

Abstract
Identifying the microbiome composition from primary tissues directly affords an opportunity to study the causative relationships between the host microbiome and disease. However, this is challenging due the low abundance of microbial DNA relative to the host. We present a systematic evaluation of microbiome profiling directly from endoscopic biopsies by whole genome sequencing. We compared our methods with other approaches on datasets with previously identified microbial composition. We applied this approach to identify the microbiome from 27 stomach biopsies, and validated the presence of Helicobacter pylori by quantitative PCR. Finally, we profiled the microbial composition in The Cancer Genome Atlas gastric adenocarcinoma cohort.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-015-0821-z) contains supplementary material, which is available to authorized users.
Background
The human microbiome is a critical constituent of normal human physiology as well as in the pathogenesis of disease [1, 2], including malignancy [3–6]. Conservative estimates are that greater than 15 % of all malignancies may be attributed to microbiota [7], though the mechanisms by which the human microbiome contribute to malignancy remain largely unknown. There are several hypotheses, including microbiota inducing oxidative stress, altering immunosurveillance, changing local metabolism, affecting stem cell dynamics, or producing mutagenic metabolites [8]. Gastric cancer is one of the most commonly diagnosed gastrointestinal malignancies worldwide. It is responsible for nearly one million new cases and over 700,000 deaths annually [9]. The fatality:case ratio is high, demonstrating that the majority of patients diagnosed with gastric cancer will die of their disease, despite advances in drug therapy [10]. Gastric cancer is divided into specific subtypes based on specific epidemiology and risk factors [11] and, more recently, molecular profiles [11–13]. Helicobacter pylori is an endemic bacterial pathogen that infects nearly half of the world’s population [14], and is a WHO class I carcinogen for the development of gastric cancer, specifically the non-cardia, non-diffuse subtype of gastric cancer with an approximately threefold increased risk of malignancy in chronically infected individuals. H. pylori infection is believed to result in changes in gastric mucosal physiology and the epithelial host immune system [15–18]. Although several studies have examined causative features of H. pylori-associated gastric cancer, including bacterial virulence factors (CagA and VacA) [19–21], and host genetic alterations [22], what determines the consequence of H. pylori infection (i.e., whether mild gastritis, more severe peptic ulcer disease, or even gastric cancer) remains unknown.
Recently, a comprehensive molecular analysis performed through The Cancer Genome Atlas (TCGA) identified four distinct gastric cancer subtypes — (1) Epstein-Barr virus (EBV)-positive, (2) microsatellite instability, (3) genomically stable, and (4) chromosomal instability — based on their characterization using six molecular platforms: array-based somatic copy number analysis, whole-exome sequencing, array-based DNA methylation profiling, messenger RNA sequencing, microRNA sequencing and reverse-phase protein array [13]. The notable finding that EBV-associated gastric cancer is molecularly distinct from other gastric cancer subtypes speaks to the importance of external pathogens in this disease. However, this study did not report on the microbial composition in these tumor samples and the annotation of H. pylori was incomplete. Data are emerging on the diversity of the bacterial population (over hundreds of phylotypes [23, 24]) that reside in the stomach [23] and its dynamic composition associated with different disease states [25–27]. The microbial community in the stomach is typically limited by the low pH of the gastric lumen, which selects for acid-resistant bacterial populations, and usually limits the colonization densities to less than 1000 colony-forming units/g [24]. Due to the rise in pH caused by H. pylori urease activity [28] as well as other factors, H. pylori infection may have important effects on the composition of the gastric microbiome. However, the effect of H. pylori on the gastric microbiome is not well studied due to inadequate numbers of tissue samples and low bacterial content. Current methodology for profiling the microbiome from fecal or environmental samples is not directly applicable for detecting the microbiome from the upper gastrointestinal tract (i.e., stomach), in part due to the high content of human DNA in the sample that confounds microbial identification. Here we report on a systematic study utilizing whole genome sequencing (WGS) data to identify the microbiome composition from small endoscopic biopsy samples. We validated this methodology by quantitative PCR (qPCR) as well as comparison with other tools using datasets from the Human Microbiome Project (HMP), HapMap, human blood serum samples, a cystic fibrosis study, and TCGA studies. We further analyzed 27 gastric biopsies collected from patients with active H. pylori infection and with and without a history of treated H. pylori infection and found significant differences in their microbial content. We also performed similar analysis on the TCGA gastric samples and found that 40 % of the analyzed samples have strong evidence for H. pylori infection. This report details the methodology, computational pipeline, and validation of this novel approach, which can now be applied over serial endoscopic biopsies to examine temporal changes in the microbiome over time.
Results
Identification of microbiome content in mucosal biopsies
Microbial profiling of human mucosal tissues, such as oral cavities, skin, gastrointestinal tract, and urogenital tract, are typically performed by sequencing variable regions of the 16S ribosomal RNA gene [29, 30]. A limitation of this approach is that species identification is dependent on the extent of evolutionary diversification in those variable 16S regions where other genomic regions may be more informative for such speciation. More recently, whole genome shotgun sequencing was used to profile viral families [31, 32], for metagenomic studies [33, 34], and for the HMP [35]. The use of 16S and whole genome approaches for profiling the microbiome in human gastric biopsy samples is limited, however, because stomach mucosal biopsies contain mostly human DNA and a low abundance of microbial DNA, in contrast to samples collected from mucosal surfaces that are highly enriched for bacteria.
To overcome these limitations of microbiome identification from stomach biopsies and to maximize detection power, we performed WGS of 27 mucosal biopsy samples at roughly 10× coverage. On average, each library produced 400 million reads, of which 97 % were mapped to the standard human reference genome (Additional file 1). Initial profiling of the microbiome content from the remaining unmapped reads using clade-specific markers [36] resulted in a number of uncultivable bacteria species in roughly equal proportions in all samples (Fig. S1 in Additional file 2), such as Candidatus Carsonella ruddii (160-kbp genome,16.5 % GC content), Candidatus Sulcia muelleri (240-kbp genome, 22.5 % GC content), Candidatus Zinderia sp. (210-kbp genome, 13.5 % GC content) and Wigglesworthia glossinidia (700-kbp genome, 22.5 % GC content). These species are symbiotic bacteria that are restricted to insect hosts [37] with no previous report of human infection. Upon closer inspection we found that their genomes are short and have a low GC content, suggesting that their identification was incorrect, likely a result of short regions homologous with other bacteria or the human genome. We therefore devised a more stringent filtering approach adapted from the PathSeq pipeline [4]. Briefly, the pipeline is composed of successive filtering steps intended to remove the human-derived genomic reads (Fig. 1; Additional file 1). The reads are first mapped to the standard hg19 genome. The remaining unmapped reads are then mapped to a number of additional human assembled genomes and finally to a database of repeat regions. The remaining unmapped reads are then mapped to the National Center for Biotechnology Information (NCBI) full set of microbial reference genomes (see "Materials and methods").
Fig. 1.
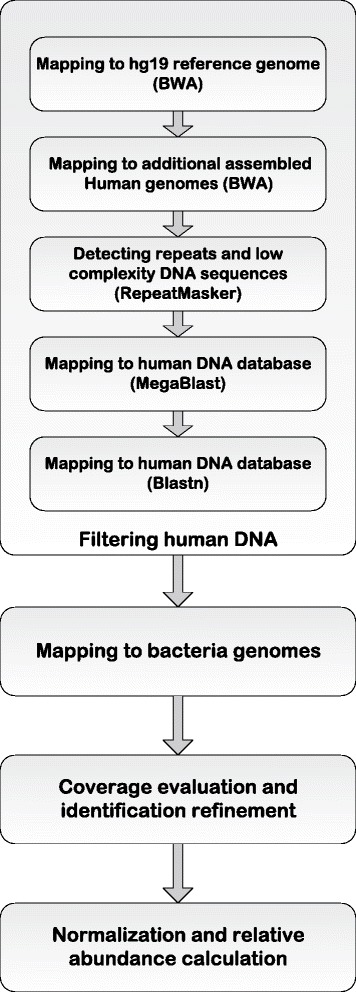
Workflow of the human DNA filtering and bacterial identification procedure. BWA Burrows-Wheeler Aligner
We used two specific mapping criteria for definitive identification of bacteria to reduce false detection. In the first, we evaluated the number of reads that map to the bacterial genome after the successive filtering steps. In many instances the number of reads that map to the bacterial genome drops sharply (often by more than 80 %) following the repeat masker and MegaBlast filtering (Fig. 2a, c). This removes the majority of reads with sequence similarities to repeat regions or human genomes. The second criterion is coverage uniformity of the bacterial genome. To that end we devised two measures of coverage variance that correct for sequencing depths and size of the genome (see "Materials and methods"). Both coverage measures were highly correlated.
