to account for multiple statistical parameters for calculating statistical significance. The Kaplan–Meier method was used to generate the survival curves. Univariate and multivariate Cox proportional hazard modeling was done to investigate association with survival with protein levels as categorized variables using the Statistica version 12 software (StatSoft, Tulsa, OK). Although some molecular markers (NPM1, FLT3-ITD, and RAS mutations) are known for this dataset other more recently discovered prognostic markers (e.g., DNMT3A, CEBPα, Wilms Tumor 1) are not; therefore, the multivariate analysis did not contain all known AML prognostic markers. Overall survival was determined based on the outcome of 415 newly diagnosed AML patients treated at UTMDACC and RD was based on the 237 patients that achieved remission.
3. Results
3.1. Phosphorylated GSK3β levels are elevated in a subset of AML patient cell samples compared to normal CD34+ cells
We have used RPPA for the analysis of protein expression from clinical samples in many hematologic malignancies [7], [8], [19], [22], [23], [24]. Analysis of proteins of interest is possible as long as a validated antibody is available. We used RPPA to study total and phosphorylated GSK3α/β (at serine 21 and serine 9, respectively) in AML blast cells obtained from 511 newly diagnosed AML patients and CD34+ cells obtained from 11 normal bone marrow donors. We compared phosphorylation status of the kinase in the AML patient set with that in normal CD34+ cells. Total levels of GSK3β protein were significantly (p = 0.0059) lower in the blast cells from the AML patients compared to normal CD34+ cells as shown in the histogram in Fig. 1A. Levels of phosphorylated GSK3α/β protein were lower in the AML cells overall (p < 0.0273; Fig. 1B) with 17.4% of cases having levels above normal and 35.8% with levels below normal CD34+ cells. There is a set of patients that show high levels of phosphorylated GSK3α/β on the histogram in Fig. 1B. Differences in phosphorylation status of the kinase among various AML sub-types determined by French–American–British (FAB) class and cytogenetic category were very pronounced (p < 0.0001 for both categories; Supplemental Fig. S1A and B). Corresponding plots for total GSK3α/B are shown in Supplementary Fig. S1C and D. Levels of total and phosphorylated GSK3α/β did not differ between diagnosis and relapse in 47 patients with paired samples (p = 0.21 for total protein and p = 0.31 for phosphorylated protein).
Fig. 1.

GSK3 α/β total and serine 21/9 phosphorylated protein expression in AML blast cells compared to normal counterpart CD34 + cells. RPPA was performed as described in the “Materials and methods” section. Expression of total (A) and phosphorylated (B) GSK3 α/Β was compared in blast cells from 511 AML patients versus CD34 + bone marrow cells from 11 normal donors.
Supplemental Fig. 1.

GSK3 α/β total and serine 21/9 phosphorylated protein expression in AML blast cells by FAB and cytogenetics. RPPA was performed as described in the “Materials and methods” section. Expression of phosphorylated and total GSK3 α/Β was compared by FAB category (A and C for phosphorylated and total, respectively) and cytogenetic category (B and D for phosphorylated and total, respectively) from AML patients versus CD34 + bone marrow cells from 11 normal donors.
3.2. Phosphorylated GSK3β levels are associated with poor survival outcome and shorter RD
The level of total GSK3α/β, divided at the median, was not predictive of remission attainment (55.5 vs. 54.8%, respectively) RD (46 vs. 46 weeks, respectively, p = 0.68), or overall survival (49 vs. 49 weeks, respectively, p = .80). For survival analysis, RPPA values for phospho-GSK3α/β were categorized into two groups (i.e., lower two-thirds and upper one-third) based on the range of expression in all 511 cases. However among all AML patients, those with higher levels of phosphorylated GSK3α/β exhibited shorter overall survival duration compared to those with low levels of the phosphorylated kinase (40 vs 54 weeks, respectively; p = 0.015; Fig. 2A). For patients with intermediate cytogenetics, phosphorylated GSK3α/β predicted poor overall survival as patients with elevated levels of phosphorylated kinase exhibited significantly shorter overall survival duration compared to patients with lower levels of phosphorylated GSK3α/β (Fig. 2B; 56 vs 77.8 weeks, respectively; p = 0.018). Phosphorylation status of GSK3α/β had no effect on overall survival duration in patients with unfavorable cytogenetics (Fig. 2C). Likewise no difference was seen among those with favorable cytogenetics though the patient sample size was small (N = 32; p = 0.39; data not shown).
