

Abstract
Amyloidosis refers to a group of widely diverse conditions characterized by the deposition of insoluble protein within the extracellular space, leading to disruption of normal organ function. AL primary amyloidosis is associated with plasma cell dyscrasias and is caused by the deposition of insoluble kappa or lambda light chains. Cardiac involvement by AL primary amyloidosis has a very poor prognosis, and patients are treated with systemic chemotherapy. Clinically, the presence of cardiac amyloidosis in patients with plasma cell disorders is usually presumed to represent AL primary amyloidosis, and they are often managed as such. We reported four cases of elderly patients with plasma cell disorders who were found to have biopsy-proven cardiac senile transthyretin amyloidosis. Our cases demonstrated that cardiac amyloidosis in patients with plasma cell disorders does not necessarily represent AL primary amyloidosis. Cardiac biopsy is important in making the correct diagnosis. Accurate subtyping of the amyloid has significant implications in the management of patients and discussion of prognosis.
INTRODUCTION
Plasma cell dyscrasia results from the abnormal proliferation and expansion of a monoclonal population of plasma cells. In the majority of patients, a monoclonal immunoglobulin protein (M spike) or elevated level of serum free light chains are present. Plasma cell dyscrasias include several related disease processes such as monoclonal gammopathy of undetermined significance (MGUS), multiple myeloma, AL primary amyloidosis, light chain disease, heavy chain disease, POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M protein and skin abnormalities), and Waldenström’s macroglobulinemia. Plasma cell disorders are common in the general population. Multiple myeloma is the second most common hematologic malignancy, and its incidence is approximately 5–7 patients per 100,000 populations. MGUS is considered to be a benign pre-malignant condition that is quite common in the elderly. MGUS occurs in 3.2% of individuals aged 50 years old and its incidence increases with age. At age 70, approximately 5.3% of the general population has MGUS[1].
AL primary amyloidosis is caused by deposition of insoluble fibrils of fragmented immunoglobulin light chain and can involve many different organs. The kidneys are the most commonly involved organ for AL primary amyloidosis. However, it is heart involvement that determines the patient’s outcome. Cardiac amyloidosis can also result from deposition of other fibrils such as serum amyloid A protein (AA amyloidosis), wild-type transthyretin (senile transthyretin amyloidosis) or mutated transthyretin (familial amyloidosis). AL primary amyloidosis and senile transthyretin amyloidosis (ATTR) are the two most common types in cardiac amyloidosis. The clinical presentation and imaging studies are quite similar between cardiac ATTR amyloidosis and cardiac AL primary amyloidosis. The management and the prognoses are markedly different. For AL primary amyloidosis the treatment is chemotherapy, and for healthy individuals with good performance status, high-dose chemotherapy followed by autologous hematopoietic stem cell support is offered. For cardiac ATTR amyloidosis, the mainstay of treatment is supportive care for optimization of cardiac function. Both conditions are considered incurable, but the outcome is quite different. The median survival from the onset of symptoms was 6.07 years in cardiac ATTR amyloidosis patients compared to only 1.7 years in cardiac AL primary amyloidosis patients[2]. Due to the marked difference in management and patient outcome, it is vital to correctly identify the amyloid subtype, and the diagnosis has a major prognostic impact that dictates treatment options.
Similar to plasma cell dyscrasias, senile transthyretin amyloidosis is also a disease of the elderly. The incidence of senile transthyretin amyloidosis in the general population is unknown. Given the high prevalence of MGUS, it is not uncommon to see the coexistence of cardiac ATTR amyloidosis and plasma cell disorders. It was reported that in 24.1% of cardiac ATTR amyloidosis patients there was evidence of plasma cell dyscrasias as an incidental finding [2]. Furthermore, in clinical practice it is a common error to assume AL primary amyloidosis in patients presenting with monoclonal gammopathy and cardiac amyloidosis. In such cases, chemotherapy may be given unnecessarily, and patients may be given a much worse prognosis. We herein presented four cases of cardiac ATTR amyloidosis in patients with plasma cell disorders. As a result of the correct diagnosis being made, chemotherapy was avoided and the prognosis was much more favorable.