Fig. 2.
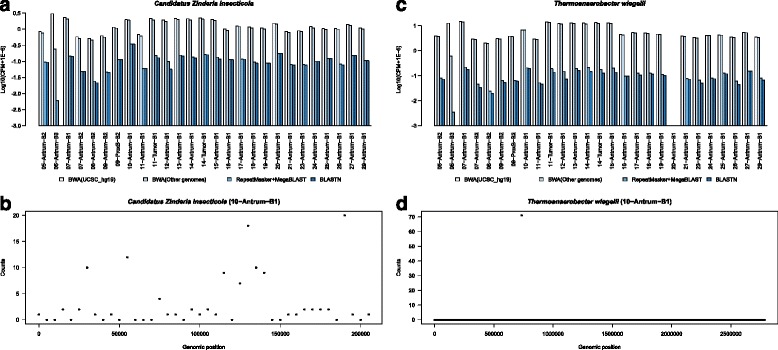
False bacteria identifications are readily identified by a drop in the number of mapped reads after filtering human sequences and inconsistent coverage. a Log-transformed counts per million (CPM) reads mapped to Candidatus Zinderia insecticola after each filtering step for each sample as outlined in Fig. 1. b Reads coverage map of Candidatus Zinderia insecticola in sample 10-Antrum-B1 after all filtering steps. Points represent the read counts in a 5-kb window. c Log-transformed CPM mapped to Thermoanaerobacter wiegelii after each filtering step for each sample. d Similar to (b), reads coverage map of Thermoanaerobacter wiegelii in sample 10-Antrum-B1. After all filtering steps, the coverage map indicates that reads map to only a single region of the genome. BWA Burrows-Wheeler Aligner
These two filtering criteria were used to distinguish between falsely and correctly identified bacteria in the samples. For example, the number of reads mapping to Candidatus Zinderia insecticola and Thermoanaerobacter wiegelii, which were initially detected in high abundance in the antrum of patient 10, falls off dramatically with successive filtering (Fig. 2a, c), and the coverage of the mapped reads across each bacterial genome is not uniform (Fig. 2b, d), suggesting that these bacteria are not likely to be present in these samples. Alternatively, the data for H. pylori and Lactococcus lactis provide strong support for positive identification. These bacteria are not present in every sample, there is no dramatic reduction in number of mapped reads with successive filtering steps (Fig. 3a, c), and the coverage across each genome is more uniform (Fig. 3b, d).
Fig. 3.
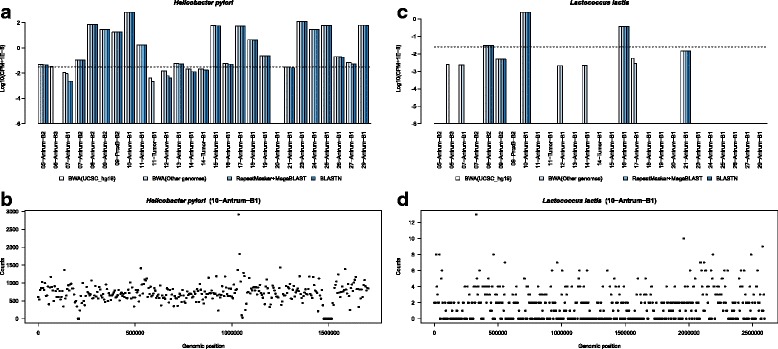
H. pylori (a, b) and Lactococcus lactis (c, d) retain consistent number of reads after each filtering step and the genomic coverage is uniform, indicating positive identifications. Similarly to Fig. 2, panels (a) and (c) indicate log-transformed counts per million (CPM) reads mapped to the respective bacteria and panels (b) and (d) contain read pileup along the genome in 5-kb segments. Dashed lines indicate minimum CPM values used in this study for positive identification of bacteria. BWA Burrows-Wheeler Aligner
Microbial content validation by qPCR in biopsy samples
To validate the results from WGS identification, we performed qPCR analysis to quantify the total bacteria content and H. pylori content by target amplification of a conserved region of 16S rDNA and the H. pylori specific ureA gene, respectively. We found excellent correlation between qPCR-amplified ureA gene and WGS H. pylori quantification (Fig. 4a; Spearman rank correlation 0.91), as well as high correlation with quantification of total bacteria using universal 16S primers and WGS (Fig. 4b; Spearman rank correlation 0.785, p value < 0.006). We note that although H. pylori accounts for the majority of the detected microbiome content in some samples, H. pylori read counts alone are poorly correlated to 16S qPCR-based total bacteria content, suggesting the presence of other (non-H. pylori) bacteria. This is further supported by the good agreement between the sequencing-based quantification and universal 16S qPCR when excluding H. pylori read counts (Fig. 4c; Spearman rank correlation 0.84).
Fig. 4.
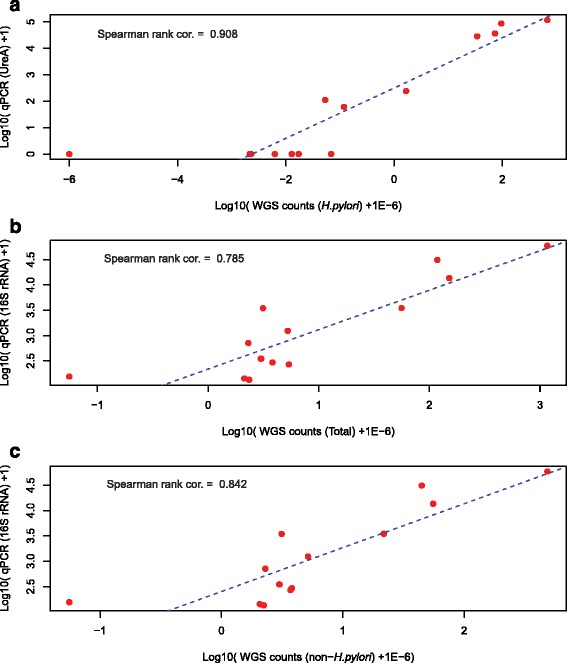
Correlation between qPCR-based bacterial quantification and WGS read counts. a Correlation plot between H. pylori-specific ureA gene-based qPCR results and WGS read counts (normalized by counts per million (CPM) reads) mapped to H. pylori. b Correlation plot between 16S rRNA gene-based qPCR results and total WGS read counts mapped to all bacterial genomes. c Correlation plot between 16S rRNA gene-based qPCR results and WGS read counts mapped to non-H. pylori genomes. In three cases qPCR measurements were derived from a sample that was adjacent to the WGS sample whereas in all other cases qPCR and WGS were performed on the same biopsy. Blue dashed lines are the linear regression models
Comparison of microbiome detection methods in human samples
To further validate our microbial detection approach, we compared our methodology with MetaPhlAn [36] and Kraken [38], commonly used programs for microbiome detection, using samples from relatively sterile blood samples to evaluate false detection and bacteria-rich samples to evaluate concordance between the methods. We also included a number of tumor samples from various tissues to demonstrate that our approach is not restricted to tissue type or biased by bacteria content.