Fig. 2.

Correlation of overall survival with serine 21/9 GSK 3α/β phosphorylation levels in AML patient cytogenetic subsets. The Kaplan–Meier curve for overall survival (OS) for the total AML patient set (A; N = 415), intermediate cytogenetic patient set (B; N = 187), and unfavorable cytogenetic patient set (C; N = 195) based on expression of phosphorylated GSK3 α/β.
The phosphorylation of GSK3 kinases also influenced RD. Among all categories of AML patients that achieved remission (N = 237), patients with higher levels of phosphorylated GSK3α/β exhibited shorter RD compared to those with low levels of phosphorylated GSK3α/β (Fig. 3A; 43 weeks vs 67 weeks respectively; p = 0.022). Among intermediate cytogenetic patients (N = 121), the influence of phosphorylation of GSK3 kinase on RD was more pronounced as patients with higher levels of phosphorylated GSK3α/β displayed much shorter RD compared to patients with low levels (Fig. 3B; 45 weeks vs 95 weeks respectively; p = 0.0075). This correlation was observed even in the intermediate cytogenetic patients (N = 35) with FLT3 mutation (Fig. 3C; 24 weeks vs 50 weeks respectively; p = 0.009).
Fig. 3.

Correlation of remission duration with serine 21/9 GSK 3α/β phosphorylation levels in AML patient cytogenetic subsets. The Kaplan–Meier curve for remission duration for the total treated AML patient set (A; N = 231), treated intermediate cytogenetic patient set (B; N = 120), and treated intermediate cytogenetic with the FLT3 mutant patient set (C; N = 35) based on expression of phosphorylated GSK3α/β.
3.3. Phosphorylation of GSK3 kinases correlates with activation of AKT and other signaling molecules in blast cells from AML patients
We have previously used RPPA to study the survival signaling pathways in AML blast cells including those that involve AKT [7], [8], [19], [22], [23], [24]. RPPA was used to examine correlations of total or phosphorylated GSK3α/β with 229 proteins in blast cells from 511 AML patients. Sixty four proteins are significantly correlated with total GSK3 expression in the AML cells (p < 0.0001; R > 030; Supplemental Fig. 2). As shown in Fig. 4, forty-four proteins showed statistically significant (p < 0.0001, R > 0.20) correlation with phosphorylated GSK3α/β. Twenty four proteins displayed a positive correlation while twenty proteins displayed a negative correlation with the phospho-kinase. The two proteins that exhibited the greatest positive correlation with phosphorylated GSK3 α/β are phospho-S6K1 (T389) and phospho-AKT (S473). AKT phosphorylated at S473 reflects the active form of the kinase. That higher levels of phosphorylated GSK3 α/β are found when AKT is active is consistent with AKT's role as a GSK3 kinase [11], [25]. S6K1 has also been shown to phosphorylate GSK3 kinases, though usually in the absence of TSC2 [26]. Interestingly, phosphorylated PKCδ (at both T507 and S645) was found to be positively correlated with phospho-GSK3 levels. PKC δ has often been viewed as a stress kinase but recent studies suggest that the enzyme can have pro-survival properties [17], [18], [27], [28]. A recent study by Kinehara and colleagues suggest that PKCδ may phosphorylate GSK3 kinases in human pluripotent stem cells as part of a mechanism to regulate stem cell renewal [28].
Supplemental Fig. 2.

Proteins correlated with total GSK3 α/β expression in AML patients. Negative and positive correlation from a list of 229 proteins also assayed compared to total GSK3 α/β expression using the same reverse phase proteomics analysis on the same samples; Pearson correlation coefficient R > 0.25, p < 0.001.
Fig. 4.

Proteins correlated with phosphorylated GSK3α/β expression in AML patients. Negative and positive correlation from a list of 229 proteins also assayed compared to phosphorylated GSK3α/β expression using the same reverse phase proteomics analysis on the same samples; Pearson correlation coefficient R > 0.25, p < 0.001.
