

Abstract
The purpose of the present study was to develop oral dispersible tablets containing prednisolone (PDS)-loaded chitosan nanoparticles using microcrystalline cellulose (MCC 101), lactose, and croscarmellose sodium (CCS). The PDS-loaded chitosan nanoparticles were formulated by ionotropic external gelation technique in order to enhance the solubility of PDS in salivary pH. Prepared nanoparticles were used for the development of oral fast disintegrating tablets by direct compression method. The prepared tablets were evaluated for disintegration time (DT), in vitro drug release (DR), thickness, weight variation, drug content uniformity, friability, and hardness. The effect of concentrations of the dependent variables (MCC, lactose, CCS) on DT and in vitro DR was studied. Fast disintegrating tablets of PDS can be prepared by using MCC, CCS, and lactose with enhanced solubility of PDS. The minimum DT was found to be 15 seconds, and the maximum DR within 30 minutes was 98.50%. All independent variables selected for the study were statistically significant. Oral fast disintegrating tablets containing PDS nanoparticles could be the better choice for the pediatric patients that would result in better patient compliance. From this study, it can be concluded that fast disintegrating tablets could be a potential drug delivery technology for the management of asthma in pediatrics.
Keywords: asthma, superdisintegrant, prednisolone, oral tablets, MCC, CCS, factorial design, ANOVA
Introduction
Asthma is a chronic inflammatory disease of the airways that is characterized by reversible airflow obstruction and bronchospasm. Common symptoms of asthma include wheezing, coughing, chest tightness, and shortness of breath.1 Asthma may also be classified as atopic (extrinsic) or nonatopic (intrinsic), where atopy refers to a predisposition toward developing type 1 hypersensitivity reactions.2,3 Asthma is the clinical condition that may occur in adult and pediatric patients. For some children with asthma, their first asthmatic experience can be frightening – heavy wheezing, a tight chest, and shortness of breath can quickly catch their active, young bodies off-guard.
Prednisolone (PDS) is a synthetic glucocorticoid that is used as anti-inflammatory or immunosuppressive agent. It is indicated in conditions where corticosteroid therapy is likely to be beneficial, including allergic disorders, asthma, leukemia, thrombocytopenic purpura, insulin resistance in diabetes mellitus, immunosuppression, liver disorders, and ulcerative colitis.4 In elderly patients, it is used in asthma as well as psoriatic arthritis. Hence, it is a choice of treatment for pediatric and geriatric patients, and hence, in conventional dosage form produces difficulty in swallowing. PDS is a Biopharmaceutics Classification System (BCS) class II drug having slight water solubility, and as a consequence, it can exhibit low and/or variable bioavailability after oral administration.5 Therefore, a well-designed formulation must be capable of presenting a therapeutically effective amount of the hydrophobic drug to the desired absorption site, in an absorbable form.
Nowadays, nanoparticle engineering processes have been developed and reported for pharmaceutical applications to increase the dissolution rate of low-soluble drugs, which in turn may lead to substantial increases in bioavailability.6 Nanoparticle engineering enables poorly soluble drugs to be formulated as particles alone or with a combination of pharmaceutical excipients. By decreasing the particle size from a micron to a nanometer scale, there is a significant increase in the surface area and related dissolution rate.7
Optimization of oral drug delivery system in case of pediatric patients is challenging task. Fifty-four percent of children aged between 6 years and 11 years are unable to easily swallow a tablet.8,9 Oral fast disintegrating tablets containing PDS nanoparticles would be the new drug delivery technology and an alternative to conventional dosage forms such as tablets, capsules, syrups, and other formulations for pediatric and geriatric use. Oral fast disintegrating tablets are easy to administer and provide better patient compliance in the elderly, pediatric, mentally retarded, nauseated, and uncooperative patients.10 Hence, the aim of the present study was to develop oral fast disintegrating tablets containing PDS nanoparticles for the management of pediatric asthma to improve the patient compliance.
Experimental
Materials
PDS was purchased from Baoji Guokang Bio-Technology Co, Ltd (Baoji, Shaanxi, People’s Republic of China). Chitosan (CS), microcrystalline cellulose (MCC), lactose, aerosil, magnesium stearate, and croscarmellose sodium (CCS) were purchased from Sigma-Aldrich (St Louis, MO, USA). Tripolyphosphate (TPP) and acetic acid were purchased from Shanghai Chemical Co (Shanghai, People’s Republic of China).
Methods
Preparation of PDS-CS nanoparticles by ionotropic external gelation technique
CS nanoparticles were prepared by ionotropic external gelation method.11 CS was dissolved in acetic acid under stirring at room temperature. The concentration of acetic acid in the aqueous media was maintained 1.5 times higher than that of CS. In a clear solution of CS, Tween-80 (1.5% v/v) was added as a surfactant. PDS was dissolved in an organic solution (methanol); this organic phase was added dropwise to the aqueous phase of CS solution. Stirring was continued for 5 minutes after the complete addition of the organic phase to the aqueous phase. Later, cross-linking of the particles was carried out by dropwise addition of TPP solutions of different concentrations into an o/w emulsion under stirring. For complete evaporation of organic solution, it was kept overnight at room temperature. The formation of nanoparticles was a result of the interaction between the negative groups of TPP and the positively charged amino groups of CS. Nanoparticles were isolated by centrifugation at 15,000 rpm for 30 minutes at −100°C using cooling centrifuge, and the supernatant was used for the measurement of free PDS by UV spectrophotometer. Prepared PDS-loaded CS nanoparticles were characterized, and optimized nanoparticles were used in the development of oral dispersible tablets (ODT). Table 1 represents the different formulations of nanoparticles.