Comparison using sterile blood samples
We first selected three random samples (DRR000615, ERR055396, ERR047873) from the human HapMap project [39] to serve as negative controls. The HapMap project sequenced blood samples from donors across a wide range of human populations for the purpose of charting human genomic diversity. Erroneous bacteria identification is introduced from incomplete filtering of human reads or incorrect mapping of reads to bacterial genomes. As indicated by our initial analysis, some bacteria may be incorrectly detected because of similarities to human genomic loci. The HapMap samples serve as reasonable controls to assess false discovery since there should be little if any bacteria in blood samples.
Similar to the results from our gastric biopsy samples, we find that both Kraken and MetaPhlAn identified a number of bacteria species, such as Candidatus Carsonella ruddii, Candidatus Sulcia muelleri, Candidatus Zinderia sp. and Wigglesworthia glossinidia, that are likely misidentified due to contaminating human reads (Fig. S2 in Additional file 2). For example, in sample ERR055396, Thermoanaerobacter wiegelii and Mycoplasma hyopneumoniae had a large number of mapped reads while their genomic coverage was restricted to a few short regions (Fig. S3a, b in Additional file 2). In contrast, no detectible microbial species passed both the minimal number of reads and coverage uniformity criteria when using our own identification pipeline. Propionibacterium acnes was found in sample DRR000615; although 219 reads supported this identification, the genomic coverage is slightly below our cutoff for positive identification (Fig. S3c in Additional file 2). It is, however, consistent with previous reports that P. acnes is a common contaminant of blood cultures [40].
Comparison using bacteria-rich samples
To evaluate the performance of our method on bacteria-rich metagenomics samples, we profiled samples from the HMP [29] to serve as positive controls. The HMP is focused on mapping microbiome diversity across the human population at different body regions that are enriched for microbial species. We selected three posterior fornix samples from the HMP (SRS052620, SRS065347, SRS024428) with WGS data that were previously analyzed for microbiome content, and analyzed their reads using our computational detection procedure.
Overall, the microbiome profile of the HMP samples generated by our analysis closely resembled the previously published microbial composition in both diversity and quantification (average cosine similarity 0.983, no statistical difference by t-test or Wilcoxon rank sum test; Fig. 5a; Fig. S4a, b in Additional file 2). The differences in species identification between the three methods are largely restricted to low abundance species. The sequence markers used by MetaPhlAn were derived from short unique bacteria regions that can miss low abundance species due to limited coverage. Kraken identifies bacteria species by matching k-mers to a database of bacteria k-mers (see "Comparison of microbiome detection methods" in Additional file 2). We identified several low abundance species that have not been reported by previous studies, and are likely true identifications as indicated by the uniform coverage of their genomes (Fig. S5 and "Comparison of microbiome detection methods" section in Additional file 2). Other previously identified species were not detected by our pipeline due to low read counts (<10) or insufficient genome coverage.
Fig. 5.

a Comparison of bacteria identified from three samples profiled by the HMP. b Comparison of bacteria identified from two lung sputa samples of cystic fibrosis patients. Both MetaPhlAn and Kraken and the procedure outlined in this study identify a consistent set of microbial species in the five samples (cosine similarity >0.95). Values represent percentage of bacteria reads found in each sample
In addition to the HMP samples, we also analyzed two sputa samples from a published cystic fibrosis study [41]. Both of these samples contain a high percentage of bacterial content relative to biopsy samples, and serve as another positive control. The three methods reported very similar microbiome profiles (cosine similarity >0.92; Fig. 5b), where, similarly to the HMP samples, the discrepancies between the methods are attributed to differences in identification of low abundance species (see "Comparison of Microbiome Detection Methods" in Additional file 2). These results indicate that our pipeline has a similar capacity to detect microbiomes from the regular metagenomics samples as MetaPhlAn and Kraken.
Evaluation of human samples with low bacteria content to demonstrate clinical applicability
To test the applicability of our methodology for microbiome detection in clinical samples we performed additional evaluation on samples from a variety of different tissues collected in different clinical studies. We first analyzed WGS data from a serum sample collected in a non-malaria febrile illness study (SRR1106126) where Haemophilus influenzae was detected in one patient [42]. Consistent with the study report, both our pipeline and Kraken identified high levels of H. influenzae (82.9 % and 75.4 %, respectively) as well as P. acnes (17.1 % and 24.6 %, respectively), whereas no bacteria were identified by MetaPhlAn.
Next we analyzed WGS data collected from three TCGA studies (one from lung adenocarcinoma, two from rectum adenocarcinoma, and two from colon adenocarcinoma). Overall, all three methods identified the same dominant species from each sample, such as P. acnes in lung adenocarcinoma, Bacteroides fragilis in rectum adenocarcinoma, and Fusobacterium nucleatum in colon adenocarcinoma (Fig. 6; Fig. S6 in Additional file 2). Both MetaPhlAn and Kraken falsely identified bacteria that were also incorrectly reported in a previous evaluation, such as Candidatus Carsonella ruddii, Candidatus Sulcia muelleri, Candidatus Zinderia sp., Thermoanaerobacter wiegelii and Achromobacter xylosoxidans. Conversely, both methods did not identify species with significant genome coverage that were marked as a positive identification by our approach (Fig. S7 in Additional file 2).
Fig. 6.
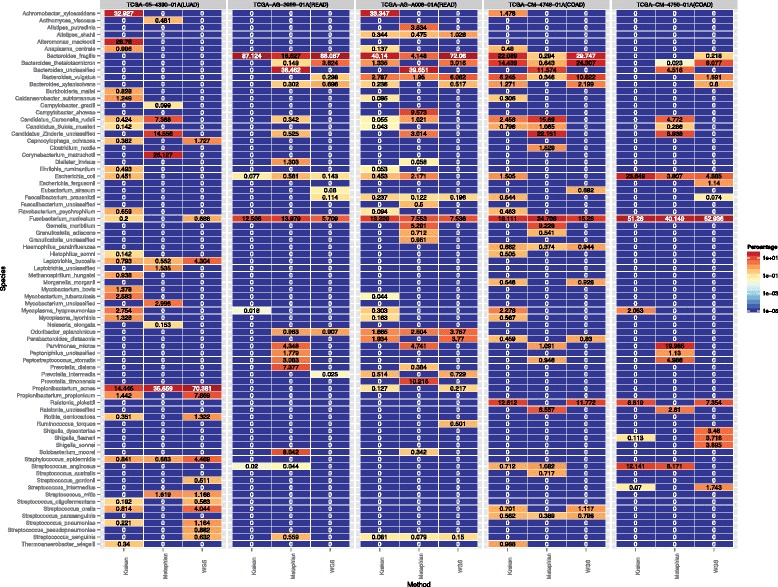
Comparison of identification by three methods from five TCGA tumor samples. Values represent percentage of bacteria reads found in each sample. COAD colon adenocarcinoma, LUAD lung adenocarcinoma, READ rectum adenocarcinoma
To specifically address the impact of additional filtering of human DNA on all detection methods, we also performed comparison among the three methods on four gastric biopsy samples with full filtering of human reads. Even after extensive human DNA filtering steps, results from MetaPhlAn and Kraken included questionable identifications as measured by bacterial genome coverage. We conclude, therefore, that genome coverage evaluation is a key step to achieve accurate identification from these samples (Fig. S8 and " Comparison of Microbiome Detection Methods" section in Additional file 2).
Collectively, these results demonstrate that our methodology is able to identify microbial species in a wide variety of tissue types and that the main advantage of this approach over MetaPhlAn and Kraken is in samples with low levels of bacteria where the abundance of human DNA confounds bacteria detection.
Clinical microbiome findings from gastric biopsy samples
Patients undergoing upper endoscopy who had no prior evidence of chronic inflammatory disease and no chronic use of nonsteroidal anti-inflammatory drugs were approached for enrollment and for research biopsies for microbiome analysis (Table 1). Twenty-two patients were enrolled, of which eight had active H. pylori infection as assessed by positive CloTest and confirmed on pathologic evaluation of the endoscopic tissue biopsy, and four of the eight actively infected patients also had prior history of H. pylori infection. The remaining 14 patients had no active H. pylori infection, although seven of them had prior history of infection. Our microbiome identification pipeline identified 18 patients with H. pylori, including all eight patients with active infection.
