

Abstract
Due to their extensive use as plasticisers in numerous consumer products, phthalates have become ubiquitous environmental contaminants. An increasing number of epidemiological studies suggest that exposure to phthalates may be associated with worsening or development of airway diseases. Peroxisome Proliferation Activated Receptors (PPAR)s, identified as important targets for phthalates in early studies in rodent liver, have been suggested as a possible mechanistic link. In this review we discuss the likelihood of an involvement of PPARs in asthma development and exacerbation due to pulmonary phthalate exposure. First, we go through the literature on indoor air levels of phthalates and pulmonary phthalate kinetics. These data are then used to estimate the pulmonary phthalate levels due to inhalation exposure. Secondly, the literature on phthalate-induced activation or modulation of PPARs is summarized. Based on these data, we discuss whether pulmonary phthalate exposure is likely to cause PPAR activation, and if this is a plausible mechanism for adverse effects of phthalates in the lung. It is concluded that the pulmonary concentrations of some phthalates may be sufficient to cause a direct activation of PPARs. Since PPARs mainly mediate anti-inflammatory effects in the lungs, a direct activation is not a likely molecular mechanism for adverse effects of phthalates. However, possible modulatory effects of phthalates on PPARs deserve further investigation, including partial antagonist effects and/or cross talk with other signalling pathways. Moreover other mechanisms, including interactions between phthalates and other receptors, could also contribute to possible adverse pulmonary effects of phthalates.
Keywords: phthalates, asthma, Peroxisome Proliferation Activated Receptors, molecular mechanism
Introduction
During the last decades, the prevalence and incidence of respiratory allergies and asthma has reached extensive proportions especially in the industrialized countries. The airway symptoms may vary from mild and temporary to severe and life threatening with urgent need for emergency treatment to restore normal breathing. Although the increase in prevalence appears to be levelling out and reaching a plateau in some countries, there is still a concern for this rise in prevalence in developing countries (Pearce and Douwes, 2006[79]; Bousquet et al., 2005[17]; Lotvall et al., 2009[66]). Moreover, the number of people affected by asthma is so high that it is considered as a major public health problem - especially for children - with global treatment costs running at billions of dollars each year (Bousquet et al., 2005[17]). A broad spectrum of factors seem to influence asthma development, ranging from genetics to life style and environmental factors, but much remains to be learned about what causes the disease and how to reduce its occurrence (Pearce and Douwes, 2006[79]; Bousquet et al., 2005[17]).
Since many children and infants spend more than 90 % of their time indoors (Leech et al., 2002[63]), indoor factors are of particular interest with respect to environmentally triggered conditions such as asthma. Reviews have concluded that indoor factors like environmental tobacco smoke (Baena-Cagnani et al., 2009[5]) as well as mold and dampness (Mendell et al., 2011[71]) are associated with asthma development or exacerbations, although there is lack of knowledge concerning the underlying mechanisms. Moreover, a recent review concluded that more research was needed to clarify the potential risks related to chemicals such as volatile and semi-volatile compounds, phthalates and chlorinated chemicals (Heinrich, 2011[36]). However, several epidemiological studies report an association between phthalate exposure and worsening or development of respiratory diseases, and a causal relationship has been suggested (as reviewed in (Bornehag and Nanberg, 2010[14]; Jaakkola and Knight, 2008[42])).
Phthalates are used as plasticisers in numerous consumer products, and since they are not covalently linked to the plastic, they leak out into the environment. Consequently, phthalates are ubiquitous environmental contaminants found in air, dust and food (Wormuth et al., 2006[110]). The general population is continuously exposed to phthalates through inhalation, dermal uptake and ingestion, as confirmed by the presence of phthalate metabolites in nearly all analysed urine samples in large population based studies (Wittassek et al., 2011[109]). When phthalates enter the human body they are rapidly hydrolysed to their primary metabolites, and then further oxidized into various secondary metabolites (Wittassek et al., 2011[109]). Generally, the low molecular weight phthalates (molecular weight < di-2-ethylhexylphthalate; DEHP) are primarily metabolised into their monoesters, whereas the heavier phthalates (≥ DEHP) may be further metabolised into oxidative secondary metabolites. The phthalate metabolites are then secreted via urine. Although the metabolic pathways of phthalates after oral intake are partly known for some phthalates, the knowledge about distribution of metabolites in the body is limited (Frederiksen et al., 2007[30]). When comparing the daily internal exposure to phthalates based on contribution of various sources, ingestion appears to be the major exposure route for many phthalates (Wormuth et al., 2006[110]). However, for diethylphthalate (DEP), di-n-butylphthalate (DnBP), butylbenzylphthalate (BBzP) and di-iso-nonylphthalate (DiNP) inhalation exposure contributed to more than 20 % of the daily internal dose, suggesting that inhalation is an important exposure route for some phthalates (Wormuth et al., 2006[110]). Interestingly, fasting did not impact on the urinary levels of light weight phthalates, thus other pathways than ingestion appeared to contribute to these levels (Wittassek et al., 2011[109]; Koch et al., 2013[52]). Moreover, a significant correlation has been reported between the metabolites of the low weight phthalates DEP, DnBP and BBzP in urine and the corresponding personal air levels (Adibi et al., 2003[2], 2008[3]), and a recent study also reports a correlation between BBzP in dust and urinary MBzP levels in children (Hsu et al., 2012[39]).
The levels of DEHP and BBzP in house dust have been associated with asthma and wheeze in children in a cross sectional study (Bornehag et al., 2004[15]; Kolarik et al., 2008[54]), and BBzP was higher in house dust in homes of subjects categorised as allergic or asthmatic (Hsu et al., 2012[39]). Both cross sectional and longitudinal studies show that use of polyvinyl chloride (PVC) flooring is related to asthma, and the correlations between PVC flooring and dust concentration of DEHP and BBzP, as well as uptake of BBzP in infants, provide further support for an association between phthalate exposure and asthma (Carlstedt et al., 2012[20]). Urinary concentrations of the metabolites of DEP and DnBP were recently associated with decreased lung function in male volunteers (Hoppin et al., 2004[37]), whereas the metabolites of DEP and BBzP were associated with an increase in fractional exhaled nitric oxide, a marker of airway inflammation (Just et al., 2012[44]). Moreover, the concentrations of metabolites of the high molecular weight phthalates di-iso-decylphthalate (DiDP) and DiNP in urine were modestly associated with current asthma in children (Bertelsen et al., 2013[11]), while the metabolites of BBzP and DnBP were associated with diagnosed asthma (Hsu et al., 2012[39]). Thus, there is emerging evidence for an association between phthalate exposure and respiratory symptoms, although no firm link has been established between the inhalation exposure route and the various airway responses.
Recent reviews of in vivo and in vitro studies of phthalates conclude that phthalates are capable of inducing an inflammatory response in lung and immune cells, and to modulate the response to a co-allergen (Bornehag and Nanberg, 2010[14]; Jaakkola and Knight, 2008[42]). There is however still an ongoing discussion as to whether phthalates can induce inflammatory and adjuvant responses at concentrations relevant for indoor air exposures (Larsen et al., 2007[60]; Hansen et al., 2007[35]; Nielsen et al., 2007[75]; Kimber and Dearman, 2010[50]). Moreover, the molecular mechanisms involved in any inflammatory response to phthalates are largely unknown. Early studies on phthalate effects identified Peroxisome Proliferation Activated Receptors (PPAR)s as important targets for phthalates and possible mediators for various effects observed in the liver of some rodents (Rusyn et al., 2006[91]; Hurst and Waxman, 2003[40]). Thus, a similar mechanism with a phthalate-induced activation of PPARs has also been suggested in the pulmonary effects of phthalates (Magliozzi et al., 2003[67]; Rosicarelli and Stefanini, 2009[88]; Just et al., 2012[45]). However, there are only few studies investigating if PPARs actually are involved in the phthalate-induced effects (Rakkestad et al., 2010[85]; Bolling et al., 2012[13]; Larsen and Nielsen, 2007[61]), these either suggest an anti-inflammatory or modulatory role for PPARs (Rakkestad et al., 2010[85]; Bolling et al., 2012[13]) or no influence on the phthalate-induced effects (Larsen and Nielsen, 2007[61]).
PPAR is a family of nuclear receptors that function as ligand-activated transcription factors. They participate in a range of cellular processes including lipid metabolism, glucose homeostasis, proliferation and differentiation, but also in positive and negative regulation of inflammation (Yessoufou and Wahli, 2010[114]). The three known PPAR isotypes α, γ and δ/β can be activated by fatty acids, fatty acid derivatives, but also by synthetic compounds like thiazolidinediones and phthalates (Yessoufou and Wahli, 2010[114]). Upon activation by an appropriate ligand, PPARs form a heterodimer with RXRs (cis-retinoic acid receptors) and recruit nuclear co-activators or co-repressors before binding to specific promoter elements. In this way ligand binding to PPAR can result in both activation and inhibition of gene expression (Ricote and Glass, 2007[87]).
The three PPARs have unique though overlapping tissue distributions and functions, but all PPARs are expressed in various cell types of the lung, including epithelium, fibroblasts and smooth muscle cells, macrophages, T lymphocytes and eosinophils (Becker et al., 2006[8]; Rehan et al., 2009[86]). As recently reviewed by Becker and co-authors, PPARs are mainly involved in anti-inflammatory responses in the lung, and they have been suggested as possible targets in the treatment of pulmonary symptoms in asthmatics (Becker et al., 2006[8]). Similarly, PPAR agonists such as Rosiglitazone, are used as treatment for Type 2 diabetes, another disease with an inflammatory component (Gross and Staels, 2007[33]). The involvement of PPARs in the development of inflammatory diseases like diabetes and asthma is not obvious, but phthalate-induced dysregulation of PPARs has been proposed as a possible mechanism (Desvergne et al., 2009[25]).
In this review we discuss the likelihood of an involvement of PPAR in the exacerbation of asthma symptoms and the development of asthma due to pulmonary exposure to phthalates. First, we review the literature on indoor air levels for the most commonly measured phthalates, and summarize the current knowledge on pulmonary deposition, adsorption and metabolism of phthalates. These data are then used to estimate the pulmonary phthalate levels due to inhalation exposure. Secondly, the literature on phthalate-induced activation or modulation of PPARs is reviewed. Based on these data we discuss if PPAR activation is likely to be induced by pulmonary phthalate exposure and if PPAR activation is a plausible mechanism of action for phthalate induced effects in the lung. Finally, other suggested mechanisms for phthalate-induced effects are briefly discussed.
Inhalation Exposure to Phthalates
Although ingestion and dermal uptake are believed to be the major exposure routes for most phthalates, exposure by inhalation is likely to cause a higher dose of phthalates in the lung than in other organs. This may be of importance for pulmonary endpoints like asthma and airway hyper responsiveness. In addition, exposure by inhalation may exclude first path metabolism/elimination of phthalates via the liver.
Most phthalates fit to the definition of semi volatile organic compounds (SVOCs) and phthalates released from consumer products in indoor environments are present in the gas phase (Weschler et al., 2008[108]). Like other SVOCs, the phthalates partition between gas phase, airborne particles, house dust and other surfaces. This partitioning process depends on their individual vapour pressures, which is related to their molecular weights (Weschler and Nazaroff, 2010[107]). In general, phthalates with low molecular weight are predominately found in the gas phase whereas heavier phthalates are associated with particles (Weschler et al., 2008[108]). For some of the high molecular weight phthalates, like DEHP, the partitioning between particle- and gas-phase is difficult to predict and depends on the physicochemical properties of the particles present in the indoor environment (Schossler et al., 2011[95]).