Table 1.
PDS-CS nanoparticles formulations
| Batch | CS | TPP |
|---|---|---|
| F1 | 0.25 | 0.5 |
| F2 | 0.25 | 0.10 |
| F3 | 0.50 | 0.10 |
| F4 | 0.50 | 0.50 |
Abbreviations: PDS, prednisolone; CS, chitosan; TPP, tripolyphosphate.
23 factorial design for development of ODT
Statistical analysis of the experimental work was carried out using Design Expert 6.0.8 portable software. A three-factor, two-level full factorial design was used to derive a second-order polynomial equation. Concentration of CCS (A), MCC (B), and compression pressure (C) were selected as independent variables, while disintegration time (DT; Y1) and in vitro drug release (DR; Y2) were selected as dependent variables. Variables and their two levels are presented in Table 2. The 23 full factorial design is used to optimize the formulation variables with basic requirement of understanding interaction of independent variables. Linear, cross-product contribution (2FI), quadratic, and cubic models were generated for the responses. The significance of the model was determined by the comparisons of statistical parameters, and the best model (suggested) was decided based on the reasonable agreement between adjusted R2 and predicted R2 (within 0.2 of each other), higher values of adjusted R2 and predicted R2, model P-value (should be <0.05), and small PRESS value of the model. PRESS is a measure of the fit model to data points in the design. The PRESS for the chosen model should be relatively small in consideration with another model. Contour plots and 3D response plots were also constructed using the Design Expert® software. The polynomial equations may be used to draw conclusions after considering the magnitude of coefficients and either the positive or negative mathematical sign.12 Results for experimental design batches and its analysis of variance were studied.
Table 2.
Variable and two levels
| Low level (−1) | High level (+1) | |
|---|---|---|
| Independent variables | ||
| A = CCS concentration (%) | 4 | 8 |
| B = MCC concentration (%) | 20 | 40 |
| C = compression force (ton) | 2 | 4 |
| Dependent variables | ||
| Y1 = DT | ||
| Y2 = %DR |
Abbreviations: CCS, croscarmellose sodium; MCC, microcrystalline cellulose; DT, disintegration time; DR, drug release.
Preparation of ODTs by direct compression method
Mouth dissolving tablets were prepared by direct compression method. All the ingredients were weighed and passed through sieve no 60, collected, and mixed in geometric order. The PDS-loaded CS nanoparticles from the optimized batch were weighed and added to the above mixture of ingredients. Then the tablets were compressed using 13 mm punch on KBR press machine to get tablets of 150 mg weight. Formula composition of all batches is represented in Table 3.
Table 3.
Evaluation of CS-PDS nanoparticles
| Batch | Yield (%) | EE (%) | DL (%) | PSD (nm) | Solubility (mg/L) |
|---|---|---|---|---|---|
| F1 | 75.85 | 63.20 | 10.20 | 450 | 425 |
| F2 | 80.25 | 72.25 | 13.25 | 320 | 569 |
| F3 | 88.90 | 80.63 | 17.59 | 255 | 735 |
| F4 | 95.30 | 88.36 | 22.36 | 130 | 970 |
Abbreviations: CS, chitosan; PDS, prednisolone; EE, encapsulation efficiency; DL, drug loading.
Evaluation of PDS-loaded CS nanoparticles Percentage yield
PDS-loaded nanoparticles were collected and weighed accurately. The yield was calculated by using the following formula:13
| (1) |
Drug entrapment efficiency and drug loading
The drug-loaded nanoparticles were centrifuged, and the amount of nonentrapped drug (free drug) was measured in the clear supernatant using UV spectrophotometer at 280 nm. The drug-loading (DL) capacity and encapsulation efficiency (EE) of the nanoparticles were calculated according to the following equations:14
| (2) |
| (3) |
Phase solubility study of PDS and PDS-loaded CS nanoparticles
Excess amount of plane PDS was added to 5 mL of water to ensure the drug reached saturation level. Similarly, saturated solution of PDS-loaded CS nanoparticles was also prepared by adding excess amount in distilled water. These solutions were mechanically shaken for 24 hours at 370°C, and then the solutions were centrifuged at 10,000 rpm for 3 minutes.15 The saturated solutions were then diluted to proper concentration, and absorbance was measured at 247.5 nm and solubility in each solution was determined.
Particle size distribution analysis
The particle size distribution of PDS-loaded CS nanoparticles was determined by using laser diffraction technique using Malvern Instruments, Malvern, UK. Sufficient amount of nanoparticles was dispersed in distilled water. Light diffraction was measured at 25°C and with an angle of 90°.