Table 1.
Patient characteristics
| Patient ID# | Gender | Age | H. pylori status | Prior H. pylori | H. pylori Pathology |
|---|---|---|---|---|---|
| 05 | F | 49 | Negative | Y (2009) | negative |
| 07 | F | 50 | Negative | Y (2010) | negative |
| 08 | M | 50 | Active Infection | Y | positive |
| 09 | M | 36 | Active Infection | Y (2003) | positive |
| 10 | F | 69 | Active Infection | Y (2009) | positive |
| 11a | F | 85 | Negative | N | N/A (pathology did not test for h. pylori) |
| 12 | F | 78 | Negative | N | negative |
| 13 | M | 57 | Negative | N | negative |
| 14a | M | 36 | Negative | Y (2013) | N/A (pathology did not test for h. pylori) |
| 15 | F | 37 | Active Infection | Y (2012) | positive |
| 16a | M | 63 | Negative | N | N/A (pathology did not test for h. pylori) |
| 17a | F | 67 | Negative | N | N/A (pathology did not test for h. pylori) |
| 18a | M | 71 | Negative | N | N/A (pathology did not test for h. pylori) |
| 19 | F | 46 | Negative | Y (2009) | negative |
| 20 | M | 56 | Negative | Y (2004) | negative |
| 21 | F | 43 | Negative | Y | negative |
| 23 | F | 51 | Active Infection | N | positive |
| 24 | F | 43 | Active Infection | N | Unknown |
| 25 | F | 26 | Active Infection | N | positive |
| 26 | F | 78 | Negative | Y (2010) | negative |
| 27 | F | 63 | Negative | Y (2014) | negative |
| 29 | F | 48 | Active Infection | N | positive |
a Patient also with gastric cancer
Hierarchical clustering of the 27 microbial profiles (from 22 patients) indicated three major groups (Fig. 7) that were largely consistent with the clinical annotation of the samples (Fig. 7).
Fig. 7.

Bacterial relative abundance heatmap of profiled gastric biopsy samples. Hierarchical clustering of the 27 microbial profiles identified three primary clusters indicated by the "Microbial profiles" bar, with sample assignment to cluster p value <0.05 determined by 10 K bootstrap sampling. This grouping largely coincides with the clinical status of the samples: red-labeled samples were collected from patients with active H. pylori infection, black-labeled samples are from patients with prior history of treated H. pylori, and green-labeled samples are from patients with no history or present H. pylori infection
The first group of samples (orange bar in Fig. 7) is characterized by a predominant signature of abundant levels of H. pylori (98.11 ± 0.96 %) that includes the nine samples from eight patients (IDs 08, 09, 10, 15, 23, 24, 25, and 29) with clinical validation of infection and two additional samples that had no prior clinical symptoms but contained similarly high levels of H. pylori (IDs 17 and 18).
The second group of samples (blue bar in Fig. 7) is characterized by a high proportion of P. acnes, as well as other less frequent species, including Haemophilus parainfluenzae, H. influenzae, and Staphylococcus epidermidis. P. acnes is the most dominant species in all nine samples and accounted for 51.2–96.0 % of relative content. Included in this group are five samples from five patients (IDs 05-Antrum-B2, 07-Antrum-B2, 19, 26, and 27) with intermediate levels of H. pylori that accounted for 4.2 %, 8.3 %, 18.2 %, 19.0 % and 4.0 % of their total bacteria content, respectively. These patients had previous history of H. pylori infection that was previously treated. None of these patients exhibited signs or symptoms of recurrent infection at the time of sampling. We also checked additional samples from two (IDs 05 and 07) of the above five patients with H. pylori read counts, but we did not detect H. pylori in additional adjacent biopsies (05-Antrum-B3, 07-Antrum-B1). Both of these adjacent biopsies had a low number of unmapped reads following the filtering of the human reads, although the starting number of sequenced reads was comparable to other samples, suggesting that these two biopsies had relatively low amounts of bacteria (Additional file 1). Although H. pylori was identified in only one biopsy collected from patient 07, another top bacterial species (P. acnes) was identified in both biopsies.
The third group of samples (grey bar in Fig. 7) is characterized by a broader bacterial diversity, in which there is no single bacteria that consistently dominates all samples, although three samples had a significant proportion of Lactobacillus gasseri and four had detectable H. influenzae. Two samples with previous infection history are included in this group (07-Antrum-B1, 05-Antrum-B3). The remaining four patients had neither history of infection nor clinical evidence for active infection, although three of them (ID 11-Antrum-B1, 13, and 16) had detectable levels of H. pylori bacteria in the gastric mucosa with 52.8 %, 2.0 % and 1.0 % H. pylori bacterial DNA, respectively. The presence of H. pylori in the above three cases may not represent active infection as their bacterial profile is distinctly different from the first group (Table 1).
Notably, we also had two gastric tumor biopsies (ID 11-Tumor-B1 and 14-Tumor-B1), neither of which contained H. pylori sequences. Patient ID 11 is of particular interest because the mucosal sample adjacent to the tumor had high levels of H. pylori that accounted for 52.8 % of mapped bacterial reads. The other top bacterial species in ID 11 (Lactobacillus gasseri and P. acnes) were identified in both tumor and mucosa, but H. pylori appears to have been excluded from the tumor.
Finally, P. acnes was identified in almost all biopsies at various levels. As a skin-enriched bacterium rather than a skin-specific bacterium, P. acnes could infect bones, joints, mouth, eye, brain, heart valves, and shunt [43]. P. acnes was also found in prostate samples and some studies suggest that inflammation caused by it contributes to prostate cancer [44, 45]. Other gastric microbiome studies also identified P. acnes in human stomach [46, 47]. In our evaluation of the detection pipeline we also identified P. acnes in samples from other studies, such as in blood serum [48] and lung adenocarcinoma [49]. Our biopsies were collected by a number of clinicians at different locations and dates, and sequenced at different dates. Therefore, contamination from a single source is not likely, although we cannot fully exclude this possibility. Because of the low number of reads mapped to P. acnes and the high similarity between P. acnes strains, we were not able to determine whether there is a single P. acnes strain common to all samples. Given the consistent low read counts of P. acnes across all samples, it might have been introduced to stomach mucosa through oral ingestion or oral contamination of the endoscope, rather than laboratory contamination.
H. pylori and EBV infection status discovery in TCGA WGS data
We next analyzed the microbiome content in previously collected and sequenced TCGA gastric tumor samples, of which 37 of the 295 samples were profiled by low-pass WGS [13]. We began by examining our pipeline to identify EBV-associated gastric cancer as a validation of our pipeline. We found strong evidence for EBV infection in both samples that were previously classified as EBV-positive by TCGA genetic and expression profiling, providing further validation for our approach for microbiome detection (Table 2).
Table 2.