RPPA revealed a negative correlation between FOXO3A and phospho-GSK3. Since FOXO3A is negatively regulated by AKT, the negative association between phospho-GSK3 and the forkhead protein is likely an indicator of AKT activity in the individual patients. Included among the proteins exhibiting negative correlation with phosphorylated GSK3 is the epidermal growth factor receptor (EGFR) phosphorylated at tyrosine 922 (Y922). EGFR is a membrane tyrosine kinase that is activated by factors such as EGF and transforming growth factor α (TGFα). EGFR can act as an upstream regulator of many kinase pathways including PI3K/AKT, so a negative correlation between phosphorylated (i.e., active) EGFR and phospho-GSK3 would be predicted. Implications of EGFR regulation of GSK3 activity, however, may be very important as Du and colleagues have recently demonstrated that the stem cell phenotype of cancer stem cells may be dependent on suppression of GSK3 via an EGFR/AKT axis [29]. The protein showing the strongest negative correlation with phospho-GSK3 was serine 127 phosphorylated Yes Associated Protein 1 (YAP1). YAP1 is a transcriptional co-activator that is part of the Hippo regulatory pathways [30], [31]. When YAP1 is phosphorylated on serine 127, it is expelled from the nucleus and YAP1 driven gene expression decreases. While EGFR supports YAP1 phosphorylation via PI3K, the mechanism is independent of AKT [31]. There is no evidence that GSK3 is a YAP1 kinase. However, it is possible that GSK3 could be promoting YAP1 phosphorylation via suppression of a protein phosphatase. GSK3 suppresses PP2A function [32] and PP2A has been shown to regulate phosphorylation of YAP1 [30].
Considering that GSK3 serine 9/serine 21 phosphorylation levels were significant to survival characteristics in the intermediate cytogenetic AML patients, we examined protein correlations between the phospho-kinase and the 229 proteins and the AML patients in this subset. Most of the proteins that were represented in all AML patients were also found in the intermediate cytogenetic group. As shown in Fig. 5, PKCδ, phosphorylated on either threonine 507 or serine 645, serine 473 phosphorylated AKT and serine 217/221 phosphorylated MEK1 (MAP2K1) are still among the proteins with the strong positive correlation with phospho-GSK3. A few proteins such as ATG7 and EIF2S1 did not correlate with phosphorylated GSK in the intermediate cytogenetic patients compared to all AML patients (Fig. 5). Also, Galectin 3 (LGALS3) was found to correlate in the Intermediate group though this protein was not in the group of total AML patients. Serine 127 phosphorylated YAP1, β-catenin (CTNNB1), KIT, and HIF1α (HIF1A) were still among the proteins with strong correlation with phospho-GSK, and two proteins not found in the total AML set, PIM1 and Asparagine Synthase (ASNS), were found in the Intermediate subset of patients (Fig. 5).
Fig. 5.

Proteins correlated with phosphorylated GSK3α/β expression in the intermediate cytogenetic group AML patients. Negative and positive correlation from a list of 229 proteins also assayed compared to phosphorylated GSK3α/β expression using the same reverse phase proteomics analysis on the same samples; Pearson correlation coefficient R > 0.25, p < 0.001.
3.4. AKT is a positive regulator of FOXO3A and PKCδ that is independent of GSK3
AKT is well known to regulate FOXO3A [4], [5]. OCI-AML3 cells exhibit basal levels of FOXO3A phosphorylated at threonine 32 (T32; Fig. 6A). As shown in Fig. 6A, treatment of OCI-AML3 cells with the specific AKT inhibitor MK-2206 suppresses T32 phosphorylation of FOXO3A indicating that AKT is likely the FOXO3A kinase in these cells. While GSK3 is known to phosphorylate a number of transcription factors including MYC, MAX, and USF1 [33], the kinase is not known to target FOXO3A. To investigate the relationship between FOXO3A and GSK3, expression and phosphorylation status of GSK3 was compared in OCI-AML3 cells expressing a lentiviral control vector targeting a non-human gene (NH; target sequence for green fluorescent protein; GFP) with cells expressing lentiviral shRNA against FOXO3A. As shown in Fig. 6B, two transductants displayed roughly 40% reduction of FOXO3A. However, there was no difference in either total GSK3α/β or phosphorylated kinase in either FOXO3A shRNA transductant suggesting that FOXO3A does not act directly on GSK3. The association between GSK3 phosphorylation status and FOXO3A levels in the AML patient samples likely reflects AKT activity in those samples (i.e. both GSK3 and FOXO3A are AKT substrates). The inhibition of FOXO3A phosphorylation by MK-2206 (Fig. 6A) supports this hypothesis.