Scanning electron microscopy
Scanning electron microscopy was obtained to characterize the surface morphology of nanoparticles, that is, the shape, size, and surface characteristics of the nanoparticles.
Evaluation of ODTs
Weight variation test
Weight variation was measured by weighing 20 tablets, and the average weight of the individual tablet should fall within specified limits. The tablets encounter the United States Pharmacopeial Convention (USP) test if no more than two tablets are outside the percentage limit and if no tablet differs by more than two times the percentage limit.16
Uniformity of thickness and diameter
The diameter and thickness of ten tablets were measured using a Vernier caliper at three different positions.16 Results were reported as the mean (± standard deviation) of three measurements.
Hardness
The hardness of tablets was determined using Monsanto hardness tester. The mean hardness of each was determined. The hardness of a tablet is indicative of its tensile strength and is measured in terms of load/pressure required to crush it when placed on its edge.16
Friability
The friability of the tablets was determined by laboratory friability tester known as Roche Friabilator; the percentage loss in tablet weight before and after 100 revolutions of ten tablets was calculated and taken as % friability. The weight loss should not be >1%. To achieve % friability within limits for an ODT is a challenge for a formulator since all methods of manufacturing of ODT are responsible for increasing the % friability values.17
Disintegration test
The test is carried out on six tablets using the apparatus. A 900 mL phosphate buffer (pH 6.8) at 37°C±0.50°C was used as a disintegration medium, and the time taken for complete disintegration of the tablet with no palpable mass remaining in the apparatus was measured in seconds.17
Dissolution test
The in vitro dissolution studies were performed using USP apparatus type II at 50 rpm. The dissolution medium used was pH 6.8 phosphate buffer (900 mL) maintained at 37°C±0.50°C. Aliquots of dissolution media were withdrawn at different intervals, and the content of PDS was measured by determining absorbance at 247.5 nm.
Results
PDS-loaded CS nanoparticles
Yield of PDS-loaded CS nanoparticles
The prepared nanoparticles were collected and weighed. The yield of the nanoparticles was found to be satisfactory and in the range of 75.85%–95.30% of total solid content employed during the formulation of nanoparticles. Table 3 represents the summary of the yield of the nanoparticles. There was no wide variation in the yield of the nanoparticles. There was minimum loss of the nanoparticles due to centrifugation at higher speed with the cooling temperature maintained at −100°C. From the results, it was found that as the concentration of both CS and TPP increases, the yield of the nanoparticles also increases linearly. The possible reason behind this is the formation of denser CS solution and counteracting by the TPP with CS that might have minimized the loss of polymeric material.
EE and DL of PDS-CS nanoparticles
The percentage of DL of nanoparticles was found to be in the range of 10.20%–22.36%, and the percentage of EE of nanoparticles was found to be in the range of 63.20%–88.36%. DL and EE of all batches of nanoparticles are shown in Table 3. The EE and DL were varied by varying the concentration of CS and TPP. Generally, the low entrapment efficiency was due to the high affinity of the drug and polymer in the used organic solvent system during the nanoparticle preparation.18 DL and EE were mainly affected by the polymer and cross-linking agent. It has been reported that increased EE and DL may be due to the higher concentration of polymer with respect to the amount of the drug.19 Higher drug encapsulation was observed for batch F4, and batch F1 has the lowest drug EE. As concentration of CS and TPP increases, EE and DL increases. Such enhancement was due to the fact that the CS has higher ability of the ionic gel formation with the TPP increasing the DL and EE.
Particle size distribution study
The particle size was found to be in the range of 130–450 nm as shown in Table 3 and was used to characterize the nanoparticles because it facilitates the understanding of the dispersion and aggregation. Due to large surface area and attractive force between the particles, more chances of aggregation are possible in small-sized particles.20 The presence of the surfactant also played an important role in reducing the particle size of nanoparticles by avoiding the aggregation of the particles that suspend immediately after formation. From the results, it can be concluded that as the concentration of CS and TPP increases, the particle size of the nanoparticles decreases. The particle size data indicate that the nanoparticles produced were of nanosize and with a low polydispersity index of 0.310, which showed a relatively narrow particle size distribution of PDS-CS nanoparticles.
Phase solubility study
According to the BCS, PDS is a class II drug. It is a poorly water-soluble agent having log P-value of 1.62. Due to poor water solubility, bioavailability is also very low.21 The intrinsic aqueous solubility of pure PDS was determined, and it was found to be 350±5 mg/L. This intrinsic aqueous solubility is nearly same as that determined by the other research groups. In order to enhance the aqueous solubility, PDS-loaded CS nanoparticles were prepared by ionotropic external gelation technique. Due to formation of the nanoparticles in the range of 130–450 nm, enhancement of the solubility was found. The solubility was enhanced in all formulations of the nanoparticles. The maximum solubility was found to be 970±6 mg/L in water as shown in Table 3. Comparative dissolution profile is represented in Figure 1A. From the results, it was clear that as the particle size of the nanoparticles was decreased, the solubility of PDS was increased. Nanoparticles are defined as a discrete pharmaceutical ingredient having physical dimensions <1 μm in an external phase. It is estimated that ~40% of active substances identified through combinatorial screening programs are difficult to formulate as a result of their lack of significant solubility in water. When these types of situations arise, a nanoparticle formulation approach has proven to be very useful and invaluable in all stages of the drug development and has opened opportunities for revitalizing marketed products with suboptimal delivery. According to the Noyes–Whitney model for dissolution kinetics, the dissolution rate is directly proportional to the surface area of the drug. When the drug molecule reduced in size from 10 μm to 100 nm, the surface area to volume ratio increased 100 fold.22 Due to increase in the surface area of the nanoparticles, there is tremendous increase in the solubility of the drug molecule. This increase in the surface area has a profound impact on the bioavailability of the molecule.
