

Abstract
Parkinson’s disease, like many other common age-related conditions, is now recognized to have a substantial genetic component. Here, I will discuss how mutations in a large complex gene, LRRK2, affect protein function and review recent evidence that LRRK2 mutations affect pathways that involve other proteins that have been implicated in Parkinson’s disease, specifically α-synuclein and tau. These concepts can be used to understand disease processes and to develop therapeutic opportunities for Parkinson’s disease.
Fifteen years ago, Parkinson’s disease, an age-related neurodegenerative disease affecting 1.5% of the population older than 65 years 1, was considered to have little or no genetic component. However, it has since been recognized that rare mutations are pathogenic for Parkinson’s disease in a number of families2, and genome-wide association studies published in late 2009 have shown that genetic variants have a less deterministic but nevertheless detectable role in sporadic Parkinson’s disease as well3, 4. Furthermore, mutations in the same genes can be involved both in familial Parkinson’s disease and as risk factors for sporadic Parkinson’s disease (Box 1), supporting the idea that inherited and sporadic Parkinson’s disease can have common pathological mechanisms.
Box 1. : Dominant genes for familial Parkinson’s disease and risk factors for sporadic Parkinson’s disease.
There are several genes associated with different forms of parkinsonism 2. The test for establishing pathogenicity of gene variants is that they should either show segregation with disease in rare families with inherited Parkinson’s disease or that they show association with sporadic Parkinson’s disease across a population.
The SNCA gene codes for the small presynaptic protein α-synuclein. Dominant mutations in SNCA, including multiplication mutations, are found in rare families with inherited Parkinson’s disease 2. The protein is also aggregated in Lewy bodies in patients with sporadic Parkinson’s disease (see Box 2) and common variants around the SNCA locus increase the risk of sporadic Parkinson’s disease in the general population 3, 4.
Mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) are pathogenic for a dominant disease 5, 6 that, clinically, strongly resembles idiopathic Parkinson’s disease 62. In addition, they are usually accompanied by α-synuclein-positive Lewy bodies. LRRK2 variants can also act as risk factors for sporadic Parkinson’s disease in some populations 3, 4.
Mutations in the gene encoding microtubule associated protein tau (MAPT) segregate with frontotemporal dementia with parkinsonism 57, and common variants in the region on chromosome 17 that harbours the MAPT gene increase the risk for sporadic Parkinson’s disease 4.
This Progress article focuses on recent studies into the gene encoding Leucine rich repeat kinase 2 (LRRK2) 5, 6, as mutations in this gene represent one of the stronger risk factors for the development of Parkinson’s disease (Box 2). Based on recent findings that suggest how mutations in LRRK2 can affect protein function 7–12, including that of newly proposed interactors of LRRK2 13, 14, together with very recently reported animal models in which LRRK2 has been overexpressed or knocked out 15–20, I will discuss how mutant LRRK2 might predispose to Parkinson’s disease, acknowledging that we still have much to learn about some aspects of LRRK2, including its effects on cellular signalling pathways. Importantly, as LRRK2-associated cases of Parkinson’s disease generally have similar neuropathology to patients with sporadic Parkinson’s disease (Box 3), I will also develop some ideas that may indicate that LRRK2, in conjunction with other Parkinson’s related genes, might be relevant to the more common sporadic form of the disease.
Box 2. Box 2: LRRK2 genetics.
In 2002, a gene defect localized to chromosome 12 was reported in a family from Japan with autosomal dominant inherited Parkinson’s disease63, and the locus was designated PARK8. Within two years after this report, several other families around the world with dominantly inherited Parkinson’s disease were identified as having mutations in the same genetic region, which mapped to the gene encoding leucine-rich repeat kinase 2 (LRRK2)5, 6. The original Japanese family was subsequently found to have an I2020T mutation in LRRK264.
Subsequently, one mutation, G2019S, was found relatively frequently in a number of different patients and families 65–70. Individuals who are heterozygous for mutant LRRK2 have a similar risk of disease and progression of disease as those who are homozygous for the mutation 71. This shows that LRRK2 mutations have a true dominant effect.
Parkinson’s disease is an age-dependent disorder and the proportion of people with a single dominant mutation in LRRK2 who show symptoms of Parkinson’s disease increases with age. The overall penetrance of the G2019S mutation is high but incomplete, meaning that there are some carriers of mutations who do not develop Parkinson’s disease in their lifetime. Some mutations have a much lower penetrance than G2019S, including risk factor variants found in Asian populations 72.
Box 3. The neuropathology of Parkinson’s disease.
The neuropathology of Parkinson’s disease has two main components:
The first is neuronal cell loss. This occurs in diverse cell populations throughout the brain, but is particularly prominent in dopaminergic neurons of the substantia nigra pars compacta in the midbrain. It is thought that neuronal cell death begins several years before the onset of clinical symptoms and it progresses throughout the course of the disease 73.