Indoor air levels of phthalates
Phthalate levels in indoor air are generally reported as the sum of the particulate and the semi-volatile fractions. In several epidemiological studies, phthalate levels in household dust (i.e. settled dust) have been used as a measure of phthalate exposure. These levels are more relevant for oral exposure since children and especially infants are known to ingest considerable amounts of dust, thus they will not be discussed in detail here. Resuspension of settled dust might also contribute to indoor air levels; however, we assume that this resuspended dust is accounted for in the levels measured in indoor air. Note also that the phthalate adsorption to house dust particles differs from that to the airborne particles both with respect to the total amount of phthalates adsorbed and the relative amount of the various types of phthalates. Generally, high molecular weight phthalates like DEHP are present in higher levels in house dust than in the gas and particle phase (Rudel et al., 2003[89]; Bergh et al., 2011[10]; Fromme et al., 2004[31]). Moreover, the phthalate levels in the gas and particle phase cannot be calculated from the levels in house dust with sufficient accuracy (Weschler et al., 2008[108]).
Indoor air levels of phthalates have been reported for a range environments, including homes, kindergartens, schools, universities, workplaces and cars (Rudel et al., 2003[89]; Fromme et al., 2004[31]; Otake et al., 2004[77]; Rakkestad et al., 2007[84]; Adibi et al., 2003[2], 2008[3]; Just et al., 2010[44]; Bergh et al., 2011[10]; Kanazawa et al., 2010[47]). Indoor air seems to be the major source for inhalation exposure to phthalates, since phthalate levels in outdoor air are low, generally below 2 ng/m3 (Otake et al., 2004[77]; Rudel and Perovich, 2009[90]). To summarize current data on indoor air levels of phthalates, the mean, median and range of levels reported in the nine identified studies are listed in Table 1a(Tab. 1) and 1b(Tab. 1). The relative abundance of the different phthalates varies between the studies, but generally either DEP or DnBP are most abundant, with reported means in the ranges 150-3000 ng/m3 and 100-2900 ng/m3, respectively. Moreover DEHP and DiBP generally seem to be present in moderate levels, with a range of mean concentrations of 0-600 and 250-1000 ng/m3, respectively, whereas BBzP and DiNP are clearly the least abundant phthalates measured (Table 1a(Tab. 1) and 1b(Tab. 1)). When the phthalate levels in homes, kindergartens and workplaces were compared, similar total levels and concentration profiles for phthalates were found in the different environments (Bergh et al., 2011[10]). The reported mean values are often higher than the median (Table 1a(Tab. 1) and 1b(Tab. 1)), suggesting that the exposure distributions for phthalates are skewed due to much higher exposure levels for some individuals (Rudel et al., 2003[89]).
Table 1. Phthalate concentrations in indoor air for (a) low and (b) high molecular weight phthalates. The levels represent the sum of vapour and particulate phases unless otherwise noted. The grey rows represent the range of reported values rounded to the nearest 50.
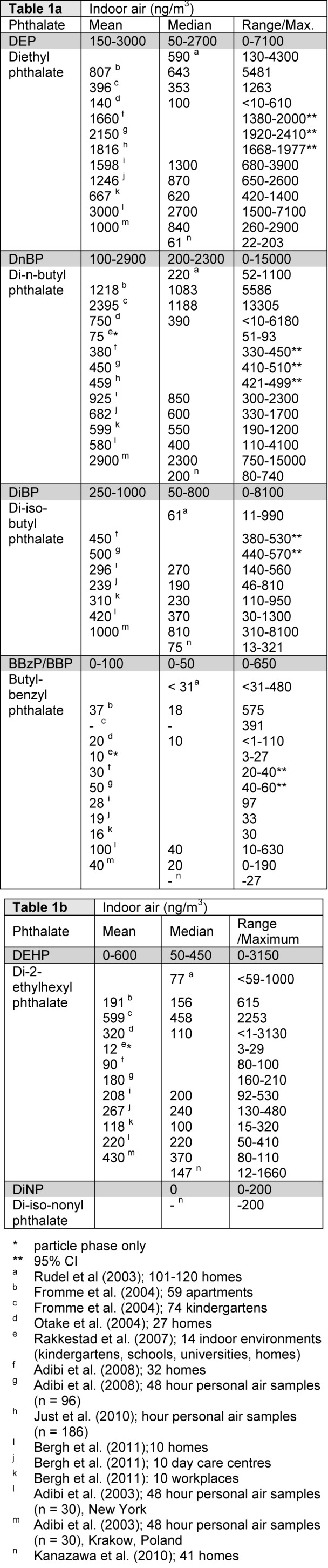
The use of DEHP has largely been phased out, but since DEHP was the most commonly used phthalate in PVC containing interior surface materials and consumer products for many decades, it may still be the most abundant phthalate in indoor dust samples, as long as these products are in use. Over the last ten years the use of phthalates with higher molecular weight has increased, but indoor concentrations for many of these phthalates have not been investigated (Schossler et al., 2011[95]). For instance DiNP and DiDP, that presently account for more than half of the overall plasticizers consumption in Europe, have only been included in few studies of indoor dust (summarized in (Abb et al., 2009[1])), whereas only one study reports levels in the gas/particle phase for DiNP (Kanazawa et al., 2010[47]). The reported mean values for DiNP and DiDP in house dust range from 30-60 μg/g dust and 70-130 μg/g dust, respectively. In comparison the mean values for DEHP seem to be considerably higher (750-2000 μg/g dust; (Fromme et al., 2004[31]; Bergh et al., 2011[10])). Based on knowledge about the physical properties of high molecular weight phthalates, these compounds are likely to be present in the particle phase, yielding low gas phase concentrations (Schossler et al., 2011[95]).
The Fate of Phthalates in the Lung
The deposition pattern of gas and particle phase phthalates is likely to differ. Since low and high molecular weight phthalates partition differently between the gas and particle phase, it is necessary to distinguish between these phthalates when considering pulmonary phthalate exposure due to inhalation.
Low molecular weight phthalates
The lungs are covered by a thin layer of lung lining fluid (LLF) consisting of water, ions, proteins and antioxidants (Lewis, 2006[64]). The thickness of the layer varies between the different parts of the lungs, from 6-10 µm in the upper part of the lungs to approximately 0.1 µm in the alveoli. Similarly, the composition differs between the various parts of the lungs; with a high mucous content in the conducting airways and a higher surfactant content in the alveoli (Ng et al., 2004[74]). Calculations and measurements based on volume markers suggest that the total volume of LLF in a human of 70 kg is approximately 25 ml (Walters, 2002)[102]. The LLF is covered by surfactant which consists of a monolayer of phospholipids, neutral lipids and surfactant associated proteins (Lewis, 2006[64]). Thus, inhaled phthalates are likely to first interact with the surfactant and then with the LLF. Low molecular weight phthalates are primarily found in the gas phase, and the deposition of gas phase molecules will depend on physicochemical properties of the molecules, such as the water solubility and molecule size. For instance, low water solubility of the molecules will result in higher deposition in the bronchiolar/alveolar regions whereas high water solubility will result in higher deposition in the upper airways (Bakand and Hayes, 2010[6]). To our knowledge, pulmonary deposition of phthalates has not been elucidated, neither with regard to deposition site nor deposition probability.
High molecular weight phthalates
Since high molecular weight phthalates are primarily particle bound, the pulmonary dose and distribution of these phthalates will be determined by the physicochemical properties of the particulate matter they are adsorbed to (Phalen, 2002[80]; Kreyling et al., 2007[55]; Löndahl et al., 2007[65]). Particle deposition is highly non-uniform in the lung, and some sites receive much higher particle doses than others. In the peripheral lung, deposition of particulate matter is particularly high in the proximal alveolar region, which is defined as the section located between the terminal bronchiole and the alveolar space (Donaldson et al., 2008[27]; Pinkerton et al., 2004[81]; Saldiva et al., 2002[92]).
The fate of particle bound phthalates after deposition in the lung has not been studied. However, partitioning between particle and aqueous phase has been studied in an environmental pollutant context for some phthalates and particle types (Julinova and Slavik, 2012[43]; Xu and Li, 2008[112]; Wang et al., 2010[103]). Since adsorption/desorption of phthalates from particles is an equilibrium process it is likely that phthalates desorb into the LLF to some extent after deposition. The degree of desorption depends on the properties of the particulate matter and the LLF, as well as the mass to volume ratio. Experimental studies are necessary to address desorption of phthalates in the context of inhaled particulate matter.
Pulmonary phthalate kinetics
Many studies have used DEHP as a model compound to investigate the absorption, distribution and metabolism as well as toxicity of phthalates. Based on data from human and animal studies, metabolism of DEHP has been shown to involve a complex series of reactions that produce a high number of metabolites, including mono-2-ethylhexylphthalate (MEHP) (Koch et al., 2006[53]).
Generally, phthalates are rapidly metabolised after absorption through a lipase mediated cleavage into hydrolytic monoesters, followed by oxidation of the alkyl chain of the monoesters causing formation of various secondary metabolites (Koch and Calafat, 2009[51]). The low molecular weight phthalates are mostly metabolised into their monoesters, whereas the heavier phthalates may be further metabolised into oxidative secondary metabolites (Koch and Calafat, 2009[51]). The lipases that mediate the hydrolytic cleavage of phthalates have been identified in a number of organs including lungs, where they have been found in several cell types such as alveolar macrophages and type 2 cells as well as in LLF (Albro and Thomas, 1973[4]; Mahoney et al., 1982[68]; Coonrod et al., 1989[22]). This suggests that pulmonary hydrolysis of phthalates, with subsequent cellular exposure to phthalate monoesters, is possible. However, it is presently not known to what extent phthalate metabolism takes place in the lung (Albro and Thomas, 1973[4]; Mahoney et al., 1982[68]; Coonrod et al., 1989[22]). Although the lipase activity as well as the metabolic rate in lung tissue has been shown to be lower compared to intestines, liver and kidney (Ito et al., 2005[41]; Choi et al., 2012[21]), this does not exclude the possibility that phthalates might be metabolised in the lung after inhalation exposure.
A recent review of DEHP toxicity conducted by United States Consumer Product Safety Commission concluded that absorption of DEHP after inhalation exposure has received limited investigations both in humans as well as rodents. Thus the kinetics of human phthalate metabolism after inhalation exposure and the site for the hydrolysis of phthalates into secondary metabolites is largely unknown. However, DEHP has been reported to translocate rapidly from the lung to the blood stream in rodents, suggesting only transient exposure of pulmonary cells (Carlson, 2010[19]). Although this might suggest rapid absorption and distribution also in the humans, it remains to be elucidated whether the absorption is slow enough to allow for pulmonary metabolism of phthalates. Interestingly, the metabolism of inhaled DEHP to MEHP in rodent lung has been suggested to be in the order of 1-3 % based on comparison of the doses of DEHP and MEHP inducing similar effects on airway irritation, airway inflammation and allergen specific IgE production in mice (Larsen et al., 2007[60]).