Figure 1.

Solubility profile and surface morphology of PDS nanoparticles.
Notes: (A) Comparative solubility of the PDS-loaded CS nanoparticles and pure PDS in pH 6.8 phosphate buffer, (B) scanning electron microscopy of the PDS-loaded CS nanoparticles with spherical shape and morphology.
Abbreviations: PDS, prednisolone; CS, chitosan.
Morphological characterization of nanoparticles
The morphological analysis of PDS-loaded CS nanoparticles were visualized by field emission scanning electron microscopy and is presented in Figure 1B. The nanoparticles were of spherical shape with smooth surface and regular without any crack and erosion. The pores were not spotted on the surface of the nanoparticles. All nanoparticles were free flowing and discrete from each other.
ODT containing PDS nanoparticles
Physical evaluation of tablets
ODT of the PDS were prepared by the direct compression method. In order to enhance the solubility and dissolution rate of the PDS, PDS-CS nanoparticles were formulated. As described earlier, these nanoparticles increased the solubility of the PDS and such solubility-enhanced nanoparticles were used in tablet formulation. Table 4 represents the physical evaluation of all the batches. The prepared tablets were directly compressed with 6 mm round punch. From the results, it can be found that the diameters of all the tablets are nearly same with small variation. The diameter of the tablets was in the range of 5.8±0.5 mm to 6.1±1 mm. From this, it is clear that all tablets had a diameter within the range. The final weight of the tablet was set at 150 mg, and all batches of the tablets were within the range of 146±3 mg to 151±3 mg. No wide variation was found in the weights of the tablets. The tablets were compressed at the two different compression forces, that is, 2 ton and 4 ton. Based on the compression force, the hardness was varied in batches. Batches with compression force 4 ton had hardness ranging from 1.8±1 kg/cm2 to 2.8±1 kg/cm2, the hardness was found to be more in this case as compared to the tablets with 2 ton compression force (1.4±1 kg/cm2 to 1.7±1 kg/cm2). Hence, it was clear from the results that as the compression force increases, the hardness also increases. The inverse relation was found in case of the thickness. Batches with 2 ton force had higher thickness ranging from 2.9±1 mm to 3.5±3 mm, while for batches with 4 ton force, the thickness was found to be 2.5±2 mm to 2.7±2 mm. All formulation batches showed the friability within the range (not >1%). Hence, overall it could be concluded that all formulation batches were pharmaceutically accepted and could be useful for the pediatric patients.
Table 4.
Evaluation parameters for the ODT
| Batch | Weight variation (mg) | Thickness (mm) | Diameter (mm) | Hardness (kg/cm2) | Friability (%) | DT (Sec) |
|---|---|---|---|---|---|---|
| F1 | 150±2 | 3.0±2 | 6.0±1 | 2.0±1 | 1.0 | 50 |
| F2 | 151±3 | 3.1±2 | 5.9±0 | 1.8±1 | 0.8 | 30 |
| F3 | 149±1 | 2.9±1 | 6.1±2 | 2.2±1 | 0.8 | 60 |
| F4 | 148±2 | 3.2±2 | 6.0±1 | 1.7±1 | 0.9 | 40 |
| F5 | 146±3 | 3.3±2 | 5.8±1 | 1.7±1 | 0.8 | 20 |
| F6 | 150±1 | 2.5±2 | 6.1±1 | 2.8±1 | 0.3 | 80 |
| F7 | 148±2 | 2.7±2 | 5.9±2 | 2.4±1 | 0.5 | 70 |
| F8 | 149±3 | 3.5±3 | 6.0±1 | 1.4±1 | 0.7 | 15 |
Abbreviations: ODT, oral dispersible tablets; DT, disintegration time.
Discussion
PDS ODT were developed with the intention to improve the patient compliance in pediatrics who suffered from asthma. PDS solubility was enhanced with the formulation of CS nanoparticles. These nanoparticles were found to be spherical, smooth, and free flowing in nature with a minimum particle size of 130 nm. The EE and DL were also maximized. PDS-loaded CS nanoparticles enhanced the solubility of the PDS by reducing the particle size of the molecule. The solubility of PDS was enhanced 6.5 times. These promising results of the nanoparticles played very important role in the development of the ODT of PDS.