Fig. 6.

Role of GSK3α/β on AKT suppression of FOXO3A and PKCδ in AML cell lines. (A) Protein lysate (200,000 cell equivalents) from OCI-AML3 cells containing control (NS) lentiviral plasmid or cells containing FOXO3A shRNA using 2 different lentiviral plasmids, and THP-1 cells or HL60 cells containing control (NS) lentiviral plasmid or cells containing PKCδ shRNA lentiviral plasmid were subject to SDS/PAGE and Western analysis performed for GSK3α/β, phosphorylated GSK3α/β, PKC d, FOXO3A, MYC, and Tubulin. Ratios were determined by densitometric analysis of the bands depicted in the figure and normalized to each NS control. (B) Protein lysate (200,000 cell equivalents) from OCI-AML3 cells treated with vehicle (-/-; 0.1 % DMS) or Calbiochem GSK3 inhibitor XVI (5 μM) or MK-2206 AKT inhibitor (1 μM) for 6 h were subject to SDS/PAGE and Western analysis performed for GSK3α/β, phosphorylated GSK3α/β, PKCδ, phosphorylated PKCδ, and Tubulin. Ratios were determined by densitometric analysis of the bands depicted in the figure and normalized to vehicle treated control.
The RPPA data suggests that there is a positive association between phospho-GSK3 and phospho-PKCδ (Fig. 4, Fig. 5). Phosphorylated GSK3 indicates that the kinase is inactive while phosphorylation of PKCδ indicates that the kinase is active. A logical assumption would be that PKCδ may act as a GSK3 kinase. Such a function for the PKC has been suggested by Kinehara and colleagues [28]. PKCδ expression was suppressed by lentiviral shRNA in two AML derived cell lines (i.e., THP-1 and HL60). As shown in Fig. 6B, there was roughly 80% reduction of the kinase in THP-1 cells and approximately 60% reduction of the PKC in HL60 cells (when compared to cells with control NH vector). There was no difference in either total GSK3α/β or phosphorylated GSK3α/β in cells with PKCδ shRNA in either cell line suggesting that PKCδ does not act directly on GSK3. The study by Kinehara and colleagues [28] involved human pluripotent stem cells so the action of the PKC on GSK3 may be different in leukemia cells compared to normal stem cells. A second possibility is that GSK3 acts on the PKC. OCI-AML3 cells were treated with 5 μM dose of a GSK3α/β inhibitor (Calbiochem Inhibitor XVI) for 6 h. As shown in Fig. 6C, the inhibitor blocked GSK3 phosphorylation though the molecule induced expression of total protein. However, inhibition of GSK3 had no effect on levels of phospho-PKCδ (Fig. 6C). MK-2206 is a specific inhibitor of AKT [35]. Treatment of OCI-AML3 cells with 1 μM dose of MK-2206 blocked phosphorylation of both GSK3 and PKCδ (Fig. 6C). This novel finding suggests that AKT positively regulates PKCδ though it remains to be determined if the kinase acts directly on the PKC. These results suggest that the correlation between phospho-GSK3 levels and phospho-PKCδ levels in the AML patient samples likely represent AKT activity in the samples (phosphorylation of both GSK3 and PKCδ are positively regulated by AKT).
4. Discussion
The GSK3 kinases are involved in diverse cellular functions regulating cell growth, proliferation, survival, adhesion, and differentiation [9], [10], [11]. These enzymes are critical for maintaining cellular homeostasis by regulating metabolic processes (e.g., glycolysis), transcription (e.g., MYC and WNT pathways), translation (e.g., via mTOR), cell death processes including apoptosis (e.g., via MCL-1) and autophagy (e.g., via Bif-1) to name a few processes [9], [10], [11], [12], [13], [14]. The GSK3 kinases are clearly important cellular rheostats. Since GSK3 phosphorylation of survival proteins such as MCL-1, β-catenin, and various cyclins results in the proteolytic destruction of these proteins, GSK3 is often viewed as a tumor suppressor [11], [13]. Indeed as AKT is viewed as a pro-survival kinase that inactivates GSK3 in many cancers including AML, this would support strategies for AML therapy to suppress AKT and if possible, support GSK3 function. Consistent with this notion, in our RPPA analysis of phosphorylated GSK3 AML patients from the intermediate cytogenetic groups fare better in overall survival and in RD when they express lower levels of phosphorylated GSK3 (see Fig. 2, Fig. 3).