The second is formation of Lewy bodies — conglomerates of lipids and proteins — in the surviving neurons 74. The main protein component of Lewy bodies is the small synaptic protein α-synuclein. This protein normally binds to curved lipid membranes, where it adopts a helical structure, but in some circumstances can aggregate with itself in structures that are rich in α-sheet conformations. Small oligomeric accumulations coalesce into larger fibrils that are insoluble and are the building blocks of Lewy bodies; whether the Lewy body is neurotoxic or represents an attempted protective response is currently debated 53.
Mutations in LRRK2 occur in functional domains
LRRK2 is widely expressed in many organs and tissues 5, 6 including the brain although, interestingly, expression is not particularly high in dopamine-producing cells of the substantia nigra, which are heavily lost in Parkinson’s disease (Box 3) 21. LRRK2 encodes a large multidomain protein that includes a central catalytic tridomain with GTPase and kinase activities surrounded by a series of potential protein–protein interaction domains (Fig. 1).
Figure 1. Domains and mutations of LRRK2.

LRRK2 is drawn schematically here as a dimer, in a likely head-to-head orientation and in a linear scheme for clarity. The domains of the protein and some of the proposed intramolecular interactions are listed above the diagram. LRRK2 is a large multidomain protein, with several potential protein–protein interaction regions surrounding a central catalytic core. This core region contains a GTP-binding Ras Of Complex protein domain (ROC), a C-terminal of ROC (COR) domain and a kinase domain. The most clearly defined pathogenic human mutations are shown in red below the diagram. Both R1441 and Y1699 mutations decrease the modest GTPase activity of LRRK2, while G2019S increases kinase activity. These two activities may be related as, for example, recent studies have indicated that the kinase domain (blue) autophosphorylates the ROC domain at several sites, although this has yet to be confirmed in vivo. Using current methods of measurement, I2020T seems to have very modest effects on kinase activity, so the relationship of that mutation to biochemical activities is unclear. Outside of the catalytic regions are several domains including the Leucine-rich repeats (LRR) and WD40 domains that are thought to provide protein-protein interaction regions, although specific binding partners for these regions are only recently starting to be identified (see text).
The various dominant mutations in LRRK2 result in changes that are concentrated in the central region of the protein. This region includes the Ras Of Complex (ROC)–GTPase protein domain and the kinase domain, as well as the C-terminal of ROC (COR) sequence that links them. Thus, the ROC domain contains one residue that can have multiple mutations (R1441C/G/H), the COR domain can have one mutation (Y1699C) and the kinase domain has two adjacent residues that can be mutated (G2019S and I2020T). By contrast, mutations outside of the enzymatic domains have not been shown to segregate in a Mendelian fashion with the disease. This implies that the enzymatic activities of LRRK2, rather than other functions of LRRK2, might be important in pathogenesis. In vitro studies have shown that mutations in the ROC–GTPase and COR domains decrease GTPase activity22, 23. It should be noted, however, that purified full-length LRRK2 has only a weak GTPase activity, suggesting that if it is active in the cell it may require accessory proteins. By contrast, the G2019S mutation in the kinase domain increases kinase activity 24–27. Thus, these mutations affect the function of the domains in which they are found. The risk factor variants G2385R in the WD40 domain and R1628P in the COR domain have no measureable effect on kinase activity 13. The I2020T mutation in the kinase domain stimulates phosphorylation of 4E binding protein (4EBP) in some models 28 but has no effect on kinase activity in other assays 26. Therefore, there may be some interesting mechanistic differences between different mutations in the same domain, with the caveat that these observations could also be due to methodological differences between assays 29. Overall, these data support the idea that the kinase and GTPase activities of LRRK2 are important in pathogenesis.
On the basis of the similar phenotypes associated with LRRK2 mutations in humans, the most parsimonious explanation of why pathogenic mutations predispose to disease might be that they all evoke similar changes in downstream signalling events that are relevant for Parkinson’s disease, even if the detailed molecular mechanisms at the level of the protein differ. LRRK2 is dimeric7, 10, 11, 30, 31, suggesting that it may regulate its own activity, as well as perhaps having a signalling role by regulating other proteins in the cell. It was initially thought that GTP binding to the ROC domain regulated the protein’s kinase activity, but experiments using wild-type LRRK2 isolated from mouse brain suggest that this is not the case32. In fact, several groups have shown that the kinase domain of wild-type LRRK2 phosphorylates several sequences within the GTP-binding ROC domain7–9, 12. Therefore, the kinase domain may regulate overall LRRK2 function.
This self-regulation provides a simple suggestion as to how different mutations increase the risk of Parkinson’s disease. A speculative possibility is that the GTP-bound form of LRRK2 is the active version in cellular signalling pathways, perhaps by virtue of increasing affinity to an as yet unidentified binding partner. Mutations that either prompt the protein to enter a GTP-bound state — for example, mutations that alter kinase regulation of GTP loading — or that slow the protein’s return to the GDP-bound state — for example, mutations in the ROC and COR domains — would then result in persistent LRRK2 activity. I would therefore propose that both the kinase and the GTPase activities of LRRK2 are affected by mutations and therefore likely to be pathogenically important. In support of this idea, in vitro 24, 33 and in vivo 18 data suggest that kinase-dead or GTP-binding-deficient LRRK2 variants are less toxic than their wild-type counterparts.