After successful development of the PDS-loaded CS nanoparticles, the optimized batch of the nanoparticles (F4) was used in the tablet development. The tablets were prepared by the direct compression method by using MCC 101 as a directly compressible material. The ODT had great pharmaceutical acceptance in terms of the shape, size, thickness, hardness, weight uniformity, DT, diameter, and friability. All these evaluation parameters were within the acceptable limit of pharmacopoeia. CCS was the key excipient in the ODT to maintain the DT at saliva pH. All selected independent variables showed statistically significant effect on DT and in vitro DR. Tablets prepared from the PDS-loaded CS nanoparticles showed >80% DR within 30 minutes, while tablets with pure PDS showed only 30.65% DR. These results clearly indicated that the nanoparticles had great effect on the enhancement of the dissolution rate of the PDS. Definitely, these ODT containing CS nanoparticles could be a novel technology for the pediatric patients for the management of asthma.
Statistical analysis of DT
In vitro DT was determined in salivary pH of 6.8 phosphate buffer (saliva pH 6.2–7.4). Directly compressed tablets showed DT between 15 seconds and 80 seconds as shown in Table 5. The lower DT of the formulations signified that the prepared tablets had fast disintegrating nature at saliva pH. The polynomial equation obtained for DT (Y1) is given by:
| (4) |
where Y1 is the DT, A is the CCS concentration, B is the MCC concentration, and C is the compression force during punching of the tablets. The model F-value 411.67 indicates that the model is significant. A positive value in Equation 4 represents the synergistic effect of the independent variable, and a negative value represents the antagonistic effect.23 The suggested model for the Y1 was found to be 2FI as shown in Table 6. The value of the correlation coefficient (R2) for the response Y1 was found to be 0.9968, which indicates the good fit for the 2FI model. The P-value for the Y1 model was found to be <0.0001, which clearly indicates that the model is statistically significant. Also among the three independent variables selected, the factors A (P<0.0001), B (P=0.0022), and C (P=0.0001) are shown in Table 7.
Table 5.
Formulation of PDS ODT using 23 factorial design
| Batches | Factor
|
Response
|
|||
|---|---|---|---|---|---|
| A | B | C | Y1 | Y2 | |
| F1 | −1 | −1 | −1 | 50 | 86.26 |
| F2 | +1 | −1 | +1 | 30 | 91.25 |
| F3 | −1 | +1 | −1 | 60 | 82.33 |
| F4 | +1 | +1 | +1 | 40 | 89.55 |
| F5 | +1 | +1 | −1 | 20 | 93.64 |
| F6 | −1 | +1 | +1 | 80 | 75.20 |
| F7 | −1 | −1 | −1 | 70 | 80.55 |
| F8 | +1 | −1 | +1 | 15 | 98.95 |
Notes: A = CCS concentration (%), B = MCC concentration (%), C = compression force (Ton), Y1 = DT, Y2 = % DR.
Abbreviations: CCS, croscarmellose sodium; DT, disintegration time; DR, in vitro drug release; MCC, microcrystalline cellulose; PDS, prednisolone; ODT, oral dispersible tablets.
Table 6.
Summary of results of regression analysis for responses Y1 and Y2
| Models | R2 | Adjusted R2 | Predicted R2 | Standard deviation | PRESS | Remarks |
|---|---|---|---|---|---|---|
| Response Y1 | ||||||
| Linear | 0.8120 | 0.8012 | 0.6214 | 3.36 | 32.20 | |
| 2FI | 0.9968 | 0.9944 | 0.9871 | 1.77 | 50.00 | Suggested |
| Quadratic | 0.7261 | 0.8172 | 0.7725 | 0.40 | 26.30 | |
| Cubic | 0.9020 | 0.7963 | 0.8912 | 3.63 | 42.20 | |
| Response Y2 | ||||||
| Linear | 0.6588 | 0.6062 | 0.7863 | 3.80 | 12.00 | |
| 2FI | 0.9890 | 0.9808 | 0.9561 | 1.07 | 18.18 | Suggested |
| Quadratic | 0.9012 | 0.8817 | 0.8588 | 4.97 | 11.00 | |
| Cubic | 0.9214 | 0.8825 | 0.8590 | 6.50 | 15.36 | |
| Regression equations of the fitted models | ||||||
| Y1=+45.63−19.38A + 4.38B +9.38C | ||||||
| Y2=+87.22+6.13A − 2.04B −3.08C | ||||||
Note: The bold values signifies the terms of the suggested model.
Table 7.
ANOVA of models for Y1 and Y2
| Source | DF | Sum of squares | Mean square | F-value | P-value |
|---|---|---|---|---|---|
| Model for Y1 | 3 | 3,859.38 | 1,286.46 | 411.67 | <0.0001 |
| A | 1 | 3,003.13 | 3,003.13 | 961.00 | <0.0001 |
| B | 1 | 153.12 | 153.12 | 49.00 | 0.0022 |
| C | 1 | 703.13 | 703.13 | 225.00 | 0.0001 |
| Model for Y2 | 3 | 409.74 | 136.58 | 120.23 | 0.0002 |
| A | 1 | 300.74 | 300.74 | 264.73 | <0.0001 |
| B | 1 | 33.17 | 33.17 | 29.20 | 0.0057 |
| C | 1 | 75.83 | 75.83 | 66.75 | 0.0012 |
Note: The bold values signifies the terms of the suggested model.