Number of reads (in counts per million) mapped to H. pylori or EBV genomes in TCGA samples identified from WGS and RNA-seq data
| Tumor sample-WGS | Tumor sample-RNASeq | Tissue sample-WGS | Tissue sample-RNASeq | |||||
|---|---|---|---|---|---|---|---|---|
| Sample | H. pylori | EBV | H. pylori | EBV | H. pylori | EBV | H. pylori | EBV |
| TCGA-BR-4183 | 0.01 | 0.00 | NA | 0.01 | 0.00 | NA | ||
| TCGA-BR-4184 | 0.00 | 0.00 | NA | 0.01 | 0.00 | NA | ||
| TCGA-BR-4187 | 0.76 | 0.00 | NA | 43.97 | 0.00 | NA | ||
| TCGA-BR-4188 | 0.00 | 0.00 | NA | 0.00 | 0.00 | NA | ||
| TCGA-BR-4191 | 0.04 | 0.00 | NA | 1.65 | 0.01 | NA | ||
| TCGA-BR-4201 | 0.09 | 0.00 | NA | 27.68 | 0.03 | NA | ||
| TCGA-BR-4253 | 0.21 | 6.36 | NA | 0.44 | 0.00 | NA | ||
| TCGA-BR-4255 | 0.00 | 0.00 | NA | 0.04 | 0.00 | NA | ||
| TCGA-BR-4256 | 0.00 | 0.01 | NA | 0.00 | 0.00 | NA | ||
| TCGA-BR-4257 | 0.02 | 0.00 | NA | 11.68 | 0.00 | NA | ||
| TCGA-BR-4267 | 0.01 | 0.00 | NA | 0.40 | 0.00 | NA | ||
| TCGA-BR-4279 | 6.99 | 0.00 | NA | 41.48 | 0.00 | NA | ||
| TCGA-BR-4280 | 0.01 | 0.00 | NA | 0.01 | 0.00 | NA | ||
| TCGA-BR-4292 | 0.04 | 0.00 | NA | 0.00 | 0.00 | NA | ||
| TCGA-BR-4294 | 0.19 | 0.00 | NA | 244.69 | 0.00 | NA | ||
| TCGA-BR-4357 | 0.34 | 0.00 | 0.03 | 0.00 | 64.43 | 0.00 | NA | |
| TCGA-BR-4363 | 0.00 | 0.00 | 0.37 | 0.00 | 0.00 | 0.00 | NA | |
| TCGA-BR-4366 | 0.01 | 0.00 | 0.01 | 0.00 | 0.07 | 0.00 | NA | |
| TCGA-BR-4367 | 0.01 | 0.00 | 0.03 | 0.00 | 0.01 | 0.00 | NA | |
| TCGA-BR-4368 | 0.01 | 0.00 | 0.01 | 0.02 | 16.97 | 0.00 | NA | |
| TCGA-BR-4369 | 0.00 | 0.00 | 0.00 | 0.17 | 0.03 | 0.00 | NA | |
| TCGA-BR-4370 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | NA | |
| TCGA-BR-6453 | 0.30 | 0.00 | 0.01 | 0.00 | 0.19 | 0.00 | 0.03 | 0.00 |
| TCGA-BR-6454 | 0.01 | 0.00 | 0.00 | 0.00 | 17.95 | 0.00 | 2.72 | 0.00 |
| TCGA-CG-5720 | 3.42 | 0.00 | 1.48 | 0.00 | 12.51 | 0.00 | 2.28 | 0.00 |
| TCGA-CG-5721 | 0.00 | 0.01 | 0.00 | 0.43 | 0.01 | 0.00 | 0.00 | 0.00 |
| TCGA-CG-5722 | 0.13 | 2.55 | 0.04 | 34.13 | 1.83 | 0.00 | 0.00 | 0.22 |
| TCGA-CG-5723 | 0.02 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | NA | |
| TCGA-CG-5724 | 0.03 | 0.00 | 0.00 | 0.00 | 92.65 | 0.00 | NA | |
| TCGA-CG-5725 | 0.00 | 0.01 | 0.00 | 0.61 | 0.01 | 0.00 | NA | |
| TCGA-CG-5726 | 0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | NA | |
| TCGA-CG-5727 | 0.00 | 0.01 | NA | 0.01 | 0.00 | NA | ||
| TCGA-CG-5728 | 0.00 | 0.00 | NA | 2.79 | 0.00 | 0.34 | 0.00 | |
| TCGA-CG-5730 | 0.01 | 0.00 | NA | 0.00 | 0.00 | 0.00 | 0.00 | |
| TCGA-CG-5732 | 0.01 | 0.00 | 0.00 | 0.25 | 0.00 | 0.00 | NA | |
| TCGA-CG-5733 | 0.01 | 0.00 | NA | 2.01 | 0.00 | 0.63 | 0.01 | |
| TCGA-CG-5734 | 1.25 | 0.01 | 0.03 | 0.17 | 0.00 | 0.00 | 0.00 | 0.00 |
Entries in bold are samples that are considered H. pylori-positive and entries in italics EBV-positive by a counts per million (CPM) reads threshold of 0.3. NA not applicable
Next, the tumor and adjacent tissue samples were examined for H. pylori. Since the H. pylori history of these samples was not available, we considered having at least 0.3 counts per million (CPM) reads mapped to H. pylori as evidence of H. pylori bacteria in the sample (with the same uniformity of coverage criteria as for our in-house samples; see "Materials and methods"). Strikingly, we found that 18 patients out of 37 (49 %) had significant evidence of H. pylori in either normal tissue or tumor. The overwhelming majority of H. pylori-positive samples were from the normal adjacent tissue. Only two patients had definitive H. pylori levels in both tumor and adjacent normal samples. Additionally, two other tumor samples (ID TCGA-BR-4187 and TCGA-BR-4357) had marginal support for H. pylori presence with 276 and 197 reads mapping to the H. pylori genome, respectively, whereas the adjacent normal tissue had an abundance of H. pylori (Table 2). We did identify one patient with H. pylori present in the tumor sample but not in the adjacent mucosa (ID TCGA-CG-5734). These microbial findings were further validated by analysis of the corresponding RNA-seq data (where available) for the presence of H. pylori and EBV transcripts with good agreement with WGS detection (Fisher’s exact test p value ≤ 2.9e-4; Table 2). The presence of H. pylori did not correlate with any clinical attributes of gender, tumor location, Lauren classification, or age, although the sample size is likely underpowered for detecting significant correlations (Fig. S9 in Additional file 2).
Discussion
In this paper we present a systematic study of the unbiased and comprehensive identification of microbial species from small endoscopic biopsies by WGS. Accurate microbiome detection in these samples requires extensive processing to remove all possible reads that originate from human DNA. Careful comparison of microbiome detection with MetaPhlAn and Kraken using our own samples as well as samples from the HapMap project, the HMP, TCGA and other studies illustrates that all methods are confounded by high abundance of host DNA (Figs. S1–S9 and " Comparison of microbiome detection methods" in Additional file 2). Furthermore, because of the low coverage of bacterial genomes and sequence similarities between organisms, bacteria are often identified based on coverage along a narrow region of their genomes. To address this we included measures of uniform genomic coverage as additional evidence for bacteria identification.
In terms of microbiome profiling of biopsy samples, our results are highly consistent with qPCR quantification of H. pylori and universal 16S bacterial quantification. Using the same methodology, we were able to identify EBV-associated gastric cancer as was previously identified in the TCGA project. Finally, we also identified H. pylori in a significant portion of the mucosal samples collected as part of the TCGA project, and further identified the microbial species in the corresponding RNA-seq sample with highly significant correlation. Together, these data support our approach and demonstrate the feasibility of identifying the microbiome from small endoscopic tissue samples.
Notably, we identified H. pylori bacteria in the mucosa of patients who had been previously treated and were presumed cleared of this infection. It is conceivable that these are new infections; however, it is also possible that this identification represents persistent low-grade bacterial content of H. pylori in the gastric mucosa of previously infected and treated patients. The clearance rate of H. pylori by current standard practice of acid suppression and antibiotic therapy is 80–85 % [50, 51], although the rate of clearance may be declining due to the development of clarithromycin resistance [52, 53]. The presence of H. pylori in mucosa samples from patients with prior history of treated infections as well as in TCGA cohort raises the possibility that the tumorigenesis risk of H. pylori infection may be associated with the fact that the infection was not fully eradicated by conventional treatment, and that long-term chronic and asymptomatic infection may be the reason that current H. pylori treatment strategies to reduce gastric cancer risk have not proven successful [54]. The induced and long-term immune response facilitated by persistence of H. pylori may be a direct contributor to neoplastic transformation. Equally intriguing is the strong bias for H. pylori colonization in healthy mucosa tissues over tumor tissues seen in our sample and more strikingly in TCGA samples. Both observations are not statistically powered to draw broad conclusions but warrant follow-up studies.