Estimates of Pulmonary Phthalate Levels
Indoor phthalate levels have been used to calculate the inhalation exposure and then estimate the relative contribution of inhalation exposure to the total phthalate exposure (Otake et al., 2004[77]; Fromme et al., 2004[31]). Inhalation exposure to DEHP, DEP and BBzP was estimated to account for approximately 1-2 % of the total exposure, whereas inhalation of DnBP may contribute with up to 20 % of the total exposure (Otake et al., 2004[77]; Fromme et al., 2004[31]). Thus, inhalation exposure seems to account for a small part of the total body burden of phthalates. However, estimates of pulmonary phthalate concentrations due to inhalation exposure have to our knowledge not been reported previously.
The calculations of daily inhalation doses done by Otake and Fromme and their co-authors assumed that all the inhaled phthalates remained in the lung. However, since phthalates are relatively small molecules, they may be likely to follow the expiratory air flow to a certain extent. Such daily inhalation doses must therefore be considered as the maximum possible exposure dose. To obtain a rough estimate of the pulmonary phthalate levels due to inhalation exposure we estimate the phthalate levels in the LLF based on the exposure estimated from the indoor air concentrations. Due to limited knowledge on pulmonary phthalate deposition, we assume in our calculations that both particle and vapour phase phthalates deposit evenly in the LLF, although an uneven distribution is probably more likely both for gas and particle phase phthalates. Moreover, we assume that all the inhaled phthalates remain in the lung, as in the previous calculations by Otake and Fromme and their co-authors. Since the clearance kinetics of phthalates from the lungs is not well known, we have chosen to calculate the amount of deposited phthalate after both 2 and 24 hours inhalation, representing rapid and slow absorption respectively. The reported phthalate concentrations summarized in Table 1a(Tab. 1) and 1b(Tab. 1) exhibit great variation, we therefore present calculations for the lowest and highest reported mean values, as well as for the maximum reported levels, for each phthalate (Table 2(Tab. 2)).
Table 2. Rough estimates of pulmonary phthalate concentrations due to inhalation exposure. The table shows the maximum levels reported for each phthalate as well as the highest and lowest mean values, for each phthalate, and the corresponding calculated concentrations of phthalates in LLF after 2 or 24 hours inhalation. See main text for assumptions and explanation of calculations.
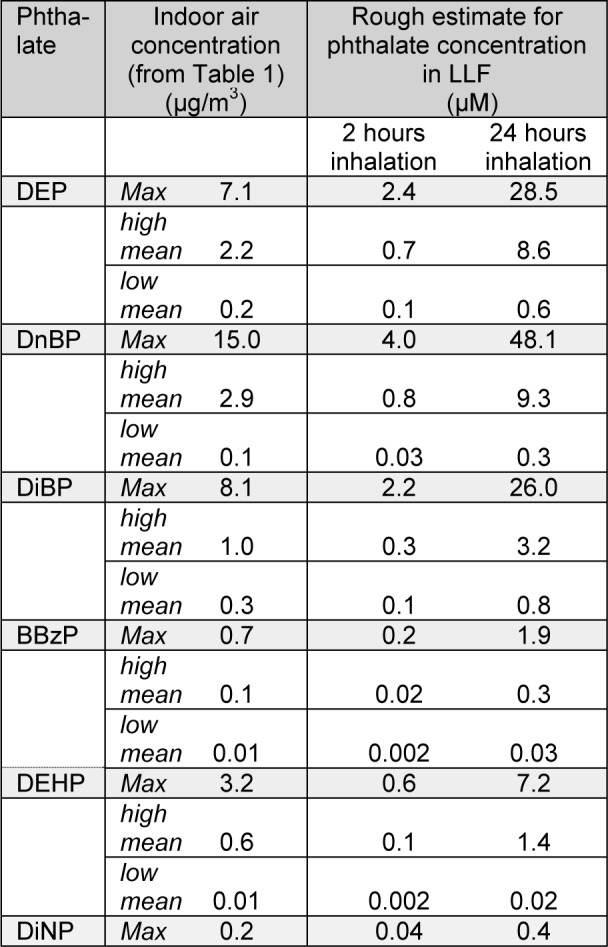
The calculations were done for adults (20-70 years) and children (0-4 years) assuming inhalation volumes for 24 hours of 23 and 5 m3 respectively (Fromme et al., 2004[31]). For the volume of LLF estimates of 25 ml was used for an adult of 70 kg and 5,6 ml for a child of 16 kg (Walters, 2002[102]). The total amount of inhaled phthalate was calculated (phthalate concentration * inhaled volume; not shown), and the corresponding phthalate concentration in the LLF was calculated (amount of inhaled phthalate / volume of LLF) to determine the molar concentration presented in Table 2(Tab. 2). The estimated pulmonary levels for adults and children are almost identical since the inhalation volume to LLF volume ratio is almost equal for the two cases, 23/25 = 0.92 for adults and 5/5.6 = 0.89 for children. We have therefore chosen to show only the data for children (Table 2(Tab. 2)).
The rough estimates presented in Table 2(Tab. 2) should be interpreted cautiously since they are based on multiple assumptions and are highly uncertain. Nevertheless they provide tentative concentration ranges for pulmonary phthalate concentrations due to inhalation exposure. If a rapid absorption of phthalates is assumed, the maximal and high mean indoor air levels will correspond to estimated pulmonary concentrations of 0.04 - 4 µM and 0.02 - 0.8 µM, respectively (Table 2(Tab. 2)). For a slow absorption, the corresponding concentrations are 0.4 - 48.1 µM and 0.3 - 9.3 µM. Keep in mind that the phthalate levels represent maximum deposition, i.e. all inhaled phthalates are assumed to deposit in the lung, suggesting that the estimates are higher than the actual exposure. In addition, a uniform pulmonary deposition was assumed, whereas a more likely scenario would be a non-uniform distribution with higher concentrations in some regions of the lung and lower in others.
Phthalates as PPAR-Ligands and -Gene Modulators/Activators
Early rodent studies of phthalate induced effects suggested PPARs as important targets for phthalates, and PPARs have also been suggested to play a role in the pulmonary effects of phthalates (Magliozzi et al., 2003[67]; Rosicarelli and Stefanini, 2009[88]). PPARs are involved in a wide range of biological processes, and are expressed in immune and lung cells involved in asthma development and exacerbation (Becker et al., 2006[8]; Di Paola and Cuzzocrea, 2007[26]). Several aspects of the interaction between phthalates and the three PPAR isotypes (α, γ and δ/β) have been studied, including molecular modelling of phthalate-PPAR interaction and co-variator recruitment in cell free assays (see chapter PPAR-ligand binding and activation in cell free systems), trans-activation studies using transfected cells and activation of constitutive PPAR in cellular models (see chapter PPAR trans-activation studies). In addition, a limited number of studies have investigated other modes of action for phthalates, including inhibitory and modulating effects (see chapter Inhibitory or modulating effects of phthalates on PPAR activity).
PPAR-Ligand Binding and Activation in Cell Free Systems
The ligand binding domains (LBD) of PPARs are well characterized, thus receptor binding of phthalates can now be studied not only in “classic” competitive receptor binding studies, but also by computer based modelling. These studies are based on calculations of the binding energies between the LBD of the PPAR and knowledge of the molecular structure of the respective phthalates (Nakagawa et al., 2008[73]; Kambia et al., 2008[46]; Kaya et al., 2006[48]; Feige et al., 2007[29]). They have revealed that the primary metabolites of phthalates (hydrolysed compounds) are more likely PPAR ligands than the parent compounds and the secondary metabolites (oxidised phthalates). For instance, computational studies suggest that MEHP can bind to PPARα and γ, whereas the parent compound DEHP is unable to bind or exhibits weak binding (Feige et al., 2007[29]; Kambia et al., 2008[46]). Similarly, in a co-variator recruitment study, MEHP induced a dose dependent increase in PPARγ activity, whereas DEHP and the oxidised metabolites showed no activation (Kusu et al., 2008[57]).
When a wider range of phthalate metabolites was investigated, the ability of phthalates to bind to PPARs differed considerably between the various phthalates, and their molecular structure affected the strength of the bond (Nakagawa et al., 2008[73]; Kaya et al., 2006[48]). The calculated free energies from a computational screening for binding of phthalate monoesters to PPARγ correlated well (R2 = 0.82) with the log EC50 values determined in an in vitro trans-activation study (Kaya et al., 2006[48]; Lampen et al., 2003[58]), suggesting that data from molecular modelling could be used to identify possible PPAR ligands (Kaya et al., 2006[48]). EC50 denotes an equivalent concentration resulting in 50 % of the maximum response level. Based on these correlations Kaya and co-authors identified 20 new phthalate monoesters that were ranked as potent PPARγ activators. Thus, a high number of phthalates, many of which have not yet been included in environmental exposure studies, could potentially activate PPARs. Although the correlation between calculations and in vitro data strengthens the reliability of data from molecular modelling, it is necessary to verify such findings in experimental model systems (Kaya et al., 2006[48]).
PPAR Trans-Activation Studies
In accordance with the molecular modelling studies, cell transfection studies report that both mouse and human PPARα and PPARγ were activated by MEHP, but not by the parent compound DEHP (Maloney and Waxman, 1999[69]; Lapinskas et al., 2005[59]). In contrast, BBzP and DnBP were able to activate all three PPAR subtypes, and to a slightly higher extent than their metabolite mono-n-butylphthalate (MnBP) (Lapinskas et al., 2005[59]). Thus, some of the parent phthalates may activate PPARs directly, rather than through their primary metabolites.
Phthalate metabolites
A range of different cell lines have been applied in transfection studies using different PPAR isotypes, mouse or human, or in some cases both, in order to compare species sensitivity. A similar degree of activation for human and mouse PPARα was reported by Lampen and co-authors, whereas two other studies reported that mouse PPARα was activated at lower concentrations than human PPARα (Hurst and Waxman, 2003[40]; Bility et al., 2004[12]; Lampen et al., 2003[58]). In contrast, mouse and human PPARγ were trans-activated at similar concentrations by a range of phthalate monoesters (Lampen et al., 2003[58]; Bility et al., 2004[12]; Hurst and Waxman, 2003[40]). A study comparing species sensitivity for PPARδ/β reported that mouse PPARδ/β was activated by several phthalate monoesters while human PPARδ/β was inactive (Bility et al., 2004[12]). Thus, the data concerning the affinity of human versus mouse PPAR for binding to phthalates is somewhat conflicting. It is, however, important to remember that the phthalate responses obtained also may depend on the specific cell type used. Feige and co-workers compared the ability of MEHP to activate PPARγ in different cell types and reported that the affinity (i.e. the concentration where the effect occurred) was rather similar for all cell types, while the efficacy (i.e. the maximal level of activation) seemed to vary (Feige et al., 2007[29]).
When comparing the sensitivity of the three PPAR isotypes α, γ and δ/β Lampen and co-authors (2003[58]) found that most of the investigated phthalate monoesters had a higher affinity (i.e. lower EC50) for PPARγ than PPARα. In contrast, a more recent study using MEHP as a model compound, reported a similar affinity for PPARα and γ, although the efficacy of MEHP was higher for PPARγ than the α-form (Feige et al., 2007[29]).