Abbreviations: ANOVA, analysis of variance; DF, degrees of freedom.
It is a well-known fact that a CCS is an excellent superdisintegrant that is most widely used in pharmaceutical formulations including tablets, capsules, and fast disintegrating films. Hence, CCS was used as a superdisintegrant in ODT to lower the DT.24 From the contour plot Figure 2A, it was clear that as the concentration of CCS increased, the DT gets decreased (from 80 seconds to 15 seconds). This is due to the presence of the higher amount of CCS in the formulation producing rapid and instant disintegration of the tablets. The results of the in vitro DT of all the batches were found to be within limit. The increase in concentration of CCS in the formulation might have reduced the wetting time of the tablets. This was happened due to the combined effect of the CCS by swelling and wicking mechanism when contact with water. The effect of B was statistically significant (P=0.0030). Minimum DT of 15 seconds was a good indicative for the ODT for the pediatric patients. Within 15 seconds, the tablet would get disintegrated and be ready for the DR, which is quite required for the rapid effect for the management of the pediatric asthma.
Figure 2.

Counter plots showing effect of independent variables on dependent variables (DT).
Notes: (A) Contour plot showing effect of CCS on the DT of tablets, (B) contour plot showing effect of MCC on the DT of tablets, (C) contour plot showing effect of compression force on the DT of tablets.
Abbreviations: CCS, croscarmellose sodium; DT, disintegration time; MCC, microcrystalline cellulose.
MCC is one of the most important and widely used of all excipients. It is a key diluent for drug formulations and an essential component for almost every kind of oral dosage, including tablets, capsules, sachets, pellets, and others. MCC 101 was used as a major excipient in the formulation due to its direct compressibility property.25 From the contour plot Figure 2B, it was clear that as the concentration of MCC 101 increased, the DT gets increased. This is due to the presence of the PDS-loaded CS nanoparticles in the tablet formulation. The presence of CS and MCC 101 led to increase in the DT of the tablets. Also its direct compressibility of CCS also imparted to increase in the DT. The effect of CCS on DT was also statistically significant (P=0.0022).
The tablets were prepared by direct compression method with MCC 101 and other excipients on KBR press with 6 mm die punch. The tablets were subjected to the 2 ton and 4 ton compression force in order to identify the effect of the compression force on the DT and DR. The effect of compression force on DT was found to be statistically significant on DT with P=0.0001. From the contour plot Figure 2C, it was clear that as the compression force increased from 2 ton to 4 ton, the DT increased. There was a direct relationship between compression force and DT of the tablet.
Statistical analysis of DR
Directly compressible tablets were tested for in vitro DR study in saliva pH 6.8 to simulate the condition. All the formulation batches prepared showed >80% of DR within 15 minutes as shown in Figure 3. The DR was found to be in the range of 75.2%–98.95% as shown in Table 4. The polynomial equation for DR is given by:
| (5) |
Figure 3.
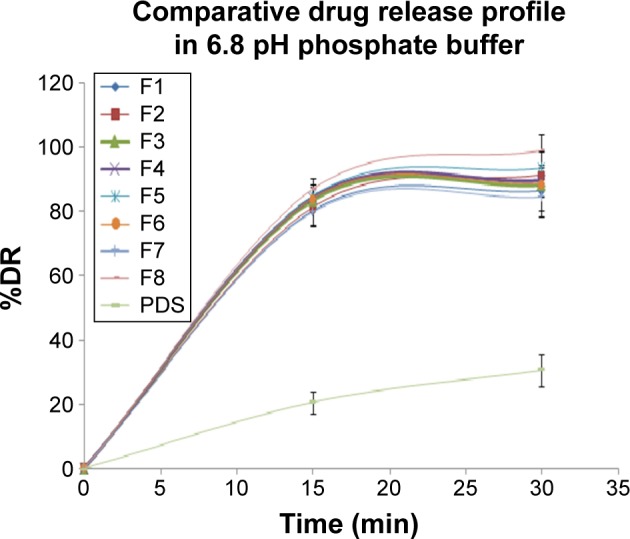
Comparative drug release profile of all tablets containing PDS-loaded CS nanoparticles and pure PDS in 6.8 pH phosphate buffer.
Abbreviations: PDS, prednisolone; CS, chitosan; DR, drug release; min, minute.
The model F-value 120.23 indicates that the model is significant. A positive value in Equation 5 represents the synergistic effect of the independent variable, and a negative value represents the antagonistic effect.14 The suggested model for the Y1 was found to be 2FI as shown in Table 6. The value of the correlation coefficient (R2) for the response Y2 was found to be 0.9890, which indicates the good fit for the 2FI model. The P-value for the Y2 model was found to be 0.0002, which clearly indicates the model is statistically significant. Also among the three independent variables selected, all independent factors A CCS (P<0.0001), B MCC (P=0.0057), and C compression pressure (P=0.0012) are shown in Table 7.