In our survey, the gastric microbiome of patients with active H. pylori infection appears to be distinct from that in other samples. Specifically, the bacterial content of the gastric mucosa in actively infected individuals was significantly higher than the bacterial content of other individuals, and was dominated by H. pylori (98.11 ± 0.96 % of the bacterial reads). We did identify occult H. pylori in several additional samples, and note that it is not clear if these individuals will go on to develop a more profound active infection, or remain colonized. Besides being a disease-causing agent and a disease-protecting agent, H. pylori also has been considered as an indicator of changing human microbe ecology [55]. The high correlation between abundance of H. pylori and overall bacteria content (Fig. S10a in Additional file 2; Spearman rank correlation 0.861) may indicate that H. pylori infection may render the stomach mucosa a more favorable environment for bacterial colonization. It could be explained by H. pylori urease activity [28], and the consequent rise in gastric pH resulting in a more favorable environment for several bacteria, such as S. epidermidis (Fig. S10b in Additional file 2). Conversely, some bacteria were less abundant with increasing H. pylori content, such as H. parainfluenzae (Fig. S10c in Additional file 2) and H. influenzae (Fig. S10d in Additional file 2). Our validation studies suggest that WGS and the computational pipeline may detect bacteria below the limits of detection by qPCR, which may have implications in the future for defining H. pylori eradiation. Also of note is that we identified fewer phylotypes in our sample set (19 total phylotypes) than previously reported [23], but consistent with another report [56], and also consistent with the understanding of the relative germ-free environment of the stomach [57]. These differences are not likely related to the small patient sample set but rather differences in technique, including a lack of an amplification step in WGS. It may be possible to identify additional bacteria by WGS with higher sequencing coverage, but this is limited by lack of ability to validate, as we have approached our detection limit by qPCR with our current DNA sequencing coverage.
Previous studies have demonstrated extensive interaction between the gastric epithelium and the immune response to bacterial infection. For example, H. pylori infection activates proinflammatory cycloxgenase (COX) enzymes which regulate immune response and development [58]. Recent genomic studies have identified Fusobacterium in colorectal carcinoma [3], fusion of Acinetobacter DNA to human mitochondrial DNA in acute myeloid leukemia samples [5], and that EBV-associated gastric tumors are a genetically distinct class of gastric cancer [13]. Collectively, these studies suggest that bacteria and viruses may contribute significantly more than previously appreciated to the progression of various tumors.
Conclusions
The microbiome, even when present in low abundance relative to human tissue, may have significant impact on human physiology. However, it has been difficult to quantify and characterize the microbiome in clinical tissue samples where the bacterial content is low. Here we present a new methodology to identify low abundant microbiome in small clinical tissue samples. The ability to directly characterize the microbiome from clinical biopsies opens a new possibility to investigate the interaction between microbial species and human tissues in a more direct way in a host of different tumor and tissue types, and longitudinally across the management of a particular condition.
Materials and methods
Gastric cancer and H. pylori research database
The Weill Cornell Medical College Gastric Cancer and H. pylori Research Database is a registry and tissue repository to examine the natural history of H. pylori infection in patients with and without gastric cancer. The primary aim of this research biobank is to provide an adequate tissue resource for the purpose of using modern molecular analytic tools to distinguish patients with chronic H. pylori infection who are at risk for subsequently developing gastric cancer from the vast majority of patients with H. pylori infection who do not develop malignancy. Subjects undergoing an upper endoscopy for clinical indications were approached for participation. All subjects provided written informed consent for participation in the Gastric cancer and H. pylori Research Database according to the Declaration of Helsinki prior to study enrollment. Samples were collected in accordance with the institutional ethical and clinical guidelines under institutional review board protocol 1203012274.
Tissue collection and processing
Gastric mucosal biopsies from the antrum, proximal body and fundus were acquired for each enrolled patient and those with gastric carcinoma had additional biopsies taken at the tumor site as well as adjacent normal tissue. Biopsies were obtained using the Bard Precisor EXL coated disposable biopsy forceps (Bard International, Murray Hill, NJ, USA) and were immediately placed into individual sterile cryovials on dry ice and flash frozen while still in the endoscopic suite. The samples were then transferred to liquid nitrogen for prolonged storage. Genomic DNA was extracted from each gastric biopsy using an AllPrep micro kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Samples were homogenized using a rotor stator homogenizer for less than 30 seconds. DNA concentration was measured for each sample using a Qubit® 2.0 Fluorometer (Life Technologies, Grand Island, NY, USA) and DNA quality was checked on a 1 % agarose gel stained with ethidium bromide. Samples were run alongside a 1-kb DNA Extension Ladder (Life Technologies, Grand Island, NY, USA).
Whole genome sequencing
Extracted genomic DNA (1 μg) from each sample was given to the Weill Cornell Medical College Epigenomics Core for library preparation and subsequent WGS using an Illumina TruSeq DNA-seq DNA sample preparation kit and the Illumina HiSeq 2500 platform. Each sample was sequenced on a single flow cell lane as 50-bp paired-end reads. Homopolymers, adapters and distribution of base quality of raw sequences from each sample were investigated using FastQC (version 0.10.1). The filtered, non-human sequenced reads are available for download at BioProject ID PRJNA297869 [59].
Quantitative PCR
Commercially available H. pylori-specific and universal bacteria16S qPCR assays were performed on patient samples according to the manufacturer's protocol and suggested cycling conditions (Primerdesign Ltd, UK). qPCR reactions were conducted in MicroAmp® 48-well optical plates (Life Technologies, Grand Island, NY, USA) in 25 μl volumes using 25 ng template genomic DNA, Perfecta® qPCR FastMix® II 10X master mix (Quanta Biosciences, Inc., Gaithersburg, MD, USA) and primers and probe supplied by Primerdesign Ltd with each assay, specific either for the ureA gene of H. pylori or the conserved region of the bacterial 16S gene. Samples were tested in triplicate along with negative and positive controls on the StepOne™ Real-Time PCR System (Life Technologies, Grand Island, NY, USA). Copy number was determined through extrapolation using the standard curve supplied with each kit.
External datasets
HapMap samples (DRR000615, ERR055396, ERR047873), HMP WGS data (SRS052620, SRS065347, SRS024428), and a serum sample from a non-malaria febrile illness patient (SRR1106126) were collected from the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) and the HMP (http://www.hmpdacc.org/catalog/grid.php?dataset=metagenomic&hmp_isolation_body_site=urogenital_tract), respectively. WGS from a gastric adenocarcinoma study, a lung adenocarcinoma study, a rectum adenocarcinoma study, and a colon adenocarcinoma study and RNA-seq data from a gastric adenocarcinoma study were downloaded from the Cancer Genomics Hub (https://cghub.ucsc.edu/).
Computational pipeline for microbial detection from WGS data
Filtering human DNA
This pipeline is based on the PathSeq [4] protocol for filtering human reads from WGS with a few modifications (Fig. 1). In the first step, the Burrows-Wheeler Aligner (BWA; version 0.6.2, with aln/sampe command) [26] was used to align the whole WGS data against the human reference genome (version hg19; http://genome.ucsc.edu/) using default parameters. In the second step, unmapped reads from the first step were aligned to three additional assembled human genomes available from the NCBI (hs_alt_CRA_TCAGchr7v2, hs_alt_HuRef, hs_ref_GRCh37.p5, build 37.3; ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens//Assembled_chromosomes/seq/, downloaded Feb. 06,2014 ) and to the Ensembl human genome reference (ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/) using BWA. RepeatMasker (version open-4.0, http://www.repeatmasker.org/ with –qq option) was used in the third step to identify repeat regions and low complexity sequences, and then any reads with three or more masked nucleotides were discarded for the next step. A combined human sequence database was generated for the last two steps and includes the following three datasets: i) the Ensembl Homo sapiens cDNA database (ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/cdna/), ii) the NCBI Homo sapiens RNA database (ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/RNA/) and, iii) the NCBI BLAST human genome database (ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/). In the fourth step, we performed alignments on the above database using MegaBlast (version 2.2.27) [60] with e-value cutoff 10−7 and word size 16. BLASTN (version 2.2.27) [60] was used as the last filtering step with the following parameters: cutoff expected value 10−7, word size 7, nucleotide match reward 1, nucleotide mismatch reward −3, gap open cost 5, gap extension cost 2. The remaining reads after all five filtering steps were used as the input for bacteria identification. All data used in this pipeline were downloaded on 6th February 2014.