GSK3 phosphorylation status had no impact on survival or RD in AML patients from the unfavorable cytogenetic category (see Fig. 2, Fig. 3). The PI3K/AKT/GSK axis may not be as potent in this population of patients or the various enzymes have different roles in these AML cells compared to leukemia cells from the favorable/intermediate cytogenetic groups. Recent studies suggest there are instances where GSK3 can function as a tumor promoter [11]. Banerji and colleagues have found that GSK3α may act as a negative regulator of differentiation in AML cells and thus targeting at least the α isoform of the kinase could have therapeutic benefits [34]. Scadden's group has evidence that AKT may not be critical in leukemia stem cells and that actually FOXO3A may play a more prominent role maintaining these cells [36]. The possibility exists that GSK may have a role in supporting these leukemic stem cells [11], [36]. Wang and colleagues have found that GSK3 supports association of CREB and MEIS1, which is critical for HOX-mediated transformation in leukemia [37]. It is likely that the role of GSK3 (or AKT for that matter) in AML is complex and likely varies from patient to patient.
Among the 229 other proteins examined by RPPA, many of the well-established GSK3 substrates including SMAD1, HIF-1α, and β-catenin were negatively correlated with phosphorylated GSK3 (Fig. 4 and Fig. 4B) as expected. This finding is consistent with having low levels of these target proteins when active (i.e., unphosphorylated) GSK3 is present. Still, a number of other GSK3 targets either showed no correlation (e.g. p21, MCL1; data not shown) or positive correlation (SMAD3, Cyclin E; Fig. 4). This variability likely reflects the complexity of the regulatory pathways at work in AML and warrants further investigation. Some of the proteins, such as FOXO3A that displayed a negative correlation with phospho-GSK3, likely do not directly interact with GSK3, but rather are substrates of AKT like GSK3. Suppression of FOXO3A by shRNA in the AML cell line OCI-AML3 had no effect on phosphorylation status of GSK3 (Fig. 6B). However, inhibition of AKT with a specific inhibitor (i.e., MK-2206) suppresses phosphorylation and reduces FOXO3A levels in the OCI-AML3 cells (Fig. 6A).
Consistent with conditions where AKT is active, RPPA revealed that phospho-GSK3 levels were positively correlated with AKT phosphorylated at serine 473, with BAD phosphorylated at serine 136, and with Ribosomal Protein S6 phosphorylated at serine 235 and 236 (Fig. 4). These events would suggest support of leukemia survival as phosphorylation of BAD at S136 inhibits its pro-apoptotic function while phosphorylation of Ribosomal Protein S6 supports protein translation [38]. Interestingly, there was also a strong correlation with phosphorylation (i.e., activation) of PKCδ at both threonine 507 and serine 645 (Fig. 4). Regulation of PKCδ by GSK3 or AKT is not clear. The reduction of phosphorylated PKCδ with the AKT inhibitor and not the GSK3 inhibitor suggests that AKT may act as a PKCδ kinase. The novel possibility suggests a new survival cascade that AKT may regulate since there is emerging evidence that the PKCδ can act as a survival kinase [17], [18]. A model is depicted in Fig. 7 showing some of the possible survival pathways that may be critical for AML cell survival that depend on activation of AKT with inactive GSK3.
Fig. 7.