CCS played a vital role for the immediate DR from the tablets by its super disintegrating activity. As the concentration of CCS was increased, the DT was decreased and that resulted in immediate bursting of the tablet and rapid release of the drug from the tablets.
This could also be concluded from the contour plot Figure 4A. This effect was statistically significant. The DR was increased from 75.2% to 98.95%. Due to swelling, it favored the release from the tablets. Due to disintegrant property of CCS, it helped to break the tablet at saliva pH and that resulted in the fast release of the PDS from the tablets. Another most important parameter for the DR enhancement is the utilization of the PDS-loaded CS nanoparticles in the formulation. Due to formation of nanoparticles, the solubility was increased as discussed in the previous section that resulted in the immediate release of the PDS from the tablet. For comparison purpose, the controlled tablet without the PDS-loaded CS nanoparticles was prepared with maintaining other excipient and parameters the same. In that case, it was found that pure PDS tablets showed very less (only 30.65%) DR within 30 minutes. But in the case of tablets prepared from the PDS-loaded CS nanoparticles, all the tablets showed >80% DR within 30 minutes. These results clearly indicated that the nanoparticles had great effect on the enhancement of the dissolution rate of the PDS.
Figure 4.

Counter plots showing effect of independent variables on dependent variables (DR).
Notes: (A) Contour plot showing effect of CCS on the DR of tablets, (B) contour plot showing effect of MCC on the DR of tablets, (C) contour plot showing effect of compression force on the DR of tablets.
Abbreviations: CCS, croscarmellose sodium; DR, drug release; MCC, microcrystalline cellulose.
The effect of MCC 101 was also found to be statistically significant with (P=0.0057). From the contour plot Figure 4B, it could be predicted that as the concentration of MCC 101 was increased from 20% to 40%, the DR profile was decreased. The probable reason for lowering of the DR was same as described in the DT section. Also the effect of the compression force was same like that of the MCC as shown in Figure 4C. It also imparted the lowering of the DR from the tablets.
Commercially variable dosage forms for pediatrics such as tablets, capsules, and oral liquid medications have lot of disadvantages, for example, patient compliance and inability to swallow a dosage form. Pediatric patients find difficulty to swallow the solid oral dosage form such as tablets and capsules, so the patient compliance is the major challenge during the development of the dosage form for such groups of the patients.26 Recently, many researchers aim to enhance safety and efficacy of drug molecule by formulating a most convenient dosage form for administration to pediatrics to achieve better patient compliance. Hence, in order to enhance the patient compliance and rapid effect of PDS in asthmatic patients, ODT were prepared using the CS nanoparticles. Nowadays, such ODT are highly accepted by specific segment of the population including pediatric, geriatric, unconscious and bed-ridden patients who face the difficulty of swallowing solid oral dosage form.27 ODT disintegrate in oral cavity and can be swallowed with a small amount of water or saliva.
Conclusion
ODT of PDS containing CS nanoparticles can be successfully prepared by using direct compression method. This method could be the potential tool for the preparation of ODT with BCS class II molecule. From this study, it can be concluded that the ODT of PDS could be the potential technology for pediatric patients who have swallowing problem of other dosage forms.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Hansen NCG, Evald T, Ibsen TB. Terbutaline inhalations y the turbuhaler® as replacement for domiciliary nebulizer therapy in severe chronic obstructive pulmonary disease. Respir Med. 1994;88(4):267–271. doi: 10.1016/0954-6111(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 2.Zhang X, Liu Q, Hu J, Xu L, Tan W. An aerosol formulation of R-salbutamol sulfate for pulmonary inhalation. Acta Pharm Sin B. 2014;4(1):79–85. doi: 10.1016/j.apsb.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.John BM, Singh D. Comparision of nebulised salbutamol and L-epinephrine in first time wheezy children. Med J Armed Forces India. 2010;66(1):9–13. doi: 10.1016/S0377-1237(10)80083-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu B, Aviya KR, Bhattacharya A. Development and characterization of mouth dissolving tablets of prednisolone. J Pharm Invest. 2014;44:79–102. [Google Scholar]
- 5.Rao VM, Haslam JL, Stella VJ. Controlled and complete release of a model poorly water-soluble drug, prednisolone, from hydroxypropyl methylcellulose matrix tablets using (SBE)7m-b-cyclodextrin as a solubilizing agent. J Pharm Sci. 2001;90:807–816. doi: 10.1002/jps.1034. [DOI] [PubMed] [Google Scholar]