It is also possible that different LRRK2 mutations fundamentally affect different physiological pathways that all happen to lead to Parkinson’s disease as a phenotype. It is certainly true that there is probably more than one way to kill dopamine neurons and that there are therefore multiple pathways to parkinsonism. Indeed, some recent data in model organisms suggests that only some mutations affect specific pathways, as discussed below. But for LRRK2, this is probably unlikely because the disease phenotypes in humans are more variable between individuals with the same mutation than between different mutations 34. Furthermore, if the scheme proposed above — that kinase regulates GTPase activity, leading to altered GTP-dependent function — is correct, then there would be a very simple explanation that would not require invoking multiple pathways. However, this does invoke an as yet unidentified GTP-dependent LRRK2 binding partner, and so it is difficult to evaluate this hypothesis at this time.
Along these lines, it is interesting that there are relatively few proven pathogenic mutations outside of the ROC-COR kinase tridomain of LRRK2. As shown in figure 1, much of the LRRK2 protein consists of regions that might have a role in protein–protein interactions. As such, LRRK2 may have a scaffolding role that could be important in cellular signalling. LRRK2 has been proposed to physically interact with assembled microtubules 35, 36, disheveled proteins 37, the co-chaperone CHIP (carboxy terminus of Hsp70-interacting protein) 38, 39 and itself 7, 10, 11, 30, 31 entirely or in part through its ROC domain. Mitogen-activated protein kinase (MAPK) kinase 6 (MKK6) binds to the COR/kinase region 40, which also mediates intramolecular interactions. LRRK2 is a phosphoprotein and, as such is bound by 14-3-3 proteins at a site that lies slightly N-terminal of the leucine-rich repeats 13, 26. Some of these interactions suggest alternate explanations for how multiple mutations affect the same protein. For example, both ROC and COR mutations decrease binding to 14-3-3 proteins. However, G2019S in the kinase domain does not have this effect26, which must mean that G2019S acts through a different mechanism from other mutations.
Although there are other purported LRRK2 interactors (eg Argonaute, discussed below), what is surprising about this list is that it contains no clearly identified effector proteins that bind LRRK2 outside of its catalytic core. This does not mean that the other regions of LRRK2 are unimportant. For example, the WD40 domain has been suggested to be important for the toxic effects of LRRK2 in vitro 41. However, this finding might also be explained by an effect on kinase activity rather than on a specific protein interactor, as deletion of the WD40 domain or even shorter C-terminal sequences renders LRRK2 kinase inactive 25, 41. What it does mean is that it is difficult to investigate the putative scaffolding role of LRRK2 without either strong mutations affecting this role or a nominated scaffold protein. This potentially important future direction for the field is discussed below.
LRRK2 in CNS physiology and pathology
In terms of normal function, there is evidence that wild-type LRRK2 can regulate neurite outgrowth in developing neurons, as knockout of endogenous LRRK2 in mouse neurons results in elongation of neuronal processes 42–44. The mechanism for this effect might involve interaction with either the disheveled proteins 37 or tubulins36. The LRRK homologue in Caenorhabditis elegans may also have a role in the specification of axons and dendrites45, 46, although the molecular details of this process remain undefined. It should also be noted that vertebrates have two homologues of LRRK (LRRK1 and LRRK2), so LRRK functions may differ across species. Therefore, despite the identification of functional regions of LRRK2, the physiological functions of the protein in the nervous system are unclear at this time.
Effects on protein translation
One way to understand the aberrant function of LRRK2 would be to identify its interactions with other proteins. Results from one study in Drosophila suggested that LRRK2 can interact with the microRNA (miR) processing protein Argonaute and thereby affect protein translation 14. In this study, only kinase domain mutations altered let-7 miR production and protein translation, whereas the one non-kinase mutation tested in this study (R1441G) did not. This suggests either that different LRRK2 mutations affect different pathways, or that there are kinase-dependent functions of LRRK2 that may or may not be relevant to the pathogenesis of Parkinson’s disease. Parsimony suggests that the latter is more likely, although both viewpoints are reasonable, and it will be important to determine whether experimental evidence indicates common or different pathways in other models (see below).
Although awaiting confirmation, this study provided evidence of changes in the regulation of Argonaute proteins in the brains of old (but not young) flies expressing mutant LRRK214. The idea that LRRK2 regulates any process in an age-dependent manner is exciting, as it is difficult to understand why people with LRRK2 mutations that are present from birth develop disease as adults (usually between 50 and 60 years old). However, the mechanistic details of how this occurs still need to be worked out. At this stage it is not known how LRRK2 function is altered during the aging process, and no specific post-translational modifications associated with aging are known. It is also possible that in the fly models, miR function is a sensitive marker for dysfunction in tissues that have not degenerated, meaning that miR function is relatively downstream of the initial signalling events. In either case, it will be important to use large-scale approaches, measuring mRNA and miR changes in an unbiased fashion in experimental models of LRRK2 pathogenesis.