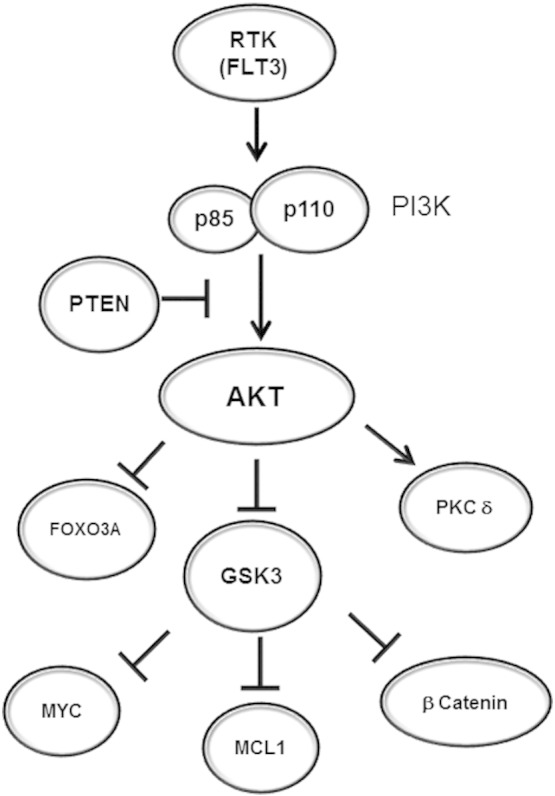
Model of AKT and GSK3α/β-mediated pathways in AML cells. Extracellular survival signals activate AKT which suppresses GSK3α/β. When AKT is suppressed, FOXO3A and PKCδ are inhibited independent of GSK3α/β. GSK3α/β when active will suppress MYC, MCL-1, and β-catenin.
While it is not yet determined if inactivation of GSK3 by AKT is required for its pro-survival function, a recent report from the Grant lab suggests that inactivation of GSK3 is at least necessary if not sufficient for apoptotic pathways involving inhibition of AKT in leukemia cells [39]. In that study, U937 cells in which GSK3 is suppressed either by a pharmacologic inhibitor such as CHIR-98,014 or by shRNA become resistant to killing by the PI3K/mTor inhibitor BEZ235 in combination with the BH3 mimetic ABT-737 [39]. A mechanism whereby GSK3 activation alters expression of targets such as MCL-1, FOXO3A, CTNNB1 (β-catenin), or MYC was suggested. While in the RPPA analysis in the present study no significant correlation between phospho-GSK3 and MCL-1 was observed, we did see expression of FOXO3A and β-catenin negatively correlated with the phosphorylated (i.e., inactive) kinase (Fig. 4). Studies are ongoing to determine the mechanistic role of GSK3 activation for apoptosis in AML cells.
In summary, the findings presented here suggest that GSK phosphorylation is likely an important event in AML patients with intermediate cytogenetics. Phosphorylation (i.e. inactivation) of the GSK3 kinases is associated with poor survival outcome in these AML patients. The GSK3 targets that may be critical for leukemia cell survival remain to be determined. Still, strategies to promote GSK3 activation by inhibiting GSK3 kinases such as AKT or by inducing protein phosphatases such as PP2A could be beneficial to improving therapy for at least some AML patients.
The following are the supplementary data related to this article.
This table shows the modified Hugo name for the protein studied, the full name from the GeneCards website, the manufacturer of the antibody used, and the catalog number from that manufacturer. Proteins often have many different names and current nomenclature systems do not account for post translational modifications. We use a modification of the Hugo nomenclature system in which we use the Hugo name followed by a dot (“.”) which is then followed by details of the post-translational modifications. The convention uses "p" for phosphorylation, "cl" for cleavage or "Me" for methylation, and this is followed by the amino acid "Y" for tyrosine, "S" for serine, "T" for threonine and/or the position number of the affected amino acid. Hence BAD.pS112 is the protein Bad, phosphorylated on serine 112, while Casp9.cl315 is caspase 9 cleaved at amino acid 315, and H3K4.Me2 is Histone3 lysine 4 di-methylated. Including the post-transnational modification after the protein name allows for alphabetical sorting, whereas putting the post translational modification before the protein name will sort all the phosphorylated antibodies together, but separate from their unmodified forms.
Transparency document
Transparency document.
Acknowledgments
This work was supported by the Leukemia Lymphoma Society (Translational Research Grant to SMK) and National Institutes of Health (PO1 grant CA-55164 to MA).
Footnotes
The Transparency document associated with this article can be found in the online version.
Contributor Information
Peter P. Ruvolo, Email: pruvolo@mdanderson.org.
Steven M. Kornblau, Email: skornblau@mdanderson.org.