Mapping to bacterial genomes
We collected 2736 bacterial whole genomes from NCBI (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/). Due to the extremely unbalanced number of available strains of different bacteria, we retained the longest strain as the representative for each bacterium, resulting in a 1421-genome database that was indexed and used for bacteria identification. Bowtie2 [61] was used as the aligner to map the read to each bacterial genome (parameters --local -D 20 -R 3 -N 1 -L 32 -i S,1,0.50). According to the mapping results, each read will be labeled as an unmapped read, a unique mapping read or a multiple mapping read. Unmapped reads are reads that cannot be aligned to any bacterial genomes, and multiple mapping reads are reads which align to two or more bacterial genomes. We marked uniquely mapped reads as those mapping to only one bacterium genome regardless of whether they mapped to multiple locations of the same genome. The count of total bacteria reads is the sum of the uniquely mapped reads (Additional file 1).
Genomic coverage
For any bacteria with more than ten unique mapped reads, we calculated the read counts for each 5-kbp window and then computed two coverage measures to normalize for differences in library sizes and for differences in genome size:
| 1 |
where Ci is the read count in window i, and n is the total number of 5-kb windows. In Eq. 1, the raw counts of each 5-kbp window are normalized by the total number of read counts mapping to corresponding bacteria to account for differences in number of reads mapped to each bacteria.
| 2 |
The second variation measure corrects for differences in genome size that can range from 139 kbp (Tremblaya princeps) to 13 Mbp (Sorangium cellulosum).
In this study positive bacteria identification was determined by ≥0.025 CPM reads, var1 ≤ 2 and var2 ≤ 1e-05.
Calculating Bacteria Abundance
In order to improve the sensitivity of relative abundance estimation, we used all reads mapped to bacterial genomes with PathoScope (version 2.0) [62], which is based on a Bayesian statistical framework to assign multiply mapped reads to the most probable bacterial source genome. To calculate relative bacteria abundance within each sample, read counts were normalized to the size of the corresponding bacterial genome to account for variation in bacterial genome size. The relative abundance of each bacterium is then calculated based on these normalized values.
Source code
Source code files are available online (https://github.com/zhangch/WGSpipeline).
H. pylori and EBV detection from RNA-seq gastric TCGA samples
BWA (version 0.6.2) was used to align the WGS data against the human reference genome (version hg19) with default parameters, and STAR aligner was used to align the RNA-Seq data against the human whole transcriptome. Unmapped reads were extracted and used for H. pylori and EBV identification. The EBV genome was added to the 1421-genome database that was generated for the analysis of biopsy WGS samples and Bowtie2 was used as the aligner with the same parameters as used in microbial detection from WGS data. H. pylori and EBV counts from each were normalized as CPM.
Acknowledgments
The authors greatly acknowledge the contribution of the Weill Cornell Epigenomics Core facility and computing support from the Applied Bioinformatics Core at Weill Cornell as well as Eric Pamer, MD PhD for his comments. This work was supported by the DeGregorio Family Foundation for Gastroesophageal Malignancies and by an internal grant from the Center for Advanced Digestive Care at Weill Cornell Medical College. We thank Gaudenz Hafen for kindly sharing the WGS data from cystic fibrosis patients.
Abbreviations
- BWA
Burrows-Wheeler Aligner
- EBV
Epstein-Barr virus
- HMP
Human Microbiome Project
- NCBI
National Center for Biotechnology Information
- qPCR
quantitative polymerase chain reaction
- TCGA
The Cancer Genome Atlas
- WGS
whole genome sequencing
Additional files
Table listing the read counts of the gastric biopsy samples after each filtering step. (XLSX 12 kb)
Supplementary figures and details of the comparison of identification results from three different methods. (PDF 663 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CZ performed all the computational analysis; KC and PT performed experimental validation; FSS collected the gastric biopsies; KC, BM, and MB coordinated sample handling; NS provided access to TCGA data; DB and MAS led the study and DB, MAS, and CZ wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Manish A. Shah, Email: mas9313@med.cornell.edu
Doron Betel, Email: dob2014@med.cornell.edu.
References
- 1.Cox MJ, Cookson WOCM, Moffatt MF. Sequencing the human microbiome in health and disease. Human Mol Genet. 2013:R1–R7. [Epub ahead of print]. [DOI] [PubMed]
- 2.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–6. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 3.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–8. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostic AD, Ojesnia AI, Pedamallu CS, Jung J, Verhaak RGW, Getz G, et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–6. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riley DR, Sieber KB, Robinson KM, White JR, Ganesan A, Nourbakhsh S, et al. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput Biol. 2013;9 doi: 10.1371/journal.pcbi.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrett WS. Cancer and the microbiota. Science. 2015;348:80–6. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M. Global burden of cancers attributable to infections in 2008: a review and sythetic analysis. Lancet Oncol. 2012;13:607–15. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 8.Abreu MT, Peek RM. Gastrointestinal malignancy and the microbiome. Gastroenterology. 2014;146:1534–1546. doi: 10.1053/j.gastro.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 10.Power DG, Kelsen DP, Shah MA. Advanced gastric cancer — slow but steady progress. Cancer Treat Rev. 2010;36:384–92. doi: 10.1016/j.ctrv.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Shah MA, Kelsen DP. Gastric cancer: a primer on the epidemiology and biology of the disease and an overview of the medical management of advanced disease. J Natl Compr Canc Netw. 2010;8:437–47. doi: 10.6004/jnccn.2010.0033. [DOI] [PubMed] [Google Scholar]
- 12.Tan IB, Ivanova T, Lim KH, Ong CW, Deng N, Lee J, et al. Intrinsic subtypes of gastric cancer, based on gene expression pattern, predict survival and respond differently to chemotherapy. Gastroenterology. 2011;141:476–85. doi: 10.1053/j.gastro.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pounder RE, Ng D. The prevalence of Helicobacter pylori infection in different countries. Aliment Pharmacol Ther. 1995;9:33–9. [PubMed] [Google Scholar]
- 15.Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol. 2006;12:354–62. doi: 10.3748/wjg.v12.i3.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15:306–16. doi: 10.1016/j.chom.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Brawner KM, Morrow CD, Smith PD. Gastric micriobiome and gastric cancer. Cancer J. 2014;20:211–6. doi: 10.1097/PPO.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu S, Zhang C, Miao Y, Gao J, Xu D. Effector prediction in host-pathogen interaction based on a Markov model of a ubiquitous EPIYA motif. BMC Genomics. 2010;11 Suppl 3:S1. [DOI] [PMC free article] [PubMed]
- 19.Basso D, Scrigner M, Toma A, Navaglia F, Di Mario F, Rugge M, et al. Helicobacter pylori infection enhances mucosal interleukin-1 beta, interleukin-6, and the soluble receptor of interleukin-2. Int J Clin Lab Res. 1996;26:207–10. doi: 10.1007/BF02592984. [DOI] [PubMed] [Google Scholar]
- 20.Zhang C, Xu S, Xu D. Risk assessment of gastric cancer caused by Helicobacter pylori using CagA sequence markers. PLoS One. 2012;7 doi: 10.1371/journal.pone.0036844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Zheng G, Xu S-F, Xu D. Computational challenges in characterization of bacteria and bacteria-host interactions based on genomic data. J Comput Sci Technol. 2012;27:225–39. doi: 10.1007/s11390-012-1219-y. [DOI] [Google Scholar]