Protein translation in general has been suggested to be relevant to LRRK2 effects in fly models. Overexpression of 4EBP, which binds the translation factor 4E, limits the detrimental effects of mutant LRRK2 28, and loss of the Drosophila LRRK homologue decreased 4EBP phosphorylation 28, 47. 4EBP is a target of mTOR (mammalian target of rapamycin) which is of interest in aging research, as recent evidence suggests that in diverse species deletion of mTOR signalling components or treatment with the mTOR inhibitor rapamycin can extend lifespan 48. Furthermore, there is evidence that the physiological outputs of mTOR signalling — protein synthesis and autophagy — show age-related changes 49. Therefore, mTOR signalling may link LRRK2 with aging, but there are a couple of caveats. For example, it still has to be confirmed whether 4EBP is a direct kinase substrate of LRRK2. Experiments in my own lab found that 4EBP is a relatively poor substrate for highly purified LRRK2, in contrast to other kinases such as p38α MAPK14) 50. Perhaps this simply illustrates how complex signalling can be; some of the changes in 4EBP phosphorylation in aging might be consequential to tissue stress resulting from experimental manipulation of Drosophila lrrk or human LRRK2 levels and not a direct effect of changes in activity of the kinases themselves, although this awaits confirmation in other systems.
Common pathways for LRRK2, α-synuclein and tau?
The pathological function of mutated LRRK2 may involve common pathways with protein products of other genes that are associated with Parkinson’s disease, such as α-synuclein and tau. Most individuals with Parkinson’s disease and mutations in LRRK2 also have Lewy bodies34 (Box 3). One of the main proteins in Lewy bodies is α-synuclein, which is encoded by the Parkinson’s disease associated gene SNCA (Box 1)2. However, not all patients with LRRK2 mutations have Lewy bodies and instead can have tau lesions or loss of dopamine neuron without a specific pathology. It can therefore be argued that LRRK2 acts upstream of α-synuclein and its aggregation in Lewy bodies51, 52.
In support of the idea that LRRK2 can regulate α-synuclein, when mice that were transgenic for mutant human LRRK2 were crossed with mice that overexpressed mutant human SNCA, deposition of α-synuclein increased compared with that seen in mice overexpressing SNCA alone. Neuronal loss occurred in the areas of the brain where expression of the transgenes was highest; in this case in the striatum. Conversely, removing endogenous Lrrk2 limited the detrimental effects of mutant α-synuclein, reducing protein deposition and neuronal loss as well as related measures 16. These data have not yet been replicated, but it is interesting that neurite extension assays in primary cultures also suggest that knockout of Lrrk2 has an opposing effect to overexpression of mutant alleles 42–44. These data would support the idea that LRRK2 mutations are gain-of-function mutations.
A more recent study reported that α-synuclein accumulates in the kidneys but not the brains of Lrrk2 knockout mice as they age17, suggesting that metabolism of α-synuclein shows tissue-dependent differences. It is debated whether deposits of α-synuclein protein always mean that the protein is in its pathological form53, so a crucial question is whether α-synuclein expression is required for the reported phenotypic effects of Lrrk2 knockout on kidneys. In addition, it is not known whether α-synuclein accumulates in the kidney in human mutant LRRK2 carriers. If it is confirmed that knockout alleles have pathological effects17, this may be because they act in a dominant-negative manner.
These two sets of experiments show that it is important clarify the effect of knocking out endogenous Lrrk2 in comparison to expression of the mutant protein. If these two manipulations have opposite effects16 then it is more likely that mutations result in a gain of function — either by increasing normal function of the protein above a given threshold or by acquiring a novel, pathogenic function. One could certainly imagine that in the case of a dimeric protein such as LRRK2, having one non-functional version could affect the activity of the overall complex. However, it should be noted that loss of activity of one LRRK2 in the dimer is not necessarily the same as having no LRRK2 at all. As argued above, loss of GTPase activity would be predicted to leave LRRK2 in a GTP-bound state, whereas knockout would mean there is no protein to interact with potential partners, including GTP, at all. The data from knockout studies therefore have to be evaluated carefully, especially when the phenotypes are seen in tissues other than the brain and the disease is predominantly neurological.
Some further clues as to how mutations in LRRK2 influence vulnerability for Parkinson’s disease may come from examining how common genetic variants influence the risk of sporadic Parkinson’s disease. At a population level, LRRK2 variants have a modest influence on the lifetime risk of Parkinson’s disease 3, 4. However, in these genome-wide association studies, the authors only tested whether common genetic variants of LRRK2 were associated with the disease and therefore it is not yet clear whether these were variants that might change LRRK2 expression and/or whether they were rare variants that are present on haplotypes represented by commonly assayed polymorphisms around the LRRK2 locus.