- 6.Elzatahry AA, Mohy Eldin MS. Preparation and characterization of metronidazole loaded chitosan nanoparticles for drugdelivery application. Polym Adv Technol. 2008;19:1787–1791. [Google Scholar]
- 7.Ahlin P, Kristl J, Kristl A, Vrecer F. Investigation of polymeric nanoparticles as carriers of enalaprilat for oral administration. Int J Pharm. 2002;239:113–120. doi: 10.1016/s0378-5173(02)00076-5. [DOI] [PubMed] [Google Scholar]
- 8.Stoltenberg I, Breitkreutz J. Orally disintegrating mini-tablets (ODMTs) A novel solid oral dosage form for pediatric use. Eur J Pharm Biopharm. 2011;78(3):462–469. doi: 10.1016/j.ejpb.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Batchelor HK, Kendall R, Brethes SD, Alex R, Ernest TB. Application of in vitro biopharmaceutical methods in development of immediate release oral dosage forms intended for pediatric patients. Eur J Pharm Biopharm. 2013;85(3 pt B):833–842. doi: 10.1016/j.ejpb.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhary H, Gauri S, Rathee P, Kumar V. Development and optimization of fast dissolving oro-dispersible films of granisetron HCl using Box–Behnken statistical design. Bull Fac Pharm Cairo Univ. 2013;51(2):193–201. [Google Scholar]
- 11.Antoniou J, Liu F, Majeed H, Qi J, Yokoyama W, Zhong F. Physicochemical and morphological properties of size-controlled chitosan– tripolyphosphate nanoparticles. Colloids Surf A Physicochem Eng Aspects. 2015;465:137–146. [Google Scholar]
- 12.Yasser EM, Sami N. Hydrophilic matrices: application of Placket– Burman screening design to model the effect of POLYOX–carbopol blends on drug release. Int J Pharm. 2006;309(1–2):163–170. doi: 10.1016/j.ijpharm.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 13.Papadimitriou S, Bikiaris D, Avgoustakis K, Karavas E, Georgarakis M. Chitosan nanoparticles loaded with dorzolamide and pramipexole. Carbohydrpolym. 2008;73:44–54. [Google Scholar]
- 14.Verma P, Ahuja M. Optimization, characterization and evaluation of chitosan-tailored cubic nanoparticles of clotrimazole. Int J Biol Macromol. 2015;73:138–145. doi: 10.1016/j.ijbiomac.2014.10.065. [DOI] [PubMed] [Google Scholar]
- 15.Yuvaraja K, Khanam J. Enhancement of carvedilol solubility by solid dispersion technique using cyclodextrins, water soluble polymers and hydroxyl acid. J Pharm Biomed Anal. 2014;96(5):10–20. doi: 10.1016/j.jpba.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 16.Bhardwaj S, Jain V, Jat RC, Mangal A, Jain S. Formulation and evaluation of fast dissolving tablet of aceclofenac. Int J Drug Deliv. 2010;2:93–97. [Google Scholar]
- 17.Manivannan R. Oral disintegrating tablets: a future compaction. Drug Invent Today. 2009;1(1):61–65. [Google Scholar]
- 18.Wang W, Chen S, Zhang L, et al. Poly(lactic acid)/chitosan hybrid nanoparticles for controlled release of anticancer drug. Mater Sci Eng C. 2015;46:514–520. doi: 10.1016/j.msec.2014.10.048. [DOI] [PubMed] [Google Scholar]
- 19.Niyas Ahamed MI, Sankar S, Mohammed Kashif P, Hayath Basha SK, Sastry TP. Evaluation of biomaterial containing regenerated cellulose and chitosan incorporated with silvernanoparticles. Int J Biol Macromol. 2015;72:680–686. doi: 10.1016/j.ijbiomac.2014.08.055. [DOI] [PubMed] [Google Scholar]
- 20.Ahmed TA, El-Say KM. Development of alginate-reinforced chitosan nanoparticles utilizing W/O nanoemulsification/internal crosslinking technique for transdermal delivery of rabeprazole. Life Sci. 2014;110(1):35–43. doi: 10.1016/j.lfs.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 21.Hashiguchi T, Yasutake T, Manako T, Otagiri M. In vitro percutaneous absorption of prednisolone derivatives based on solubility parameter. Int J Pharm. 1997;158(1):11–18. [Google Scholar]
- 22.Li XS, Wang JX, Shen ZG, Zhang PY, Chen JF, Yun J. Preparation of uniform prednisolone microcrystals by a controlled microprecipitation method. Int J Pharm. 2007;342(1–2):26–32. doi: 10.1016/j.ijpharm.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 23.El-Malah Y, Nazzal S. Hydrophilic matrices: application of Placket–Burman screening design to model the effect of POLYOX–carbopol blends on drug release. Int J Pharm. 2006;309(1–2):163–170. doi: 10.1016/j.ijpharm.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 24.Elkhodairy KA, Hassan MA, Afifi SA. Formulation and optimization of orodispersible tablets of flutamide. Saudi Pharm J. 2014;22(1):53–61. doi: 10.1016/j.jsps.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattsson T, Sedin M, Theliander H. Filtration properties and skin formation of micro-crystalline cellulose. Sep Purif Technol. 2012;96(21):139–146. [Google Scholar]
- 26.Sevil AY, Cahide Y, Avni K, et al. A case of congenital hypothyroidism presented with dysmyelinization findings. J Acute Dis. 2014;3(1):74–76. [Google Scholar]
- 27.Walsh J, Mills S. Formulating better medicines for children: 4th European Paediatric Formulation Initiative Conference. Ther Deliv. 2013;4:21–25. doi: 10.4155/tde.12.135. [DOI] [PubMed] [Google Scholar]