A number of in vitro studies report differential PPAR activation for the various phthalate monoesters, and the range of the lowest activation concentrations and the maximum fold increase are given in Table 3(Tab. 3). A direct comparison of the different studies is difficult since different measures of PPAR activation have been used; some studies report EC50 values, while others report the lowest concentration causing significant response (EClow). Thus, both these measures are included in Supplementary material to allow for inclusion of data from all available studies. As seen in Table 3(Tab. 3) there is a large span in the ability of the various phthalates to activate PPARs. The lowest activation concentrations range from 0.1-300 μM, with maximal fold inductions from 2-32. Generally, the metabolites from low molecular weight phthalates (< MEHP) show no PPAR activation, or only activation at moderate to high concentrations. On the other hand, metabolites from phthalate monoesters with higher molecular weight (≥ MEHP) generally cause activation of PPARs in transfection studies, with the exception of human PPARδ/β. Accordingly, the potency and efficacy of phthalate monoesters to activate PPARα and PPARγ has been reported to increase with increasing side chain length (Lampen et al., 2003[58]; Bility et al., 2004[12]), suggesting that high molecular weight phthalates are more potent PPAR ligands than low molecular weight phthalates. For PPARδ/β however, the lowest activation concentrations are in the same range both for low and high molecular weight phthalates.
Table 3. Phthlate-induced activation of PPARs in transfection studies. Summary of the lowest activation range and the maximum fold increase in PPAR activation reported in the literature (Hurst and Waxman, 2003; Maloney and Waxman, 1999; Lampen et al., 2003; Gopisetty Venkata et al., 2006; Feige et al., 2007; Lapinskas et al., 2005; Bility et al., 2004), with the full data provided in Supplementary material. The dotted line represents the division between metabolites originating from low and high molecular weight phthalates.

Some of these studies compared the efficacy of the phthalates to induce PPAR activation to that of known PPAR agonists like Rosiglitazone, Troglitazone and Wy14643. Regarding MEHP, two studies reported that the efficacy was lower than that of the PPAR agonists used as positive controls (Lapinskas et al., 2005[59]; Maloney and Waxman, 1999[69]). In contrast, a third study reported that MEHP could have a similar efficacy as the known PPAR agonists, but that the efficacy depended on PPAR isotype, species (human vs. mouse) and applied cell type (Feige et al., 2007[29]). Bility and co-authors compared the efficacy of a range of different phthalate metabolites to positive controls, and found that metabolites of the high molecular weight phthalates generally gave a similar or higher maximum fold increase as the positive control for PPARα and γ (Bility et al., 2004[12]). Thus, the efficacy of phthalate metabolites to activate PPARs may be lower than the efficacy of known PPAR agonists, but many of the high molecular weight phthalates appear to be able to act as full agonists. Combined exposure to phthalates and other PPAR agonists were rarely conducted (Gopisetty Venkata et al., 2006[32]).
Parent phthalates
Although the light molecular weight phthalates DnBP and BBzP have been reported to be able to bind and activate PPARs, several of the transfection studies have only included the metabolites of these phthalates rather than the parent phthalates (Gopisetty Venkata et al., 2006[32]; Hurst and Waxman, 2003[40]; Bility et al., 2004[12]). Both DnBP and BBzP caused a significant increase in luciferase activity for all three PPAR isotypes, but the efficacy was considerably lower than for MEHP (Lapinskas et al., 2005[59]). Accordingly, Lampen and co-authors also reported that BBzP could interact with all three PPAR isotypes, with EC2x values of 60, 125 and 175 µM for PPARα, γ and δ/β, respectively, where EC2x is defined as the concentration that induced a reporter gene response twice as high as the control (Lampen et al., 2003[58]). Since neither of these two papers provided a measure of lowest activation concentration or maximum fold increase, DnBP and BBzP are not included in Table 3(Tab. 3). Interestingly, Lapinskas et al. (2005[59]) also reported that both these low weight phthalates could bind to hPPARs, with lowest binding concentrations for DnBP and BBzP of 34 and 27 µM for PPARα and 10 and 10 µM for PPARγ, respectively. As a reference, the lowest binding concentrations for MEHP were 15 and 12 µM for PPARα and γ. Thus, DnBP and BBzP might bind and activate PPARs at relatively low concentrations, although their efficacy may be low compared to MEHP and other known PPAR agonists. Moreover, the parent phthalates appear to be more likely PPAR ligands than their primary metabolites for these low molecular weight phthalates.
Activation of endogeneous PPARs
Activation of endogenous PPARs in cellular models where PPAR is naturally present rather than transfected, has been studied for PPARα and γ in either in vitro or in vivo model systems, in relation to effects in liver and adipose tissue. Thus, the investigated cell types or organs are of limited relevance for the lung. For the liver, relatively high concentrations of both DEHP and DEP increased expression of lipid metabolizing enzymes in the liver of wild-type mice, while no response could be seen in PPARα-null mice (Lapinskas et al., 2005[59]). Moreover, activation of constitutive PPARα by phthalate monoesters was observed in a rat liver cell line as increased transcription of endogenous genes, but at slightly higher concentrations than in a luciferace transfection assay. However, no phthalate monoesters activated PPARα target genes in a human liver cell line (Bility et al., 2004[12]). With respect to adipose tissue, differentiation of 3T3-L1 fibroblasts into adipocytes has been found to depend on PPARγ, and this model is thus frequently used to investigate activation of constitutive PPARγ on endogenous genes (Bility et al., 2004[12]; Hurst and Waxman, 2003[40]; Feige et al., 2007[29]). MEHP promoted adipogenesis in a PPARγ dependent manner at concentrations between 10 and 100 µM in this model system, with a sensitivity and efficacy similar to that reported in various luciferase reporter systems (Feige et al., 2007[29]; Bility et al., 2004[12]).
Summary
Phthalates have been demonstrated to bind and activate PPARs, although the potency varies between the different phthalates, and the activation depends on the isoform, species and cell model used. Generally, the primary metabolites i.e. the phthalate monoesters are the most potent PPAR agonists. However, for some of the low molecular weight phthalates the parent compounds have been reported to be more potent than their primary metabolites. PPARα and γ seem to have a similar sensitivity to activation by phthalate monoesters, whereas PPARδ/β has a lower sensitivity to some monoesters. Using transfection assays, the difference between mouse and man is most evident for PPARα; and interestingly a similar difference in species sensitivity was also reported for endogenous model systems. However, activation of endogenous PPARγ by phthalates has not been tested in a model system of human origin, thus further research is necessary to elucidate this issue.
Inhibitory or Modulating Effects of Phthalates on PPAR Activit
As summarized in the previous chapter, a range of studies show that phthalates can bind and activate PPARs resulting in an agonistic effect. With respect to inhibitory effects, MnBP was reported to reduce the PPARα activation in a luciferase assay, suggesting an antagonistic effect of MnBP (Gopisetty Venkata et al., 2006[32]). Based on combined exposure to MnBP and known PPAR agonists, the same study suggested that MnBP is an antagonist for PPARγ and a weak antagonist for PPARδ/β (Gopisetty Venkata et al., 2006[32]). To our knowledge, no other studies report antagonistic effects of phthalates in PPAR transfection studies. However, MnBP, DnBP and MEHP were reported to have antagonistic effects with regard to co-variator recruitment by PPARγ, whereas only the DnBP metabolite DnBP-4OH had antagonistic effects on co-variator recruitment by PPARα (Kusu et al., 2008[57]). Another possibility for inhibitory effects of phthalates on PPARs is that phthalates could display an indirect inhibitory effect, by acting as partial agonists, i.e. by causing lower activity at saturating concentrations than the activity of a full agonist (Zhu, 2005[115]). Interestingly, for some phthalates and in some cell types, the efficacy of phthalate metabolites to activate PPARs appears to be lower than the efficacy of known PPAR agonists (see chapter PPAR trans-activation studies), indicating a sub-optimal activation of PPARs by phthalates. However, possible implications of partial agonist effects of phthalates on PPAR activation have not been extensively studied.
The concept of 'selective nuclear receptor modulators', i.e., that a modulating ligand can induce a specific conformational change of the receptor followed by recruitment of only a subset of co-regulators, emerged from studies on the tissue-specific modulation of the estrogen receptor (Shang and Brown, 2002[96]). Since the promoters of target genes have specific requirements to regulate transcriptional activation this will induce activation of only a selection of target genes (Shang and Brown, 2002[96]). For phthalates and PPARs, Feige and co-authors demonstrated that MEHP and the synthetic PPAR ligand Rosiglitazone activated different subsets of genes in adipocyte differentiation, a process known to depend on PPARγ (Feige et al., 2007[29]). The selective activity correlated with the recruitment of a specific subset of PPARγ co-regulators. The authors suggested that this weak dysregulation could cause small changes in regulatory pathways that are not easily detected and might not be evident as clinical effects/symptoms before after an extended period of time. However, the phthalate-induced gene activation has not yet been compared to the activation induced by endogenous PPAR ligands.
One study investigated if phthalates could influence the effects of the endogenous PPARγ ligand 15d-PGJ2 in B-cells, representing another possible modulatory mode of phthalates (Schlezinger et al., 2004[93]). MEHP induced an additive decrease in the proliferation and increase in the apoptosis induced by 15d-PGJ2 in B-cell lines at MEHP concentrations between 25 and 100 µM, whereas similar effects were observed at lower concentrations (10-15 µM) in primary B-cells. The 15d-PGJ2 levels applied by Schlezinger and co-authors were reported to be relevant for the levels in the bone marrow micro-environment, but not for the estimated pulmonary levels. Thus the study has limited relevance for the pulmonary situation except for demonstrating a possible modulatory mode of action for phthalates on PPAR activation.
Interestingly, MEHP induced activation of PPARα and γ has been found to cause inhibition of the transcription factor STAT5 at much lower concentrations than those required for a more “classic” direct PPAR activation (Shipley and Waxman, 2004[97]). For inhibition of STAT5, in kidney fibroblast cells, the EC50 value was as low as 1.1 µM, whereas the corresponding value for PPAR activation was 10 µM. STAT5 is involved in several immune processes, including proliferation, survival, and release of inflammatory mediators in mast cells, development and maintenance of T regulatory (Treg) cells, negative regulation of T helper 17 (Th17) cell differentiation, and possibly differentiation into M2 macrophages (Morales et al., 2010[72]; Wei et al., 2008[105]; Pullen et al., 2012[83]; Xiao et al., 2008[111]). Thus an inhibition of STAT5 due to phthalate-induced PPAR effects might also have relevance for the lung. Moreover, the crosstalk with other signalling pathways due to phthalate-induced PPAR activation reported by Shipley and Waxman (2004[97]) occurred at much lower concentrations than those required for direct PPAR activation, and represents a mode of action for phthalates that could be relevant for other cell types, and possibly also other signalling pathways.
Are the Pulmonary Phthalate Concentrations Sufficient to Activate Human PPARs?
Human PPARs seem to be activated only by a selection of phthalate metabolites (Table 3(Tab. 3)), and predominantly by the metabolites of the high molecular weight phthalates like DEHP, DiNP and DiDP. Generally, concentrations sufficient for PPAR activation in Table 3(Tab. 3) appear to be highest for the low weight phthalates, suggesting that a higher concentration of their metabolites is necessary for a “classic” PPAR activation. Although the inhalation exposure (Table 1a(Tab. 1) and 1b(Tab. 1)) and the estimated pulmonary phthalate concentrations (Table 2(Tab. 2)) appear to be higher for the low weight phthalates, it is not obvious that the concentrations of phthalates that are able to activate PPARs will be reached by inhalation exposure only.