- 22.Wei J, Noto JM, Zaika E, Romero-Gallo J, Piazuelo MB, et al. Bacterial CagA protein induces degradation of p53 protein in a p14ARF-dependent manner. Gut. 2014. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 23.Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F, et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci U S A. 2006;103:732–7. doi: 10.1073/pnas.0506655103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheh A, Fox JG. The role of the gastrointestinal microbiome in Helicobacter pylori pathogenesis. Gut Microbes. 2013;4:505–31. doi: 10.4161/gmic.26205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dicksved J, Lindberg M, Rosenquist M, Enroth H, Jansson JK, Engstrand L. Molecular characterization of the stomach microbiota in patients with gastric cancer and in controls. J Med Microbiol. 2009;58:509–16. doi: 10.1099/jmm.0.007302-0. [DOI] [PubMed] [Google Scholar]
- 26.Li XX, Wong GL, To KF, Wong VW, Lai LH, Chow DK, et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS One. 2009;4 doi: 10.1371/journal.pone.0007985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maldonado-Contreras A, Goldfarb KC, Godoy-Vitorino F, Karaoz U, Contreras M, Blaser MJ, et al. Structure of the human gastric bacterial community in relation to Helicobacter pylori status. ISME J. 2011;5:574–9. doi: 10.1038/ismej.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, et al. Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc Natl Acad Sci U S A. 2009;106:14321–6. doi: 10.1073/pnas.0903438106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Human Microbiome Project C A framework for human microbiome research. Nature. 2012;486:215–21. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Y, Madupu R, Karaoz U, Nossa CW, Yang L, Yooseph S, et al. Human papillomavirus community in healthy persons, defined by metagenomics analysis of human microbiome project shotgun sequencing data sets. J Virol. 2014;88:4786–97. doi: 10.1128/JVI.00093-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimon MT, Wood HM, Rabbitts PH, Arron ST. IMSA: integrated metagenomic sequence analysis for identification of exogenous reads in a host genomic background. PLoS One. 2013;8 doi: 10.1371/journal.pone.0064546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014;32:822–8. doi: 10.1038/nbt.2939. [DOI] [PubMed] [Google Scholar]
- 34.Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331:463–7. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Yang F, Ren L, Xiong Z, Wu Z, et al. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J Clin Microbiol. 2011;49:3463–9. doi: 10.1128/JCM.00273-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9:811–4. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- 38.Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.International HapMap C, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–8. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park HJ, Na S, Park SY, Moon SM, Cho OH, Park KH, et al. Clinical significance of Propionibacterium acnes recovered from blood cultures: analysis of 524 episodes. J Clin Microbiol. 2011;49:1598–601. doi: 10.1128/JCM.01842-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hauser PM, Bernard T, Greub G, Jaton K, Pagni M, Hafen GM. Microbiota present in cystic fibrosis lungs as revealed by whole genome sequencing. PLoS One. 2014;9 doi: 10.1371/journal.pone.0090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crump JA, Morrissey AB, Nicholson WL, Massung RF, Stoddard RA, Galloway RL, et al. Etiology of severe non-malaria febrile illness in Northern Tanzania: a prospective cohort study. PLoS Negl Trop Dis. 2013;7 doi: 10.1371/journal.pntd.0002324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perry A, Lambert P. Propionibacterium acnes: infection beyond the skin. Expert Rev Anti Infect Ther. 2011;9:1149–56. doi: 10.1586/eri.11.137. [DOI] [PubMed] [Google Scholar]
- 44.Alexeyev OA, Marklund I, Shannon B, Golovleva I, Olsson J, Andersson C, et al. Direct visualization of Propionibacterium acnes in prostate tissue by multicolor fluorescent in situ hybridization assay. J Clin Microbiol. 2007;45:3721–8. doi: 10.1128/JCM.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cohen RJ, Shannon BA, McNeal JE, Shannon T, Garrett KL. Propionibacterium acnes associated with inflammation in radical prostatectomy specimens: a possible link to cancer evolution? J Urol. 2005;173:1969–74. doi: 10.1097/01.ju.0000158161.15277.78. [DOI] [PubMed] [Google Scholar]
- 46.Delgado S, Suarez A, Mayo B. Identification, typing and characterisation of Propionibacterium strains from healthy mucosa of the human stomach. Int J Food Microbiol. 2011;149:65–72. doi: 10.1016/j.ijfoodmicro.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 47.Delgado S, Cabrera-Rubio R, Mira A, Suarez A, Mayo B. Microbiological survey of the human gastric ecosystem using culturing and pyrosequencing methods. Microb Ecol. 2013;65:763–72. doi: 10.1007/s00248-013-0192-5. [DOI] [PubMed] [Google Scholar]
- 48.Lee CC, Lin WJ, Shih HI, Wu CJ, Chen PL, Lee HC, et al. Clinical significance of potential contaminants in blood cultures among patients in a medical center. J Microbiol Immunol Infect. 2007;40:438–44. [PubMed] [Google Scholar]
- 49.Ishige I, Eishi Y, Takemura T, Kobayashi I, Nakata K, Tanaka I, et al. Propionibacterium acnes is the most common bacterium commensal in peripheral lung tissue and mediastinal lymph nodes from subjects without sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22:33–42. [PubMed] [Google Scholar]
- 50.Yoon JH, Baik GH, Sohn KM, Kim DY, Kim YS, Suk KT, et al. Trends in the eradication rates of Helicobacter pylori infection for eleven years. World J Gastroenterol. 2012;18:6628–34. doi: 10.3748/wjg.v18.i45.6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buzas GM. First-line eradication of Helicobacter pylori: are the standard triple therapies obsolete? A different perspective. World J Gastroenterol. 2010;16:3865–70. doi: 10.3748/wjg.v16.i31.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heo J, Jeon SW. Optimal treatment strategy for Helicobacter pylori: Era of antibiotic resistance. World J Gastroenterol. 2014;20:5654–9. doi: 10.3748/wjg.v20.i19.5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim N, Kim JM, Kim CH, Park YS, Lee DH, Kim JS, et al. Institutional difference of antibiotic resistance of Helicobacter pylori strains in Korea. J Clin Gastroenterol. 2006;40:683–7. doi: 10.1097/00004836-200609000-00004. [DOI] [PubMed] [Google Scholar]
- 54.Ford AC, Forman D, Hunt RH, Yuan Y, Moayyedi P. Helicobacter pylori eradication therapy to prevent gastric cancer in healthy asymptomatic infected individuals: systematic review and meta-analysis of randomized controlled trials. BMJ. 2014:348. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 55.Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7:956–60. doi: 10.1038/sj.embor.7400812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monstein HJ, Tiveljung A, Kraft CH, Borch K, Jonasson J. Profiling of bacterial flora in gastric biopsies from patients with Helicobacter pylori-associated gastritis and histologically normal control individuals by temperature gradient gel electrophoresis and 16S rDNA sequence analysis. J Med Microbiol. 2000;49:817–22. doi: 10.1099/0022-1317-49-9-817. [DOI] [PubMed] [Google Scholar]
- 57.Clyne M, Labigne A, Drumm B. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect Immun. 1995;63:1669–73. doi: 10.1128/iai.63.5.1669-1673.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konturek PC, Kania J, Burnat G, Hahn EG, Konturek SJ. Prostaglandins as mediators of COX-2 derived carcinogenesis in gastrointestinal tract. J Physiol Pharmacol. 2005;56:57–73. [PubMed] [Google Scholar]
- 59.NCBI Sequence Read Archive. http://www.ncbi.nlm.nih.gov/bioproject/PRJNA297869.
- 60.McGinnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004;32:W20–25. doi: 10.1093/nar/gkh435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong C, Manimaran S, Shen Y, Perez-Rogers JF, Byrd AL, Castro-Nallar E, et al. PathoScope 2.0: a complete computational framework for strain identification in environmental or clinical sequencing samples. Microbiome. 2014;2:33. [DOI] [PMC free article] [PubMed]