Normal variation around SNCA also affects the risk of sporadic Parkinson’s disease3, 4. This genetic evidence together with the influence of wild-type LRRK2 on α-synuclein in mice 16 suggests that there may be a common pathway by which these two genes influence vulnerability for Parkinson’s disease. Understanding the nature of this interaction will be an important next step for the field. One established link between the two proteins is that α-synuclein and LRRK2 are both associated with membrane components, including synaptic vesicles 54, 55.
A second important risk factor for sporadic Parkinson’s disease, at least in some populations, is the gene encoding the microtubule associated protein tau, MAPT 4. As with α-synuclein, it is unclear whether there is a link between MAPT and LRRK2, although some people with Parkinson’s disease and mutations in LRRK2 have tau pathology 6, 56 and mouse models of Lrrk2 mutations have altered tau expression patterns 15, 20, 42. Both LRRK235, 36 and tau57 are associated with microtubules and it is therefore possible that they have some common function.
Therefore — and this will be discussed more in the next section — there are potential functional links between LRRK2 and α-synuclein on the one hand and LRRK2 and tau on the other. One important question to resolve is whether we can consider LRRK2, α-synuclein and tau to be in the same pathway or to have a more distant relationship (Figure 2); perhaps each separately affects a function that is important in the maintenance of dopaminergic neurons in the adult substantia nigra. Overexpressing two proteins (e.g. mutant LRRK2 and mutant α-synuclein) does not distinguish common pathways from additive or synergistic effects. But findings from experiments in which one gene is knocked out, e.g. in recent LRRK2−/− mouse models 16, at least suggest a pathway effect, as in this study LRRK2 was required for full α-synuclein pathology.
Figure 2. LRRK2, α-synuclein and tau.
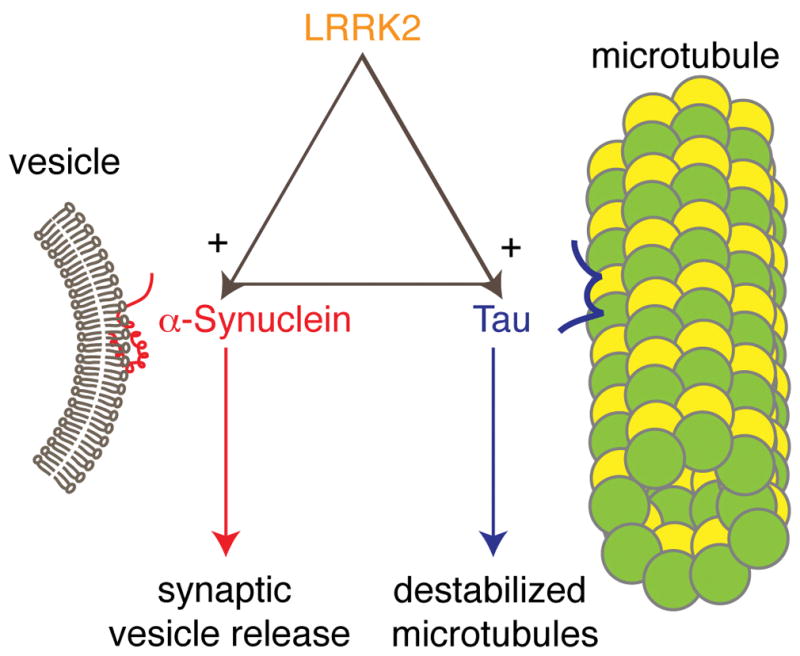
Human genetic, animal model and biochemical data support the idea that there are relationships between three gene products important in the genetic risk of familial and sporadic Parkinson’s disease, LRRK2, α-synuclein and tau. Here, I have put LRRK2 at the ‘top’ of the pathway, based on the observation that human cases with mutations in LRRK2 can have either α-synuclein or tau positive pathology and also that LRRK2 can accelerate (+ sign) α-synuclein pathology in a mouse model. Whether the same is true for LRRK2 and tau is not known, but I have made the assumption here that it could be. Finally, whether there is a truly triangular relationship in that Tau dysfunction can influence α-synuclein pathology or vice versa is unknown, and so this is indicated by a dotted line. Finally, the likely outputs of α-synuclein and tau dysfunction, namely changes in synaptic vesicle function and microtubule stability are indicated at the bottom of the diagram
Future perspectives
I have discussed that the biochemical activities of LRRK2 are fundamental and, in my opinion, can be affected by mutations in ways that are straightforward to understand. From here, things become more difficult. There is a clear gap in our understanding that prevents us from being able to tie together the biochemical activities of LRRK2 with cellular signalling pathways.
Identifying interactors
It is important to develop reliable functional assays for LRRK2 that can be related to cell signalling, preferably through an immediate interactor of the LRRK2 protein. As an example of the type of data that is needed, the C-terminus of α-synuclein has recently been shown to bind to synaptobrevin-2, a SNARE complex component 58. Exploring cellular processes that are important in neurons might help to understand how widespread genes can cause predominantly neurological diseases. The idea that maintenance of axonal integrity and/or vesicle transport along those axons is affected by mutant LRRK2 is extremely attractive. It is also important to understand whether a normal functional interactor of LRRK2 is crucially important for the effects of mutations or whether the detrimental effects are due to a new function that is restricted to mutant protein. By identifying the normal binding partners of LRRK2, we should be able to design mutations to disrupt protein–protein interactions and thereby establish whether pathogenic mutations remain pathogenic in the absence of these interactions or whether protein–protein interactions are required for their pathogenic effect.