CASE PRESENTATION
Case 1
A 79 year-old Caucasian male presented with shortness of breath, orthopnea, anasarca, and a 40 lb. weight gain over three months. His past medical history was positive for pericardial effusion, carpal tunnel syndrome, hypothyroidism, and prostate cancer. Physical examination revealed rales in bilateral lower lobes, irregular heartbeat (EKG confirmed atrial fibrillation), murmur, and generalized edema. Laboratory tests showed hemoglobin 11.0 g/dl, creatinine 1.4 mg/dl, albumin 2.8 g/dl, calcium 8.9 mg/dl, troponin 0.14 ng/ml and B-type natriuretic peptide (BNP) 564 pg/ml. His serum protein electrophoresis (SPEP) and immunofixation (IFE) revealed a monoclonal IgG kappa M-protein at 0.51 g/dl. Twenty-four hour urine protein electrophoresis (UPEP) demonstrated 0.171g/vol protein with IgG kappa M-protein at a level too low to quantitate. Kappa serum free light chain was 2.84 (normal range: 0.33 to 1.94) and lambda serum free light chain was 1.64 (normal range: 0.57 to 2.63) with kappa/lambda ratio at 1.73. Echocardiogram (ECHO) estimated the 2D global left ventricular ejection fraction at greater than 50%. There was concentric left ventricular hypertrophy with increased wall thickness and myocardial mass index. There were echocardiography features suggesting cardiac amyloidosis. The 2D left atrial end-systolic volume index was severely abnormal (>40ml/m2). There was a large circumferential pericardial effusion. Left pleural effusion and ascites were noted. His bone skeletal survey revealed a single lytic lesion on the left proximal humerus near the attachment of the deltoid muscle, which was later determined by MRI to be a non-specific change. Cardiac MRI demonstrated diffuse delayed hyperenhancement encompassing the myocardium of both the left and right ventricles, which was very compatible with cardiac amyloidosis. The patient underwent bone marrow biopsy, fat pad biopsy, and endocardial biopsy. The bone marrow biopsy revealed mild plasmacytosis with scatted CD138+ plasma cells at 5–10%. Congo red staining on the bone marrow biopsy and fat pad biopsy were negative. Endocardial biopsy demonstrated Congo red positive amyloid deposition. The sample was sent to Mayo Clinic for liquid chromatography tandem mass spectrometry (LS/MS) subtyping and the results confirmed transthyretin protein deposition. The patient was diagnosed with senile cardiac ATTR amyloidosis and MGUS. He was treated with aggressive diuresis and optimization of his cardiac medications, and his shortness of breath and anasarca was much improved. For his MGUS, he was followed every 6 months with stable SPEP and serum free light chains. His last follow-up was 2 years from his initial diagnosis and he was well.
Case 2
A 77 year-old Caucasian male was admitted to the hospital with a two-week history of cough, lower extremity edema, shortness of breath, and orthopnea. Before admission, he was seen in the ED and was found to have new onset of atrial fibrillation with wide-complex tachycardia. His past medical history was significant for only hypertension and prostate hypertrophy. Physical examination revealed enlarged jugular vein, scattered rales in bilateral lungs, tachycardia with irregularly irregular rhythm, and 3+ bilateral lower extremity edema. The lab tests returned a white blood cell count 16.88 × 109/ml, hemoglobin 9.5 g/dl, and platelet count 183 × 109/ml; creatinine 2.6 mg/dl, calcium 7.8 mg/dl and albumin 2.2 g/dl; troponin 0.54 ng/ml and BNP 753.7 pg/ml. SPEP and IFE revealed 1.49g/dl IgG Kappa M protein. UPEP also showed the same monoclonal protein in the urine. Kappa serum free light chain level was 4.28, and lambda serum free light chain was 2.25 with the kappa/lambda ratio at 1.5. Echocardiogram showed severe biventricular failure with an estimated left ventricular ejection fraction at 20–30%. The 2D left atrial end-systolic volume index was severely abnormal (>40ml/m2). The right atrial size was dilated based on the end systolic area. The right ventricle was hypokinetic at the base. The bone skeletal survey showed mottled bone lytic lesions in the skull. He underwent cardiac catheterization and cardiac biopsy that was positive for Congo red amyloid. Subtyping of amyloid with LS/MS spectrometry confirmed unmutated transthyretin amyloidosis. His bone marrow biopsy showed light chain restricted plasma cell neoplasm with plasma cells of 14% on the aspirate. Based on the presence of monoclonal plasmacytosis and the evidence of end organ damage such as anemia, renal insufficiency and bone lytic lesions, he was diagnosed with multiple myeloma and cardiac ATTR amyloidosis. He was treated with a combination of bortezomib, cyclosphosphide, and dexamethasone and closely followed by his cardiologist for the optimization of his congestive heart failure management. He was well at 1 year follow-up.