References
- 1.Martelli A.M., Evangelisti C., Chiarini F., McCubrey J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 2010;1:89–103. doi: 10.18632/oncotarget.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fathi A.T., Grant S., Karp J.E. Exploiting cellular pathways to develop new treatment strategies for AML. Cancer Treat. Rev. 2010;36:142–150. doi: 10.1016/j.ctrv.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samudio I., Konopleva M., Carter B., Andreeff M. Apoptosis in leukemias: regulation and therapeutic targeting. Cancer Treat. Res. 2010;145:197–217. doi: 10.1007/978-0-387-69259-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martelli A.M., Evangelisti C., Chiarini F., Grimaldi C., Manzoli L., McCubrey J.A. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin. Investig. Drugs. 2009;18:1333–1349. doi: 10.1517/14728220903136775. [DOI] [PubMed] [Google Scholar]
- 5.Martelli A.M., Nyåkern M., Tabellini G., Bortul R., Tazzari P.L., Evangelisti C. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911–928. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 6.Scholl C., Gilliland D.G., Fröhling S. Deregulation of signaling pathways in acute myeloid leukemia. Semin. Oncol. 2008;35:336–345. doi: 10.1053/j.seminoncol.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Kornblau S.M., Tibes R., Qiu Y.H., Chen W., Kantarjian H.M., Andreeff M. Functional proteomic profiling of AML predicts response and survival. Blood. 2009;113:154–164. doi: 10.1182/blood-2007-10-119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kornblau S.M., Womble M., Qiu Y.H., Jackson C.E., Chen W., Konopleva M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108:2358–2365. doi: 10.1182/blood-2006-02-003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rayasam G.V., Tulasi V.K., Sodhi R., Davis J.A., Ray A. Glycogen synthase kinase 3: more than a namesake. Br. J. Pharmacol. 2009;156:885–898. doi: 10.1111/j.1476-5381.2008.00085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi-Yanaga F. Activator or inhibitor? GSK-3 as a new drug target. Biochem. Pharmacol. 2013;86:191–199. doi: 10.1016/j.bcp.2013.04.022. [DOI] [PubMed] [Google Scholar]
- 11.McCubrey J.A., Steelman L.S., Bertrand F.E., Davis N.M., Abrams S.L., Montalto G. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. 2014;28:15–33. doi: 10.1038/leu.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linding R., Jensen L.J., Ostheimer G.J., van Vugt M.A., Jørgensen C., Miron I.M. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu C., Kim N.G., Gumbiner B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle. 2009;8:4032–4039. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J., Takahashi Y., Cheng E., Liu J., Terranova P.F., Zhao B. GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death. J. Cell Sci. 2010 Mar 15;123(Pt 6):861–870. doi: 10.1242/jcs.060475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazemi Z., Chang H., Haserodt S., McKen C., Zachara N.E. O-linked beta-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3beta-dependent manner. J. Biol. Chem. 2010 Dec 10;285(50):39096–39107. doi: 10.1074/jbc.M110.131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin T., George Fantus I., Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cell. Signal. 2008;20:1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Ruvolo V.R., Karanjeet K.B., Schuster T.F., Brown R., Deng Y., Hinchcliffe E. Role for PKC δ in fenretinide-mediated apoptosis in lymphoid leukemia cells. J. Signal. Transduct. 2010 Jan 1;2010:584657. doi: 10.1155/2010/584657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basu A., Pal D. Two faces of protein kinase Cδ: the contrasting roles of PKCδ in cell survival and cell death. Sci. World J. 2010;10:2272–2284. doi: 10.1100/tsw.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kornblau S.M., Singh N., Qiu Y., Chen W., Zhang N., Coombes K.R. Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin. Cancer Res. 2010;16:1865–1874. doi: 10.1158/1078-0432.CCR-09-2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu J., He X., Baggerly K.A., Coombes K.R., Hennessy B.T., Mills G.B. Non-parametric quantification of protein lysate arrays. Bioinformatics. 2007;23:1986–1994. doi: 10.1093/bioinformatics/btm283. [DOI] [PubMed] [Google Scholar]