However, LRRK2 is a large protein and thus is likely to have multiple interacting partners. Speculatively, I would suggest that there could be more than one co-complex, each with different binding partners. Two specific areas are worth more investigation. First, the N-terminal region of LRRK2 is extremely interesting as a candidate for LRRK2-specific interactors as this region differentiates LRRK2 from LRRK1. As the latter is not associated with Parkinson’s disease, proteins that are scaffolded by LRRK2 specifically may be more relevant to pathogenesis. Second, if the GTP-bound state of LRRK2 is involved in cell signalling pathways, then identifying the proteins or complexes that bind to activated LRRK2 would probably point to functionally important pathways.
Genetic approaches in model organisms may also be helpful to identify pathways that are influenced by LRRK2. Given that there are phenotypes in Drosophila and C. elegans that result from mutations in LRRK2 homologues and that forward genetic screens are well-established for these organisms, we should be able to discover genetic modifiers of those phenotypes. It would be interesting to know if modifiers of, for example, LRRK2-related alterations in axonal maintenance 45, 46 overlap with those involved in responses to aging, including in processes such as translation or cell death in dopamine neurons 14, 28. Such an approach would begin to distinguish whether there is a single underlying pathway for all LRRK2-related effects or whether there are multiple ways in which LRRK2 mutations affect cell physiology.
Does LRRK2-associated pathogenesis require α-synuclein and tau?
Another question that requires experimental attention is whether LRRK2-associated pathogenesis requires α-synuclein and/or tau expression. LRRK2 transgenic mice reported to date have not shown phenotypes of cell loss or Lewy body formation16, but there is evidence of more subtle phenotypes in these mice, such as changes in dopamine release 15, 19, 20 and some alterations in tau staining 15, 20. Therefore, these phenotypes could be useful to examine what happens when LRRK2 transgenic models are crossed with Snca and Mapt knockout animals. Another tactic would be to use viral vectors to express LRRK2 at high levels, which has recently been shown to result in dopaminergic cell loss in wild-type mice 18. This could be applied to Snca and Mapt knockout mice as well. The endpoint for either experiment would be to see whether α-synuclein or tau or both are required for LRRK2-mediated toxicity or other phenotypes. By identifying the genes that are necessary for phenotypes that we might reasonably relate to Parkinson’s disease, and by producing multiple combinations of knockouts, we may be able to define some of the relationships between the gene products.
Because the results of such experiments are not yet available, it may be foolishly early to attempt a synthesis of the roles of LRRK2, tau and α-synuclein but, in my opinion, this is something that should be explored. If there is a common theme between these three proteins it would seem to involve either microtubules or vesicles (Figure 2). As both of these are important for neuronal function this could explain why mutations in the genes encoding LRRK2, tau and α-synuclein result in a disease that is predominantly neurological. In addition, microtubule dynamics are important in axon and dendrite specification 59 and this is consistent with the finding that wild-type LRRK2 regulates the sorting of organelles in polarized cells45 and the effects on axon outgrowth under culture conditions 42–44 (which might be thought of as a form of axotomy). This might tie together the reported direct interaction of LRKR2 with tubulin 35, 36 and the reported effect of overexpression of LRRK2 on depolymerization of microtubules 16, with the effect of LRRK2 on neurite phenotypes.
Whether variations in LRRK2, tau and α-synuclein have a similar direct effect on synaptic vesicle function is less obvious. It is possible that LRRK2 directly interacts with membranes (and therefore with vesicles) and that α-synuclein dysfunction is consequential to this. Tau would not be directly affected by lipid dynamics, but is possible that changes in synaptic vesicle function are secondary to the effects of LRRK2 mutations on microtubules. Both are reasonable possibilities, but the former suggests a triangular effect of LRRK2 on α-synuclein and tau somewhat independently, whereas the latter suggests a more linear pathway. One aspect that may be important is that neither LRRK2 nor α-synuclein nor tau are required nervous system components, in the sense that there are organisms (including Drosophila and C. elegans) that lack α-synuclein or tau and have different LRRK proteins but have functioning neurons. I am not sure what this means, but I suspect that these proteins are more important in maintenance of neurons under conditions of plasticity and/or stress, which could explain why mutations in these proteins are associated with late-onset diseases.