Case 3
A 74 year-old African American male was admitted to the hospital for worsening shortness of breath and abdominal tightness. He had several prior hospitalizations due to coronary artery disease status post CABG and bare metal stent placement, congestive heart failure, atrial fibrillation status post cardioversion, and abdominal aortic aneurysm repair. His past medical history also included chronic kidney disease, chronic anemia, and prostate hypertrophy. Physical examination was relatively unremarkable. His laboratory tests revealed white blood cell count 5.45 × 109/ml, hemoglobin 8.8, and platelet count 257 × 109/ml; creatinine 1.6 mg/dl, calcium 8.9 mg/dl and albumin 3.5 g/dl. His BNP was 1107.5 pg/ml and troponin 9.24 ng/ml. His echocardiogram showed concentric left ventricular hypertrophy with increased wall thickness and myocardial mass index. Speckle tracking tissue Doppler of the left ventricle confirmed the wall motion abnormality. The 2D left ventricular ejection fraction was calculated to be 51%. The myocardial texture was suggestive of amyloidosis. Cardiac MRI demonstrated diffusely increased wall thickness in the left ventricle. There was patchy delayed hyperenhancement in a nonischemic pattern throughout the left ventricle that was primarily mid-myocardium and within the interventricular septum, most consistent with an infiltrative process such as amyloidosis. The patient underwent endocardiac biopsy that revealed Congo red deposition, and LS/MS spectrometry subtyping confirmed ATTR amyloidosis. The patient was also found to have 0.31 g/dl of IgG lambda M protein on SPEP. His UPEP showed 0.293 g/vol of protein with the presence of lambda free light chain. His kappa serum free light chain level was 1.82 and lambda serum free light chain 4.22 with the kappa/lambda ratio at 0.42. He declined further workup for his monoclonal gammopathy. Bone marrow biopsy was not performed.
Case 4
A 69 year-old Caucasian male presented with a 2–3 month history of generalized edema, 30 lb. weight gain, shortness of breath, and orthopnea. His past medical history was significant for asthma, hypertension, hyperlipidemia, erectile dysfunction, type II diabetes, and bilateral carpal tunnel syndrome. Physical exam revealed edema from his lower extremity up to his abdomen, scattered rales in bilateral lungs, irregularly irregular heart rhythm (EKG confirmed atrial fibrillation), and a palpable liver and spleen below the costal margins. His labs showed hemoglobin of 14 g/dl, calcium 9.1 mg/dl, creatinine of 2.0 mg/dl, albumin 3.9 g/dl and beta-2 microglobulin of 4.6. His troponin was 1.16 ng/ml, BNP 148.8 pg/ml and Pro-NT BNP >2800 pg/ml. His SPEP showed a restricted protein band identified by concurrent IFE as monoclonal IgA kappa (the level was too low to quantitate in the beta zone). His UPEP showed two restricted protein bands identified by concurrent IFE as monoclonal IgA kappa and free kappa light chain at 0.5 mg/dL (6.7 mg/TV) and 0.5 mg/dL (6.7 mg/TV) in the beta and gamma zones, respectively. His serum kappa light chain was 6.03 and lambda light chain of 2.67 with the kappa/lambda ratio at 2.26. The echocardiogram showed a left ventricular ejection fraction of 35–40%, severe concentric left ventricular hypertrophy, and sparking myocardial pattern infiltrative cardiomyopathy concerning for amyloidosis. He underwent bone marrow biopsy positive for amyloid deposition with 10% plasma cells that were not light chain restricted. The right ventricular endomyocardial biopsy demonstrated diffuse interstitial and perivascular pink amorphic infiltrates positive for Congo red (Figure 1). Bone marrow biopsy sample was also positive for amyloid deposition (Figure 2). MS Subtyping of both cardiac amyloid and bone marrow amyloid was consistent with unmutated ATTR senile amyloidosis. The patient was diagnosed with senile ATTR cardiac amyloidosis and MGUS. He was managed with cardiac conversion, aggressive diuresis, and optimization of cardiac medications.
Figure 1. Amyloid deposition in the heart (right ventricle endomyocardial biopsy).