In order to discuss whether the pulmonary phthalate concentrations due to inhalation exposure are likely to activate human PPARs, we will compare the lowest activation concentrations presented in Table 3(Tab. 3) to the maximal estimated pulmonary phthalate levels in Table 2(Tab. 2). Since the rate of phthalate metabolism in the lung is not known we look at two highly diverging scenarios (i) 1 % metabolite formation as suggested by Larsen et al (2007[60]) representative of low pulmonary metabolism and (ii) 50 % metabolite formation representative for a high pulmonary phthalate metabolism (Table 4(Tab. 4)). Comparison of the estimated metabolite levels and the lowest activation concentrations for PPARs, assuming 1 % metabolic rate, suggests that PPAR activation is not likely due to inhalation exposure. Assuming 50 % metabolic rate, activation of PPARα and γ may be possible for individuals highly exposed to DEHP, while PPARα activation may occur in individuals highly exposed to DnBP. Remember though that the maximal pulmonary metabolite levels in Table 4(Tab. 4) represent maximum exposure, maximum deposition, minimal absorption to the blood and maximal pulmonary metabolism.
Table 4. Comparison of maximal estimated metabolite levels and lowest activation concentrations. The table compares the lowest activation concentrations presented in Table 3 to the maximal estimated pulmonary phthalate levels in Table 2. Two different metabolic rates (met. rate) are included since the rate of phthalate metabolism in the lung is not known, see main text for detailed description.

Since the parent phthalates DnBP and BBzP have also been reported to bind and activate PPARs (Lampen et al., 2003[58]; Lapinskas et al., 2005[59]), a scenario involving a direct interaction between these phthalates and the three PPAR isotypes should also be considered. No measure of lowest activation concentration has been reported in the literature. However, the estimated pulmonary phthalate concentrations in the first column of Table 4(Tab. 4) may be compared to the lowest binding concentrations for DnBP and BBzP of 34 and 27 µM for hPPARα and 10 and 10 µM for hPPARγ, respectively (Lapinskas et al., 2005[59]). For DnBP, but not BBzP, the estimated pulmonary concentration exceeds the lowest binding concentrations, suggesting that a direct activation of PPARα and γ is more likely for DnBP than BBzP. Further studies are however necessary to study PPAR activation due to DnBP binding and determine the lowest activation concentration.
Although the metabolites of the high molecular weight phthalates generally appear to be more potent than the low molecular weight phthalates, the information regarding the levels of these phthalates in indoor air is scarce. We could only identify one study reporting data for DiNP, and based on these data the estimated pulmonary metabolite levels were 50 times lower than the lowest reported PPAR activation concentration, even when assuming 50 % metabolic rate. Thus, a direct activation of PPARs does not appear to be likely. Keep in mind though, that high weight phthalates like DiNP and DiDP are likely to partition to the particle phase rather than the gas phase (Schossler et al., 2011[95]), suggesting uneven pulmonary deposition and higher concentrations of particles and thus phthalates in some regions than others (see chapter The fate of phthalates in the lung).
PPARs in Asthma Development and Exacerbatio
Asthma is a heterogeneous and complex disease caused by multiple factors (Kim et al., 2010[49]), with genetic predispositions and environmental exposures in early life as the major identified risk factors for asthma development (Sly, 2011[99]). There are several phenotypes of asthma, including allergic, non-allergic and intrinsic asthma, and the cellular and molecular pathways involved in the development and pathogenesis of these different asthma phenotypes differs (Kim et al., 2010[49]). In allergic asthma, antigen presenting cells like dendritic cells induce antigen-specific responses in T helper type 2 (TH2) cells, resulting in IgE production and sensitisation of basophils and mast cells. Basophils, eosinophils, mast cells and natural killer cells also contribute to allergic asthma by antigen presentation or cytokine release. In non-allergic asthma, pathways independent of TH2 are activated. These involve other cell types like neutrophils, alveolar macrophages but also natural killer cells (Kim et al., 2010[49]). Airway epithelial cells also contribute to asthma development, for instance as producers of inflammatory and anti-inflammatory cytokines involved in development of both allergic and non-allergic asthma (Kim et al., 2010[49]; Wang et al., 2008[104]).
The three PPAR isotypes are expressed in many of the lung and immune cells believed to be involved in asthma development and exacerbation, including airway epithelium, smooth muscle cells, macrophages, T lymphocytes and eosinophils (Becker et al., 2006[8]; Di Paola and Cuzzocrea, 2007[26]). PPARs are involved in a variety of biological processes in these cell types. For instance, PPARα and γ ligands inhibit production of a number of inflammatory mediators and cytokines in several tissues and cell types including both lung cells and immune cells, with inhibition of transcriptional activity of inflammatory genes as a proposed mechanism (Di Paola and Cuzzocrea, 2007[26]; Standiford et al., 2005[100]; Becker et al., 2006[8]). Compared to PPARα and γ, relatively little is known about the role of PPARδ/β in the regulation of inflammatory responses (Zingarelli et al., 2010[116]), although some studies report that PPARδ/β agonists inhibit inflammatory responses both in vivo and in vitro (Schnegg and Robbins, 2011[94]; Cuzzocrea, 2006[23]).
In addition to these anti-inflammatory effects, PPARα and γ are involved in regulation of apoptosis, chemotaxis, proliferation and differentiation in lung and immune cells (Becker et al., 2006[8]). Moreover, PPARγ mediates an important role in surfactant homeostasis in alveolar type 2 cells and in surfactant catabolism in alveolar macrophages (Yang et al., 2008[113]; Baker et al., 2010[7]). A recent study reported that PPARγ, but not PPARα and δ/β, promoted monocyte differentiation towards alternatively activated macrophages (AAMs or M2) (Bouhlel et al., 2009[16]). A role of AAMs in chronic lung disease like asthma and COPD has been suggested, but it is still unclear whether these macrophages are the cause or the effect of these lung diseases (Byers and Holtzman, 2010[18]).
Thus, PPARs are mainly involved in anti-inflammatory and beneficial responses in the lung. Since asthma development involves various inflammatory processes, PPARs do not appear to play an obvious role in asthma development. Moreover PPAR agonists have been suggested as treatment of inflammatory lung diseases (Becker et al., 2006[8]; Di Paola and Cuzzocrea, 2007[26]; Cuzzocrea, 2006[23]), further supporting that PPAR activation appears to have a beneficial effect in the airways rather than contributing to development of pulmonary diseases like asthma.
Is PPAR Activation a Likely Mechanism for Pulmonary Effects of Phthalates?
As summarized in the introduction, present epidemiological data support an association between phthalate exposure and respiratory symptoms including asthma, while experimental studies report that phthalates may induce an inflammatory response in lung- and immune cells. Furthermore, recent studies have shown that phthalates may have a direct effect on airway epithelial cells and contribute to airway remodeling, which is the cardinal pathologic characteristic of chronic asthma, with a high correlation with disease severity (Tsai et al., 2012[101]).
PPARs have been identified as important targets for phthalates in the liver of some rodents and have also been suggested as a possible mechanism for phthalate-induced effects in the lungs (Magliozzi et al., 2003[67]; Rosicarelli and Stefanini, 2009[88]; Just et al., 2012[45]). The present literature review of inhalation exposure to phthalates and the phthalate-induced activation of PPARs, suggests that for highly exposed individuals, metabolites of DnBP and DEHP may be present in the lungs at sufficient concentrations for a direct “classical” activation of PPARs, but only when assuming a high extent of pulmonary metabolism (hydrolysis). The extent of phthalate metabolism in human lungs is presently unknown, but a rapid absorption to the blood stream as well as a low pulmonary metabolism has been suggested (Carlson, 2010[19]). Based on the limited data available for a direct activation of PPARs by DnBP (lowest binding concentrations, Lapinskas et al., 2005[59]), a direct activation PPARα and γ also appears to be possible since the estimated pulmonary phthalate concentrations were higher than the reported lowest binding concentrations. For the “new” high molecular weight phthalates like DiNP and DiDP, the inhalation exposure is not well characterised and it is therefore not possible to draw any conclusions regarding the likelihood of phthalate-induced activation of PPAR due to inhalation exposure. For the low molecular phthalates, other than DnBP, the pulmonary concentrations of metabolites or parent compound do not appear to be sufficient for a direct activation of PPARs. Overall, some phthalates may be inhaled in sufficient amounts to cause a direct “classical” activation of PPARs, however, when considering that PPARs mainly mediate anti-inflammatory effects, it seems unlikely that a direct activation of these nuclear receptors are involved in adverse pulmonary effects of phthalates.
In contrast to a direct activation of PPARs, other modes of action may occur at lower concentrations and could be more relevant for eventual pulmonary effects of phthalates through PPAR. These modes may include modulation of PPAR activity e.g. via inhibition due to partial agonist effects, cross talk between pathways and/or possibly more direct effects through other signalling pathways (see chapter Inhibitory or modulating effects of phthalates on PPAR activity). As activation of PPARs predominantly has anti-inflammatory effects, an inhibitory effect due to a partial agonist would interrupt the anti-inflammatory signalling and therefore possibly contribute to inflammatory effects. Compounds like phthalates, inducing lower PPAR activity than full agonists, could thus interfere with the anti-inflammatory and beneficial effects of endogenous PPAR activation and be a possible mechanism for pro-inflammatory effects of phthalates in the lung (see chapter Inhibitory or modulating effects of phthalates on PPAR activity). So far, the efficacy of phthalates has only been compared to that of the synthetic PPAR agonists, not to the efficacy of the endogenous PPAR ligands. Moreover, a partial agonist effect might be more likely to occur for the low- than the high-molecular weight phthalates since the former generally have a lower efficacy (Table 3(Tab. 3)). For the high molecular weight phthalates, that are more potent activators of PPARs, a more likely mode of action may be cross talk with other signalling pathways upon PPAR activation and/or dysregulation of gene transcription after phthalate-induced activation of PPARs causing a different transcriptional activation as compared to the endogenous PPAR ligands (Feige et al., 2007[29]; Desvergne et al., 2009[25]). Interestingly, polymorphisms of the PPARG gene may be linked to an increased risk for asthma development and airway hyper-reactivity (Lee et al., 2011[62]; Oh et al., 2009[76]; Palmer et al., 2007[78]); supporting that PPAR dysfunction could contribute to pulmonary effects. However, more studies are necessary to clarify if modulatory or inhibitory effects could contribute to phthalate-induced pulmonary effects including development and exacerbation of asthma.
Other possible mechanisms for Phthalate Induced Pulmonary Effects
Other exposure routes than inhalation may contribute to pulmonary effects. In line with this, a recent experimental study reported that ingestion of DEHP caused changes in inflammatory biomarkers in the lungs of mice, as well as in pulmonary histology and function, suggesting both adjuvant effect on airway allergy and a pulmonary inflammation (Guo et al., 2012[34]). The applied concentrations were within the maximal estimated exposure range for human DEHP intake (Wormuth et al., 2006[110]). Thus, ingestion may also be a relevant exposure route for induction of effects in the airways and the immune system. Likewise, several epidemiological studies report associations between urinary metabolite levels of phthalates and various pulmonary effects; some of these studies report associations for phthalates where ingestion is the major exposure route. Recent studies have indicated that dermal exposure may account for more than 50 % of the phthalate uptake from indoor air, thus this exposure route also deserves further investigations with regard to possible airway effects of phthalates (Weschler and Nazaroff, 2012[106]; Beko et al., 2013[9]).