One issue that has not received much attention is whether LRRK2 mutations lead to neuronal dysfunction in a simple, cell-autonomous fashion. Expression of LRRK2 is high in striatal neurons and in some B-cell lymphocyte lineages 60, and so non-cell autonomous mechanisms could contribute to LRRK2-mediated loss of dopaminergic neurons in the substantia nigra. However, high-level expression of LRRK2 in the striatum using a CAMKII promoter does not result in obvious nigral cell loss 16, arguing against a simple retrograde toxicity mechanism, and expression in nigral neurons seemed to be sufficient for nigral cell loss in one model 18. I think these observations together support the idea that LRRK2, α-synuclein and tau have a pathway relationship but allows for the possibility that some of their effects may occur at a systems level, perhaps by altering function in more than one cell type. If the latter possibility is correct, then the most important experiments should involve manipulating LRRK2 function in different cell types in the brain.
One motivation for the work described in this article is to try and identify tractable therapeutic targets. Recent papers have focused on the kinase activity of LRRK2 and reported potent and somewhat selective inhibitors of LRRK2 kinase that are likely competitors for ATP binding in the active site of the kinase domain 18, 32, 61. Furthermore, some of these inhibitors have been suggested to be neuroprotective in vivo 18, although it is unclear whether this is due entirely to direct effects of the inhibitors on LRRK2 or whether they might have some effects on other kinases. If the scheme discussed above suggesting — that kinase activity of LRRK2 influences its GTP-bound state — has any value, it would predict that competitive kinase inhibitors might be only one way to limit LRRK2 dysfunction; molecules that block the GTP-bound state of LRRK2 would also be predicted to be neuroprotective.
Acknowledgments
I would like to thank Elisa Greggio and Jean-Marc Taymans for critically reading the manuscript. This research was supported by the Intramural Research Program of the NIH, National Institute on Aging.
Biography
Dr. Mark R. Cookson is a Senior investigator at the National Institute on Aging (NIA), Bethesda, MD, USA. He is a cell biologist whose current research interests include the effects of mutations in the genes associated with neurodegeneration at the cellular and molecular level. His laboratory efforts are directed at finding the underlying pathways that lead to Parkinson’s disease and related disorders. Dr. Cookson received both his B.Sc. and Ph.D. degrees from the University of Salford, UK in 1991 and 1995, respectively. His postdoctoral studies included time spent at the Medical Research Council laboratories and at the University of Newcastle, Newcastle, UK. He joined the Mayo Clinic, Jacksonville, Florida, as an Assistant Professor in 2000 and moved to the NIA in February 2002. Within the Laboratory of Neurogenetics, Dr. Cookson’s group works on the effects of mutations associated with Parkinson’s disease on protein function.
Footnotes
Suggested online links:
Online Mendelian Inheritance in Man: http://www.ncbi.nlm.nih.gov/omim/168600
PDGene: http://www.pdgene.org/
PDOnline research: http://www.pdonlineresearch.org/
Author website: http://www.grc.nia.nih.gov/branches/irp/mcookson.htm
References
- 1.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–66. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 2.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 3.Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 4.Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paisan-Ruiz C, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 6.Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Greggio E, et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–14. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greggio E, et al. The Parkinson’s disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochem Biophys Res Commun. 2009;389:449–54. doi: 10.1016/j.bbrc.2009.08.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamikawaji S, Ito G, Iwatsubo T. Identification of the autophosphorylation sites of LRRK2. Biochemistry. 2009;48:10963–75. doi: 10.1021/bi9011379. [DOI] [PubMed] [Google Scholar]
- 10.Klein CL, et al. Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J Neurochem. 2009;111:703–15. doi: 10.1111/j.1471-4159.2009.06358.x. [DOI] [PubMed] [Google Scholar]
- 11.Berger Z, Smith KA, Lavoie MJ. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry. 2010 doi: 10.1021/bi100157u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gloeckner CJ, et al. Phosphopeptide analysis reveals two discrete clusters of phosphorylation in the N-terminus and the Roc domain of the Parkinson-disease associated protein kinase LRRK2. J Proteome Res. 2010;9:1738–45. doi: 10.1021/pr9008578. [DOI] [PubMed] [Google Scholar]
- 13.Dzamko N, et al. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–13. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–41. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12:826–8. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin X, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–27. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong Y, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of {alpha}-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci U S A. 2010;107:9879–84. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee BD, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, et al. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J Neurosci. 2010;30:1788–97. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melrose HL, et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis. 2010 doi: 10.1016/j.nbd.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galter D, et al. LRRK2 expression linked to dopamine-innervated areas. Ann Neurol. 2006;59:714–9. doi: 10.1002/ana.20808. [DOI] [PubMed] [Google Scholar]
- 22.Guo L, et al. The Parkinson’s disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–70. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis PA, et al. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–71. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greggio E, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–41. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Jaleel M, et al. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J. 2007;405:307–17. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nichols RJ, et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West AB, et al. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–7. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai Y, et al. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–43. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson’s disease: three questions. ASN Neuro. 2009;1 doi: 10.1042/AN20090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu B, et al. Expression, purification and preliminary biochemical studies of the N-terminal domain of leucine-rich repeat kinase 2. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbapap.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Sen S, Webber PJ, West AB. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem. 2009;284:36346–56. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Dobson B, Glicksman MA, Yue Z, Stein RL. Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry. 2010;49:2008–17. doi: 10.1021/bi901851y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith WW, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–3. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 34.Cookson MR, Hardy J, Lewis PA. Genetic neuropathology of Parkinson’s disease. Int J Clin Exp Pathol. 2008;1:217–31. [PMC free article] [PubMed] [Google Scholar]