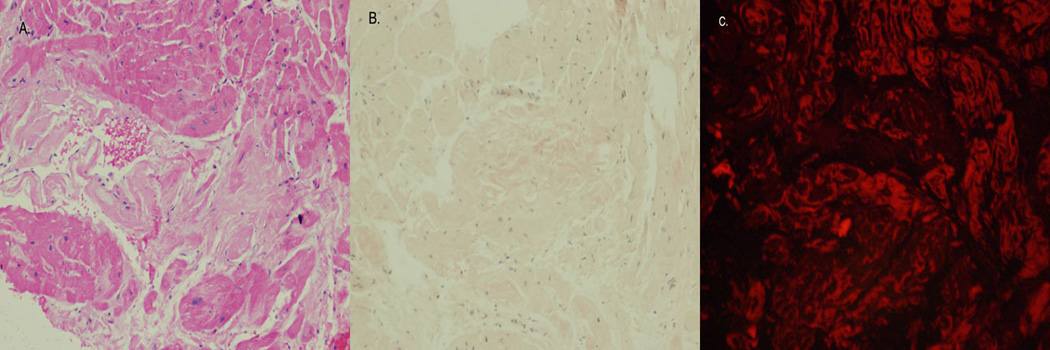
A. Amorphous infiltrate is present between heart muscle fibers (H&E 200×). B. Congo Red stain shows areas of orange-red staining consistent with amyloid infiltrate (H&E 200×). C. Using polarized light microscopy, the infiltrate is diagnostic of amyloid deposition.
Figure 2. Amyloid deposition in the bone marrow (bone marrow biopsy).
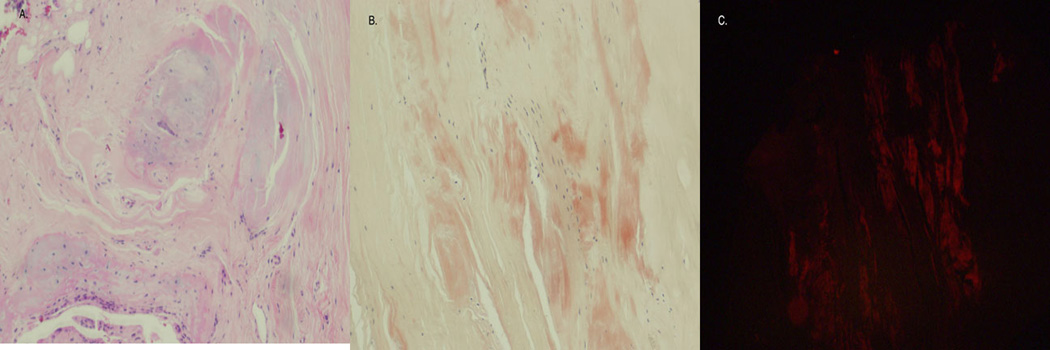
A. the bone marrow biopsy exhibited normal hematopoietic cells (not shown) with extensive fibrous tissue present adjacent to the marrow. (H&E 200×). B. Congo Red stain shows multiple amorphous orange-red deposits within the fibrous tissue. (H&E 200×). C. Polarized light microscopy confirmed amyloid deposition in the fibrous tissue.
DISCUSSION
We presented four cases with co-existing senile cardiac ATTR amyloidosis and plasma cell disorders (please see Table for the summary of patient characteristics and presentation). The main take-home message is the presence of cardiac amyloidosis in elderly patients with plasma cell disorders such as MGUS or multiple myeloma does not necessarily mean the cardiac amyloidosis is AL primary amyloidosis. This recognition is important in clinical practice. Without cardiac biopsy, all four patients could easily have been mistakenly diagnosed with cardiac AL primary amyloidosis and managed incorrectly. For instance, our Case 1 and Case 4 patients were told to prepare for systemic chemotherapy before the cardiac biopsy results were available. Our Case 2 patient was contemplating hospice care because of the presumed stage III cardiac AL amyloidosis in the setting of multiple myeloma. Stage III cardiac AL amyloidosis has a median survival of only a few months[3, 4]. After the diagnosis of senile ATTR cardiac amyloidosis, the patient changed the prospect of his prognosis and agreed to chemotherapy for multiple myeloma. He is now well at 1 year after the diagnosis of cardiac amyloidosis. It is critical to have a cardiac biopsy and confirmatory amyloid subtyping with liquid chromatography tandem mass spectrometry for making the correct diagnosis of cardiac amyloidosis.