The molecular mechanisms of phthalate toxicity are likely to vary depending not only on health endpoint and organ, but also on the species investigated. In support of this, a recent study of interactions between 16 phthalates and genes/proteins from the Comparative Toxicogenomics Database (CDT) identified that the interactions differed between rodents and humans (Singh and Li, 2011[98]). Moreover, when grouping the gene-protein interactions for DBP/BBP/ MBP and DEHP/MEHP; DEHP/MEHP had many interactions with PPARs, whereas DBP/BBP/MBP interacted more frequently with estrogen and androgen receptors. This implies that the effects of the individual phthalates are likely to be related to different molecular mechanisms, rather than being related to one common mechanism.
Several recent studies have addressed other mechanisms than PPAR activation with regard to phthalate induced effects. For instance, both DnBP and its metabolite MnBP were reported to bind directly to antioxidant superoxide dismutase (SOD) and thereby interfere with the free radical scavenging capacity of this enzyme, with DnBP as the most potent inhibitor (Prasanth et al., 2009[82]). Direct binding to cyclooxygenase enzymes (COX) has also been reported for DnBP and other low molecular weight phthalates, with inhibition of the prostaglandin pathway as a possible consequence. This could have implications for immunological disorders (Kristensen et al., 2011[56]). Moreover, Phthalate diesters were reported to interact with the two nuclear receptors Constitutive Androstane Receptor (CAR) and Pregnane X Receptor (PXR), that are known to be involved in the response to endobiotics and xenobiotics, including transcription and activation of detoxification enzymes (DeKeyser et al., 2011[24]; Eveillard et al., 2009[28]). DEHP and DiNP were however more potent activators of CAR than PXR. Interestingly, DEHP activated CAR2 at very low concentrations, down to 10 nM in transactivation experiments (DeKeyser et al., 2011[24]). The ligand-activated transcription factor Aryl hydrocarbon Receptor (AhR) has also been reported to be stimulated by parent phthalates (Mankidy et al., 2013[70]; Hsieh et al., 2012[38]). Although these enzymes and receptors represent possible molecular mechanisms for biological effects of phthalates, their involvement in phthalate effects in the lung and immune system has not been elucidated. Since the pulmonary metabolism of phthalates might be low, it is however interesting to note that the majority of these other molecular mechanisms appear to be activated by parent phthalates rather than their metabolites.
Conclusion
Current epidemiological and experimental knowledge suggests that phthalates could contribute to the development and exacerbation of asthma. If PPARs provide a mechanistic link between phthalate exposure and asthma, the most likely scenario would be through antagonistic actions or modulatory effects since a direct PPAR activation results in anti-inflammatory events. Other molecular mechanisms have been suggested for adverse effects of phthalates in other organs and cell types. These mechanisms may be equally relevant candidates in the development and exacerbation of asthma and airway hypersensitivity. Overall, further studies are still required to elucidate the mechanisms involved in phthalate-induced pulmonary effects. The role of other phthalate exposure routes such as ingestion and dermal exposure for airway related effects should also be addressed in future studies.
Acknowledgement
We thank J. Ovrevik for discussions on early versions of the manuscript. Financial support from the Norwegian research council (NFR) is gratefully acknowledged.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary Material
References
- 1.Abb M, Heinrich T, Sorkau E, Lorenz W. Phthalates in house dust. Environ Int. 2009;35:965–970. doi: 10.1016/j.envint.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Adibi JJ, Perera FP, Jedrychowski W, Camann DE, Barr D, Jacek R, et al. Prenatal exposures to phthalates among women in New York City and Krakow, Poland. Environ Health Perspect. 2003;111:1719–1722. doi: 10.1289/ehp.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adibi JJ, Whyatt RM, Williams PL, Calafat AM, Camann D, Herrick R, et al. Characterization of phthalate exposure among pregnant women assessed by repeat air and urine samples. Environ Health Perspect. 2008;116:467–473. doi: 10.1289/ehp.10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albro PW, Thomas RO. Enzymatic hydrolysis of di-(2-ethylhexyl) phthalate by lipases. Biochim Biophys Acta. 1973;306:380–390. doi: 10.1016/0005-2760(73)90176-8. [DOI] [PubMed] [Google Scholar]
- 5.Baena-Cagnani CE, Gomez RM, Baena-Cagnani R, Canonica GW. Impact of environmental tobacco smoke and active tobacco smoking on the development and outcomes of asthma and rhinitis. Curr Opin Allergy Clin Immunol. 2009;9:136–140. doi: 10.1097/ACI.0b013e3283294038. [DOI] [PubMed] [Google Scholar]
- 6.Bakand S, Hayes A. Troubleshooting methods for toxicity testing of airborne chemicals in vitro. J Pharmacol Toxicol Meth. 2010;61:76–85. doi: 10.1016/j.vascn.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Baker AD, Malur A, Barna BP, Kavuru MS, Malur AG, Thomassen MJ. PPARgamma regulates the expression of cholesterol metabolism genes in alveolar macrophages. Biochem Biophys Res Commun. 2010;393:682–687. doi: 10.1016/j.bbrc.2010.02.056. [DOI] [PubMed] [Google Scholar]
- 8.Becker J, Delayre-Orthez C, Frossard N, Pons F. Regulation of inflammation by PPARs: a future approach to treat lung inflammatory diseases? Fundam Clin Pharmacol. 2006;20:429–447. doi: 10.1111/j.1472-8206.2006.00425.x. [DOI] [PubMed] [Google Scholar]
- 9.Beko G, Weschler CJ, Langer S, Callesen M, Toftum J, Clausen G. Children's phthalate intakes and resultant cumulative exposures estimated from urine compared with estimates from dust ingestion, inhalation and dermal absorption in their homes and daycare centers. PLoS One. 2013;8:e62442. doi: 10.1371/journal.pone.0062442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergh C, Torgrip R, Emenius G, Ostman C. Organophosphate and phthalate esters in air and settled dust - a multi-location indoor study. Indoor Air. 2011;21:67–76. doi: 10.1111/j.1600-0668.2010.00684.x. [DOI] [PubMed] [Google Scholar]
- 11.Bertelsen RJ, Carlsen KC, Calafat AM, Hoppin JA, Haland G, Mowinckel P, et al. Urinary biomarkers for phthalates associated with asthma in Norwegian children. Environ Health Perspect. 2013;121:251–256. doi: 10.1289/ehp.1205256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bility MT, Thompson JT, McKee RH, David RM, Butala JH, Vanden Heuvel JP, et al. Activation of mouse and human peroxisome proliferator-activated receptors (PPARs) by phthalate monoesters. Toxicol Sci. 2004;82:170–182. doi: 10.1093/toxsci/kfh253. [DOI] [PubMed] [Google Scholar]
- 13.Bolling AK, Ovrevik J, Samuelsen JT, Holme JA, Rakkestad KE, Mathisen GH, et al. Mono-2-ethylhexylphthalate (MEHP) induces TNF-alpha release and macrophage differentiation through different signalling pathways in RAW264.7 cells. Toxicol Lett. 2012;209:43–50. doi: 10.1016/j.toxlet.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 14.Bornehag CG, Nanberg E. Phthalate exposure and asthma in children. Int J Androl. 2010;33:333–345. doi: 10.1111/j.1365-2605.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- 15.Bornehag CG, Sundell J, Weschler CJ, Sigsgaard T, Lundgren B, Hasselgren M, et al. The association between asthma and allergic symptoms in children and phthalates in house dust: a nested case-control study. Environ Health Perspect. 2004;112:1393–1397. doi: 10.1289/ehp.7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouhlel MA, Brozek J, Derudas B, Zawadzki C, Jude B, Staels B, et al. Unlike PPARgamma, PPARalpha or PPARbeta/ delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem Biophys Res Commun. 2009;386:459–462. doi: 10.1016/j.bbrc.2009.06.047. [DOI] [PubMed] [Google Scholar]
- 17.Bousquet J, Bousquet PJ, Godard P, Daures JP. The public health implications of asthma. Bull World Health Org. 2005;83:548–554. [PMC free article] [PubMed] [Google Scholar]
- 18.Byers DE, Holtzman MJ. Alternatively activated macrophages as cause or effect in airway disease. Am J Respir Cell Mol Biol. 2010;43:1–4. doi: 10.1165/rcmb.2009-0407ED. [DOI] [PubMed] [Google Scholar]
- 19.Carlson KR. Bethesda, MB, USA: United States Consumer Product Safety Commision; 2010. Toxicity review of Di/2-ethylhexyl) Phthalate (DEHP) [Google Scholar]
- 20.Carlstedt F, Jönsson BA, Bornehag CG. PVC flooring is related to human uptake of phthalates in infants. Indoor Air. 2013;23(1):32–39. doi: 10.1111/j.1600-0668.2012.00788.x. [DOI] [PubMed] [Google Scholar]
- 21.Choi K, Joo H, Campbell JL, Jr, Clewell RA, Andersen ME, Clewell HJ., III In vitro metabolism of di(2-ethylhexyl) phthalate (DEHP) by various tissues and cytochrome P450s of human and rat. Toxicol In Vitro. 2012;26:315–322. doi: 10.1016/j.tiv.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Coonrod JD, Karathanasis P, Lin R. Lipoprotein lipase: a source of free fatty acids in bronchoalveolar lining fluid. J Lab Clin Med. 1989;113:449–457. [PubMed] [Google Scholar]
- 23.Cuzzocrea S. Peroxisome proliferator-activated receptors and acute lung injury. Curr Opin Pharmacol. 2006;6:263–270. doi: 10.1016/j.coph.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 24.DeKeyser JG, Laurenzana EM, Peterson EC, Chen T, Omiecinski CJ. Selective phthalate activation of naturally occurring human constitutive androstane receptor splice variants and the pregnane X receptor. Toxicol Sci. 2011;120:381–391. doi: 10.1093/toxsci/kfq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desvergne B, Feige JN, Casals-Casas C. PPAR-mediated activity of phthalates: A link to the obesity epidemic? Mol Cell Endocrinol. 2009;304:43–48. doi: 10.1016/j.mce.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Di Paola R, Cuzzocrea S. Peroxisome proliferator-activated receptors and acute lung injury. PPAR Res. 2007;2007:63745. doi: 10.1155/2007/63745. Available from: http://dx.doi.org/10.1155/2007/63745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donaldson K, Borm PJ, Oberdorster G, Pinkerton KE, Stone V, Tran CL. Concordance between in vitro and in vivo dosimetry in the proinflammatory effects of low-toxicity, low-solubility particles: the key role of the proximal alveolar region. Inhal Toxicol. 2008;20:53–62. doi: 10.1080/08958370701758742. [DOI] [PubMed] [Google Scholar]