- 21.Neeley E.S., Kornblau S.M., Coombes K.R., Baggerly K.A. Variable slope normalization of reverse phase protein arrays. Bioinformatics. 2009;25:1384–1389. doi: 10.1093/bioinformatics/btp174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tibes R., Qiu Y., Lu Y., Hennessy B., Andreeff M., Mills G.B. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol. Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 23.Ruvolo P.P., Qui Y.H., Coombes K.R., Zhang N., Ruvolo V.R., Borthakur G. Low expression of PP2A regulatory subunit B55α is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia. 2011;25:1711–1717. doi: 10.1038/leu.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kornblau S.M., Qutub A., Yao H., York H., Qiu Y.H., Graber D. Proteomic profiling identifies distinct protein patterns in acute myelogenous leukemia CD34+ CD38− stem-like cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0078453. e78453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H.H., Lipovsky A.I., Dibble C.C., Sahin M., Manning B.D. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell. 2006;24:185–197. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emoto Y., Manome Y., Meinhardt G., Kisaki H., Kharbanda S., Robertson M. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinehara M., Kawamura S., Tateyama D., Suga M., Matsumura H., Mimura S. Protein kinase C regulates human pluripotent stem cell self-renewal. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0054122. e54122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du W.W., Fang L., Yang X., Sheng W., Yang B.L., Seth A. The role of versican in modulating breast cancer cell self-renewal. Mol. Cancer Res. 2013;11:443–455. doi: 10.1158/1541-7786.MCR-12-0461. [DOI] [PubMed] [Google Scholar]
- 30.Johnson R., Halder G. The two faces of hippo: targeting the hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan R., Kim N.G., Gumbiner B.M. Regulation of hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2569–2574. doi: 10.1073/pnas.1216462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao X.Q., Zhang X.X., Yin Y.Y., Liu B., Luo D.J., Liu D. Glycogen synthase kinase-3β regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not Src. Biochem. J. 2011;437:335–344. doi: 10.1042/BJ20110347. [DOI] [PubMed] [Google Scholar]
- 33.Terragni J., Nayak G., Banerjee S., Medrano J.L., Graham J.R., Brennan J.F. The E-box binding factors max/Mnt, MITF, and USF1 act coordinately with FoxO to regulate expression of proapoptotic and cell cycle control genes by phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3 signaling. J. Biol. Chem. 2011;286:36215–36227. doi: 10.1074/jbc.M111.246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerji V., Frumm S.M., Ross K.N., Li L.S., Schinzel A.C., Hahn C.K. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Invest. 2012;122:935–947. doi: 10.1172/JCI46465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirai H., Sootome H., Nakatsuru Y., Miyama K., Taguchi S., Tsujioka K. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 36.Sykes S.M., Lane S.W., Bullinger L., Kalaitzidis D., Yusuf R., Saez B. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z., Iwasaki M., Ficara F., Lin C., Matheny C., Wong S.H. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell. 2010;17:597–608. doi: 10.1016/j.ccr.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martelli A.M., Evangelisti C., Chappell W., Abrams S.L., Bäsecke J., Stivala F. Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia. 2011 Jul;25(7):1064–1079. doi: 10.1038/leu.2011.46. [DOI] [PubMed] [Google Scholar]
- 39.Rahmani M., Aust M.M., Attkisson E., Williams D.C., Jr., Ferreira-Gonzalez A., Grant S. Dual inhibition of Bcl-2 and Bcl-xL strikingly enhances PI3K inhibition-induced apoptosis in human myeloid leukemia cells through a GSK3- and Bim-dependent mechanism. Cancer Res. 2013;73:1340–1351. doi: 10.1158/0008-5472.CAN-12-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table shows the modified Hugo name for the protein studied, the full name from the GeneCards website, the manufacturer of the antibody used, and the catalog number from that manufacturer. Proteins often have many different names and current nomenclature systems do not account for post translational modifications. We use a modification of the Hugo nomenclature system in which we use the Hugo name followed by a dot (“.”) which is then followed by details of the post-translational modifications. The convention uses "p" for phosphorylation, "cl" for cleavage or "Me" for methylation, and this is followed by the amino acid "Y" for tyrosine, "S" for serine, "T" for threonine and/or the position number of the affected amino acid. Hence BAD.pS112 is the protein Bad, phosphorylated on serine 112, while Casp9.cl315 is caspase 9 cleaved at amino acid 315, and H3K4.Me2 is Histone3 lysine 4 di-methylated. Including the post-transnational modification after the protein name allows for alphabetical sorting, whereas putting the post translational modification before the protein name will sort all the phosphorylated antibodies together, but separate from their unmodified forms.
Transparency document.