- 35.Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res. 2008;86:1711–20. doi: 10.1002/jnr.21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability--a point of convergence in parkinsonian neurodegeneration? J Neurochem. 2009;110:1514–22. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- 37.Sancho RM, Law BM, Harvey K. Mutations in the LRRK2 Roc-COR tandem domain link Parkinson’s disease to Wnt signalling pathways. Hum Mol Genet. 2009;18:3955–68. doi: 10.1093/hmg/ddp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding X, Goldberg MS. Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS One. 2009;4:e5949. doi: 10.1371/journal.pone.0005949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ko HS, et al. CHIP regulates leucine-rich repeat kinase-2 ubiquitination, degradation, and toxicity. Proc Natl Acad Sci U S A. 2009;106:2897–902. doi: 10.1073/pnas.0810123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsu CH, et al. MKK6 binds and regulates expression of Parkinson’s disease-related protein LRRK2. J Neurochem. 2010;112:1593–604. doi: 10.1111/j.1471-4159.2010.06568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen ND, et al. The WD40 domain is required for LRRK2 neurotoxicity. PLoS One. 2009;4:e8463. doi: 10.1371/journal.pone.0008463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacLeod D, et al. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–93. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Parisiadou L, et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009;29:13971–80. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–56. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–8. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 46.Samann J, et al. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J Biol Chem. 2009;284:16482–91. doi: 10.1074/jbc.M808255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tain LS, et al. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–35. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kapahi P, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11:453–65. doi: 10.1016/j.cmet.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hands SL, Proud CG, Wyttenbach A. mTOR’s role in ageing: protein synthesis or autophagy? Aging (Albany NY) 2009;1:586–97. doi: 10.18632/aging.100070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar A, et al. The Parkinson’s disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E-BP compared to autophosphorylation. PLoS One. 2010;5:e8730. doi: 10.1371/journal.pone.0008730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wider C, Dickson DW, Wszolek ZK. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. 2010;7:175–9. doi: 10.1159/000289232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taymans JM, Cookson MR. Mechanisms in dominant parkinsonism: The toxic triangle of LRRK2, alpha-synuclein, and tau. Bioessays. 2010;32:227–35. doi: 10.1002/bies.200900163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cookson MR. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 2009;4:9. doi: 10.1186/1750-1326-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Auluck PK, Caraveo G, Lindquist S. alpha-Synuclein: Membrane Interactions and Toxicity in Parkinson’s Disease. Annu Rev Cell Dev Biol. 2010 doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- 55.Biskup S, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–69. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 56.Rajput A, et al. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–8. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 57.Matenia D, Mandelkow EM. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci. 2009;34:332–42. doi: 10.1016/j.tibs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 58.Burre J, et al. {alpha}-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science. 2010 doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Conde C, Caceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 2009;10:319–32. doi: 10.1038/nrn2631. [DOI] [PubMed] [Google Scholar]
- 60.Kubo M, et al. LRRK2 is expressed in B-2 but not in B-1 B cells, and downregulated by cellular activation. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 61.Nichols RJ, et al. Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem J. 2009;424:47–60. doi: 10.1042/BJ20091035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haugarvoll K, Wszolek ZK. Clinical features of LRRK2 parkinsonism. Parkinsonism Relat Disord. 2009;15(Suppl 3):S205–8. doi: 10.1016/S1353-8020(09)70815-6. [DOI] [PubMed] [Google Scholar]
- 63.Funayama M, et al. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- 64.Funayama M, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol. 2005;57:918–21. doi: 10.1002/ana.20484. [DOI] [PubMed] [Google Scholar]
- 65.Di Fonzo A, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet. 2005;365:412–5. doi: 10.1016/S0140-6736(05)17829-5. [DOI] [PubMed] [Google Scholar]
- 66.Gilks WP, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–6. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 67.Kachergus J, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–80. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lesage S, et al. LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med. 2006;354:422–3. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 69.Nichols WC, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365:410–2. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- 70.Ozelius LJ, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–5. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 71.Ishihara L, et al. Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch Neurol. 2006;63:1250–4. doi: 10.1001/archneur.63.9.1250. [DOI] [PubMed] [Google Scholar]
- 72.Kumari U, Tan EK. LRRK2 in Parkinson’s disease: genetic and clinical studies from patients. FEBS J. 2009;276:6455–63. doi: 10.1111/j.1742-4658.2009.07344.x. [DOI] [PubMed] [Google Scholar]
- 73.Halliday GM, McCann H. The progression of pathology in Parkinson’s disease. Ann N Y Acad Sci. 2010;1184:188–95. doi: 10.1111/j.1749-6632.2009.05118.x. [DOI] [PubMed] [Google Scholar]
- 74.Braak H, et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]