- 28.Eveillard A, Mselli-Lakhal L, Mogha A, Lasserre F, Polizzi A, Pascussi JM, et al. Di-(2-ethylhexyl)-phthalate (DEHP) activates the constitutive androstane receptor (CAR): a novel signalling pathway sensitive to phthalates. Biochem Pharmacol. 2009;77:1735–1746. doi: 10.1016/j.bcp.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 29.Feige JN, Gelman L, Rossi D, Zoete V, Metivier R, Tudor C, et al. The endocrine disruptor monoethyl-hexyl-phthalate is a selective peroxisome proliferator-activated receptor gamma modulator that promotes adipogenesis. J Biol Chem. 2007;282:19152–19166. doi: 10.1074/jbc.M702724200. [DOI] [PubMed] [Google Scholar]
- 30.Frederiksen H, Skakkebaek NE, Andersson AM. Metabolism of phthalates in humans. Mol Nutr Food Res. 2007;51:899–911. doi: 10.1002/mnfr.200600243. [DOI] [PubMed] [Google Scholar]
- 31.Fromme H, Lahrz T, Piloty M, Gebhart H, Oddoy A, Ruden H. Occurrence of phthalates and musk fragrances in indoor air and dust from apartments and kindergartens in Berlin (Germany) Indoor Air. 2004;14:188–195. doi: 10.1111/j.1600-0668.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- 32.Gopisetty Venkata N, Robinson JA, Cabot PJ, Davis B, Monteith GR, Roberts-Thomson SJ. Mono(2-ethylhexyl)phthalate and mono-n-butyl phthalate activation of peroxisome proliferator activated-receptors α and γ in breast. Toxicol Lett. 2006;163:224–234. doi: 10.1016/j.toxlet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Gross B, Staels B. PPAR agonists: multimodal drugs for the treatment of type-2 diabetes. Best Pract Res Clin Endocrinol Metabol. 2007;21:687–710. doi: 10.1016/j.beem.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 34.Guo J, Han B, Qin L, Li B, You H, Yang J, et al. Pulmonary toxicity and adjuvant effect of di-(2-exylhexyl) phthalate in ovalbumin-immunized BALB/c mice. PLoS One. 2012;7:e39008. doi: 10.1371/journal.pone.0039008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansen JS, Larsen ST, Poulsen LK, Nielsen GD. Adjuvant effects of inhaled mono-2-ethylhexyl phthalate in BALB/cJ mice. Toxicology. 2007;232:79–88. doi: 10.1016/j.tox.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 36.Heinrich J. Influence of indoor factors in dwellings on the development of childhood asthma. Int J Hyg Environ Health. 2011;214:1–25. doi: 10.1016/j.ijheh.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Hoppin JA, Ulmer R, London SJ. Phthalate exposure and pulmonary function. Environ Health Perspect. 2004;112:571–574. doi: 10.1289/ehp.6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsieh TH, Tsai CF, Hsu CY, Kuo PL, Lee JN, Chai CY, et al. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012;26:778–787. doi: 10.1096/fj.11-191742. [DOI] [PubMed] [Google Scholar]
- 39.Hsu NY, Lee CC, Wang JY, Li YC, Chang HW, Chen CY, et al. Predicted risk of childhood allergy, asthma, and reported symptoms using measured phthalate exposure in dust and urine. Indoor Air. 2012;22:186–199. doi: 10.1111/j.1600-0668.2011.00753.x. [DOI] [PubMed] [Google Scholar]
- 40.Hurst CH, Waxman DJ. Activation of PPARα and PPARγ by environmental phthalate monoesters. Toxicol Sci. 2003;74:297–308. doi: 10.1093/toxsci/kfg145. [DOI] [PubMed] [Google Scholar]
- 41.Ito Y, Yokota H, Wang R, Yamanoshita O, Ichihara G, Wang H, et al. Species differences in the metabolism of di(2-ethylhexyl) phthalate (DEHP) in several organs of mice, rats, and marmosets. Arch Toxicol. 2005;79:147–154. doi: 10.1007/s00204-004-0615-7. [DOI] [PubMed] [Google Scholar]
- 42.Jaakkola JJ, Knight TL. The role of exposure to phthalates from polyvinyl chloride products in the development of asthma and allergies: a systematic review and meta-analysis. Environ Health Perspect. 2008;116:845–883. doi: 10.1289/ehp.10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Julinova M, Slavik R. Removal of phthalates from aqueous solution by different adsorbents: a short review. J Environ Manage. 2012;94:13–24. doi: 10.1016/j.jenvman.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Just AC, Adibi JJ, Rundle AG, Calafat AM, Camann DE, Hauser R, et al. Urinary and air phthalate concentrations and self-reported use of personal care products among minority pregnant women in New York city. J Expo Sci Environ Epidemiol. 2010;20:625–633. doi: 10.1038/jes.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Just AC, Whyatt RM, Miller RL, Rundle AG, Chen Q, Calafat AM, et al. Children's urinary phthalate metabolites and fractional exhaled nitric oxide in an urban cohort. Am J Respir Crit Care Med. 2012;186:830–837. doi: 10.1164/rccm.201203-0398OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kambia N, Renault N, Dilly S, Farce A, Dine T, Gressier B, et al. Molecular modelling of phthalates - PPARs interactions. J Enzyme Inhib Med Chem. 2008;23:611–616. doi: 10.1080/14756360802205059. [DOI] [PubMed] [Google Scholar]
- 47.Kanazawa A, Saito I, Araki A, Takeda M, Ma M, Saijo Y, et al. Association between indoor exposure to semi-volatile organic compounds and building-related symptoms among the occupants of residential dwellings. Indoor Air. 2010;20:72–84. doi: 10.1111/j.1600-0668.2009.00629.x. [DOI] [PubMed] [Google Scholar]
- 48.Kaya T, Mohr SC, Waxman DJ, Vajda S. Computational screening of phthalate monoesters for binding to PPARgamma. Chem Res Toxicol. 2006;19:999–1009. doi: 10.1021/tx050301s. [DOI] [PubMed] [Google Scholar]
- 49.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol. 2010;11:577–584. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimber I, Dearman RJ. An assessment of the ability of phthalates to influence immune and allergic responses. Toxicology. 2010;271:73–82. doi: 10.1016/j.tox.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 51.Koch HM, Calafat AM. Human body burdens of chemicals used in plastic manufacture. Philos Trans R Soc Lond B Biol Sci. 2009;364:2063–2078. doi: 10.1098/rstb.2008.0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koch HM, Lorber M, Christensen KL, Palmke C, Koslitz S, Bruning T. Identifying sources of phthalate exposure with human biomonitoring: Results of a 48h fasting study with urine collection and personal activity patterns. Int J Hyg Environ Health. 2013 doi: 10.1016/j.ijheh.2012,12.002.. Available from: http://dx.doi.org/10.1016/j.ijheh.2012,12.002. [DOI] [PubMed] [Google Scholar]
- 53.Koch HM, Preuss R, Angerer J. Di(2-ethylhexyl)phthalate (DEHP): human metabolism and internal exposure- an update and latest results. Int J Androl. 2006;29:155–165. doi: 10.1111/j.1365-2605.2005.00607.x. [DOI] [PubMed] [Google Scholar]
- 54.Kolarik B, Naydenov K, Larsson M, Bornehag CG, Sundell J. The association between phthalates in dust and allergic diseases among Bulgarian children. Environ Health Perspect. 2008;116:98–103. doi: 10.1289/ehp.10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kreyling W, Moller W, Semmler-Behnke M, Oberdörster G. Particle dosimetry: deposition and clearance from the respiratory tract and translocation towards extrapulmonary sites. In: Donaldson K, Borm P, editors. Particle toxicology. Boca Raton, FL: CRC Press; 2007. pp. 47–74. [Google Scholar]
- 56.Kristensen DM, Skalkam ML, Audouze K, Lesne L, Desdoits-Lethimonier C, Frederiksen H, et al. Many putative endocrine disruptors inhibit prostaglandin synthesis. Environ Health Perspect. 2011;119:534–551. doi: 10.1289/ehp.1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kusu R, Oishi A, Kakizawa K, Kimura T, Toda C, Hashizume K, et al. Effects of phthalate ester derivatives including oxidized metabolites on coactivator recruiting by PPARα and PPARγ. Toxicol in Vitro. 2008;22:1534–1538. doi: 10.1016/j.tiv.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 58.Lampen A, Zimnik S, Nau H. Teratogenic phthalate esters and metabolites activate the nuclear receptors PPARs and induce differentiation of F9 cells. Toxicol Appl Pharmacol. 2003;188:14–23. doi: 10.1016/s0041-008x(03)00014-0. [DOI] [PubMed] [Google Scholar]
- 59.Lapinskas PJ, Brown S, Leesnitzer LM, Blanchard S, Swanson C, Cattley RC, et al. Role of PPARα in mediating the effects of phthalates and metabolites in the liver. Toxicology. 2005;207:149–163. doi: 10.1016/j.tox.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 60.Larsen ST, Hansen JS, Hansen EW, Clausen PA, Nielsen GD. Airway inflammation and adjuvant effect after repeated airborne exposures to di-(2-ethylhexyl) phthalate and ovalbumin in BALB/c mice. Toxicology. 2007;235:119–129. doi: 10.1016/j.tox.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 61.Larsen ST, Nielsen GD. The adjuvant effect of di-(2-ethylhexyl) phthalate is mediated through a PPARalpha-independent mechanism. Toxicol Lett. 2007;170:223–228. doi: 10.1016/j.toxlet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 62.Lee SH, Jang AS, Woo PS, Park JS, Kim YK, Uh ST, et al. Genetic effect of single-nucleotide polymorphisms in the PPARGC1B gene on airway hyperreactivity in asthmatic patients. Clin Exp Allergy. 2011;41:1533–1544. doi: 10.1111/j.1365-2222.2011.03801.x. [DOI] [PubMed] [Google Scholar]
- 63.Leech JA, Nelson WC, Burnett RT, Aaron S, Raizenne ME. It's about time: a comparison of Canadian and American time-activity patterns. J Expo Anal Environ Epidemiol. 2002;12:427–432. doi: 10.1038/sj.jea.7500244. [DOI] [PubMed] [Google Scholar]
- 64.Lewis J. Surfactant in lung injury: How important is it? J Organ Dysfunc. 2006;2:20–23. [Google Scholar]
- 65.Löndahl J, Massling A, Pagels J, Swietlicki E, Vaclavik E, Loft S. Size-resolved respiratory-tract deposition of fine and ultrafine hydrophobic and hygroscopic aerosol particles during rest and exercise. Inhal Toxicol. 2007;19:109–116. doi: 10.1080/08958370601051677. [DOI] [PubMed] [Google Scholar]
- 66.Lotvall J, Ekerljung L, Ronmark EP, Wennergren G, Linden A, Ronmark E, et al. West Sweden Asthma Study: prevalence trends over the last 18 years argues no recent increase in asthma. Respir Res. 2009;10:94. doi: 10.1186/1465-9921-10-94. Available from: http://dx.doi.org/10.1186/1465-9921-10-94,94-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magliozzi R, Nardacci R, Scarsella G, Di Carlo V, Stefanini S. Effects of the plasticiser DEHP on lung of newborn rats: catalase immunocytochemistry and morphometric analysis. Histochem Cell Biol. 2003;120:41–49. doi: 10.1007/s00418-003-0543-2. [DOI] [PubMed] [Google Scholar]
- 68.Mahoney EM, Khoo JC, Steinberg D. Lipoprotein lipase secretion by human monocytes and rabbit alveolar macrophages in culture. Proc Natl Acad Sci USA. 1982;79:1639–1642. doi: 10.1073/pnas.79.5.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maloney EK, Waxman DJ. Trans-activation of PPARα and PPARγ by structurally diverse environmental chemicals. Toxicol Appl Pharmacol. 1999;161:209–218. doi: 10.1006/taap.1999.8809. [DOI] [PubMed] [Google Scholar]
- 70.Mankidy R, Wiseman S, Ma H, Giesy JP. Biological impact of phthalates. Toxicol Lett. 2013;217:50–58. doi: 10.1016/j.toxlet.2012.11.025. [DOI] [PubMed] [Google Scholar]
- 71.Mendell MJ, Mirer AG, Cheung K, Tong M, Douwes J. Respiratory and allergic health effects of dampness, mold, and dampness-related agents: a review of the epidemiologic evidence. Environ Health Perspect. 2011;119:748–756. doi: 10.1289/ehp.1002410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morales JK, Falanga YT, Depcrynski A, Fernando J, Ryan JJ. Mast cell homeostasis and the JAK-STAT pathway. Genes Immun. 2010;11:599–608. doi: 10.1038/gene.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nakagawa T, Kurita N, Kozakai S, Iwabuchi S, Yamaguchi Y, Hayakawa M, et al. Molecular mechanics and molecular orbital simulations on specific interactions between peroxisome proliferator-activated receptor PPARα and plasticizer. J Molec Graphics Modell. 2008;27:45–58. doi: 10.1016/j.jmgm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 74.Ng AW, Bidani A, Heming TA. Innate host defense of the lung: effects of lung-lining fluid pH. Lung. 2004;182:297–317. doi: 10.1007/s00408-004-2511-6. [DOI] [PubMed] [Google Scholar]
- 75.Nielsen GD, Larsen ST, Olsen O, Lovik M, Poulsen LK, Glue C, et al. Do indoor chemicals promote development of airway allergy? Indoor Air. 2007;17:236–255. doi: 10.1111/j.1600-0668.2006.00468.x. [DOI] [PubMed] [Google Scholar]
- 76.Oh SH, Park SM, Lee YH, Cha JY, Lee JY, Shin EK, et al. Association of peroxisome proliferator-activated receptor-gamma gene polymorphisms with the development of asthma. Respir Med. 2009;103:1020–1024. doi: 10.1016/j.rmed.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 77.Otake T, Yoshinaga J, Yanagisawa Y. Exposure to phthalate esters from indoor environment. J Expo Anal Environ Epidemiol. 2004;14:524–528. doi: 10.1038/sj.jea.7500352. [DOI] [PubMed] [Google Scholar]
- 78.Palmer CN, Doney AS, Ismail T, Lee SP, Murrie I, Macgregor DF, et al. PPARG locus haplotype variation and exacerbations in asthma. Clin Pharmacol Ther. 2007;81:713–718. doi: 10.1038/sj.clpt.6100119. [DOI] [PubMed] [Google Scholar]
- 79.Pearce N, Douwes J. The global epidemiology of asthma in children. Int J Tuberc Lung Dis. 2006;10:125–132. [PubMed] [Google Scholar]
- 80.Phalen RF. The fates of inhaled particles. In: Phalen RF, editor. The particulate air pollution controversy - A case study and lessons learned. Boston, MA: Kluwer Academic Publishers; 2006. pp. 55–68. [Google Scholar]
- 81.Pinkerton KE, Zhou YM, Teague SV, Peake JL, Walther RC, Kennedy IM, et al. Reduced lung cell proliferation following short-term exposure to ultrafine soot and iron particles in neonatal rats: key to impaired lung growth? Inhal Toxicol. 2004;16(Suppl 1):73–81. doi: 10.1080/08958370490443123. [DOI] [PubMed] [Google Scholar]
- 82.Prasanth GK, Divya LM, Sadasivan C. Effects of mono and di(n-butyl) phthalate on superoxide dismutase. Toxicology. 2009;262:38–42. doi: 10.1016/j.tox.2009.04.036. [DOI] [PubMed] [Google Scholar]
- 83.Pullen NA, Falanga YT, Morales JK, Ryan JJ. The Fyn-STAT5 pathway: A new frontier in IgE- and IgG-mediated mast cell signaling. Front Immunol. 2012;3:117. doi: 10.3389/fimmu.2012.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rakkestad KE, Dye CJ, Yttri KE, Holme JA, Hongslo JK, Schwarze PE, et al. Phthalate levels in Norwegian indoor air related to particle size fraction. J Environ Monit. 2007;9:1419–1425. doi: 10.1039/b709947a. [DOI] [PubMed] [Google Scholar]
- 85.Rakkestad KE, Holme JA, Paulsen RE, Schwarze PE, Becher R. Mono(2-ethylhexyl)phthalate induces both pro- and anti-inflammatory responses in rat alveolar macrophages through crosstalk between p38, the lipoxygenase pathway and PPARα. Inhal Toxicol. 2010;22:140–150. doi: 10.3109/08958370903019885. [DOI] [PubMed] [Google Scholar]
- 86.Rehan VK, Asotra K, Torday JS. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung. 2009;187:281–289. doi: 10.1007/s00408-009-9158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rosicarelli B, Stefanini S. DEHP effects on histology and cell proliferation in lung of newborn rats. Histochem Cell Biol. 2009;131:491–500. doi: 10.1007/s00418-008-0550-4. [DOI] [PubMed] [Google Scholar]
- 89.Rudel RA, Camann DE, Spengler JD, Korn LR, Brody JG. Phthalates, alkylphenols, pesticides, polybrominated diphenyl ethers, and other endocrine-disrupting compounds in indoor air and dust. Environ Sci Technol. 2003;37:4543–4553. doi: 10.1021/es0264596. [DOI] [PubMed] [Google Scholar]
- 90.Rudel RA, Perovich LJ. Endocrine disrupting chemicals in indoor and outdoor air. Atmos Environ. 2009;43:170–181. doi: 10.1016/j.atmosenv.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev Toxicol. 2006;36:459–479. doi: 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saldiva PH, Clarke RW, Coull BA, Stearns RC, Lawrence J, Murthy GG, et al. Lung inflammation induced by concentrated ambient air particles is related to particle composition. Am J Respir Crit Care Med. 2002;165:1610–1617. doi: 10.1164/rccm.2106102. [DOI] [PubMed] [Google Scholar]
- 93.Schlezinger JJ, Howard GJ, Hurst CH, Emberley JK, Waxman DJ, Webster T, et al. Environmental and endogenous peroxisome proliferator-activated receptor gamma agonists induce bone marrow B cell growth arrest and apoptosis: interactions between mono(2-ethylhexyl)phthalate, 9-cis-retinoic acid, and 15-deoxy-Delta12,14-prostaglandin J2. J Immunol. 2004;173:3165–3177. doi: 10.4049/jimmunol.173.5.3165. [DOI] [PubMed] [Google Scholar]
- 94.Schnegg CI, Robbins ME. Neuroprotective mechanisms of PPARdelta: Modulation of oxidative stress and inflammatory processes. PPAR Res. 2011:373560. doi: 10.1155/2011/373560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schossler P, Schripp T, Salthammer T, Bahadir M. Beyond phthalates: gas phase concentrations and modeled gas/particle distribution of modern plasticizers. Sci Total Environ. 2011;409:4031–4038. doi: 10.1016/j.scitotenv.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 96.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 97.Shipley JM, Waxman DJ. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicol Appl Pharmacol. 2004;199:275–284. doi: 10.1016/j.taap.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 98.Singh S, Li SS. Phthalates: toxicogenomics and inferred human diseases. Genomics. 2011;97:148–157. doi: 10.1016/j.ygeno.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 99.Sly PD. The early origins of asthma: who is really at risk? Curr Opin Allergy Clin Immunol. 2011;11:24–28. doi: 10.1097/ACI.0b013e328342309d. [DOI] [PubMed] [Google Scholar]
- 100.Standiford TJ, Keshamouni VG, Reddy RC. Peroxisome proliferator-activated receptor-{gamma} as a regulator of lung inflammation and repair. Proc Am Thorac Soc. 2005;2:226–231. doi: 10.1513/pats.200501-010AC. [DOI] [PubMed] [Google Scholar]
- 101.Tsai MJ, Kuo PL, Ko YC. The association between phthalate exposure and asthma. Kaohsiung. J Med Sci. 2012;28:S28–S36. doi: 10.1016/j.kjms.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 102.Walters DV. Lung lining liquid - the hidden depths. The 5th Nils W. Svenningsen memorial lecture. Biol Neonate. 2002;81(Suppl 1):2–5. doi: 10.1159/000056764. [DOI] [PubMed] [Google Scholar]
- 103.Wang F, Yao J, Sun K, Xing B. Adsorption of dialkyl phthalate esters on carbon nanotubes. Environ Sci Technol. 2010;44:6985–6991. doi: 10.1021/es101326j. [DOI] [PubMed] [Google Scholar]
- 104.Wang Y, Bai C, Li K, Adler KB, Wang X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir Med. 2008;102:949–955. doi: 10.1016/j.rmed.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 105.Wei L, Laurence A, O'Shea JJ. New insights into the roles of Stat5a/b and Stat3 in T cell development and differentiation. Semin. Cell Dev Biol. 2008;19:394–400. doi: 10.1016/j.semcdb.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Weschler CJ, Nazaroff WW. SVOC exposure indoors: fresh look at dermal pathways. Indoor Air. 2012;22:356–377. doi: 10.1111/j.1600-0668.2012.00772.x. [DOI] [PubMed] [Google Scholar]
- 107.Weschler CJ, Nazaroff WW. SVOC partitioning between the gas phase and settled dust indoors. Atmos Environ. 2010;44:3609–3620. [Google Scholar]
- 108.Weschler CJ, Salthammer T, Fromme H. Partitioning of phthalates among the gas phase, airborne particles and settled dust in indoor environments. Atmos Environ. 2008;42:1449–1460. [Google Scholar]
- 109.Wittassek M, Koch HM, Angerer J, Bruning T. Assessing exposure to phthalates - The human biomonitoring approach. Mol Nutr Food Res. 2011;55:7–31. doi: 10.1002/mnfr.201000121. [DOI] [PubMed] [Google Scholar]
- 110.Wormuth M, Scheringer M, Vollenweider M, Hungerbuhler K. What are the sources of exposure to eight frequently used phthalic acid esters in Europeans? Risk Anal. 2006;26:803–824. doi: 10.1111/j.1539-6924.2006.00770.x. [DOI] [PubMed] [Google Scholar]
- 111.Xiao W, Hong H, Kawakami Y, Lowell CA, Kawakami T. Regulation of myeloproliferation and M2 macrophage programming in mice by Lyn/Hck, SHIP, and Stat5. J Clin Invest. 2008;118:924–934. doi: 10.1172/JCI34013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu XR, Li XY. Adsorption behaviour of dibutyl phthalate on marine sediments. Mar Pollut Bull. 2008;57:403–408. doi: 10.1016/j.marpolbul.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 113.Yang Y, Gocke AR, Lovett-Racke A, Drew PD, Racke MK. PPAR Alpha regulation of the immune response and autoimmune encephalomyelitis. PPAR Res. 2008:546753. doi: 10.1155/2008/546753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yessoufou A, Wahli W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Med Wkly. 2010;140:w13071. doi: 10.4414/smw.2010.13071. [DOI] [PubMed] [Google Scholar]
- 115.Zhu BT. Mechanistic explanation for the unique pharmacologic properties of receptor partial agonists. Biomed Pharmacother. 2005;59:76–89. doi: 10.1016/j.biopha.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 116.Zingarelli B, Piraino G, Hake PW, O'Connor M, Denenberg A, Fan H, et al. Peroxisome proliferator-activated receptor δ regulates inflammation via NF-κB signaling in polymicrobial sepsis. Am J Pathol. 2010;177:1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.