Table.
Patient Characteristics and Presentation
| Case 1 | Case 2 | Case 3 | Case 4 | |
|---|---|---|---|---|
| Age (years) | 79 | 77 | 74 | 69 |
| Gender | Male | Male | Male | Male |
|
Presenting symptoms |
Shortness of breath and weight gain |
Shortness of breath and edema |
Shortness of breath |
Shortness of breath and weight gain |
| A-Fib | Present | Present | Present | Present |
|
Troponin (ng/ml) |
0.14 | 0.54 | 9.24 | 1.16 |
| BNP (pg/ml) | 564 | 753.7 | 1107.5 | 148.8 |
|
SPEP (M protein) |
IgG Kappa (0.51g/dl) |
IgG Kappa (1.47g/dl) |
IgG Lambda (0.31g/dl) |
IgA Kappa |
|
Kappa/Lambda ratio |
1.73 | 1.5 | 0.42 | 2.26 |
| ECHO | Concentric left ventricular hypertrophy |
Biventricular failure |
Concentric left ventricular hypertrophy |
Concentric left ventricular hypertrophy |
| Cardiac MRI | Diffuse delayed hyperenhancement |
Not done | Diffusely increased wall thickness |
Diffusely increased wall thickness |
|
Bone marrow biopsy |
Mild plasmacytosis (5– 10% plasma cells) |
Light chain restricted plasma cells (>14%) |
Not done | Plasmacytosis without light chain restriction (10%) |
| Cardiac biopsy | Amyloid deposition |
Amyloid deposition |
Amyloid deposition |
Amyloid deposition |
|
LS/MS typing of amyloid |
Transthyretin (unmutated) |
Transthyretin (unmutated) |
Transthyretin (unmutated) |
Transthyretin (unmutated) |
| Diagnosis | Senile cardiac ATTR amyloidosis and MGUS |
Senile cardiac ATTR amyloidosis and multiple myeloma |
Senile cardiac ATTR amyloidosis and plasma cell dyscrasia |
Senile cardiac ATTR amyloidosis and MGUS |
| Management | Cardiac optimization |
Chemotherapy for myeloma and cardiac optimization |
Cardiac optimization |
Cardiac optimization |
It is noteworthy that all four of our patients presented with cardiac symptoms such as edema, weight gain, shortness of breath, or atrial fibrillation and were seen initially by a primary care physician or cardiologist. ECHO and/or MRI suggested cardiac amyloidosis and the patients had already planned for a myocardial biopsy before hematology/oncology was consulted and the workup for plasma cell dyscrasias was performed. In retrospect, if the patients would have been seen by a hematologist/oncologist and were found to have a plasma cell dyscrasia (MGUS or myeloma) and evidence of cardiac amyloidosis by ECHO or MRI, it is very likely they may have been diagnosed with AL primary cardiac amyloidosis. The patients would have been treated with chemotherapy and given a very dismal prognosis. Should all patients with MGUS or myeloma and evidence of cardiac amyloidosis undergo cardiac biopsy for accurate subtyping of the amyloid? The cases presented here suggested that we should consider cardiac biopsy for patients with plasma cell dyscrasia and evidence of cardiac amyloidosis before we commit them to systemic chemotherapy for presumed “AL primary amyloidosis”. Cardiac biopsy is an invasive procedure and carries certain risks; however, systemic chemotherapy is toxic and also has many side effects. We believe the benefit of accurate diagnosis from cardiac biopsy outweighs the risks of toxicities from unnecessary chemotherapy.
Cardiac biopsy is the only way to unequivocally make the diagnosis. There are some clinical, imaging, and laboratory characteristics that may assist in distinguishing senile ATTR cardiac amyloidosis from AL primary cardiac amyloidosis. For instance, senile ATTR has a male predominance (89% of the patients were male with a median age at presentation of 73 years old). In AL primary cardiac amyloidosis, male patients account for 69% and the median age at presentation was 63 years old[2]. The presenting symptoms are similar in both groups, but they are more severe in AL cardiac amyloidosis. Atrial fibrillation occurs more frequently in senile ATTR cardiac amyloidosis. Carpal tunnel syndrome is more common in senile ATTR cardiac amyloidosis patients (49%) than in AL primary cardiac amyloidosis patients (8.3%). In the study with 102 senile ATTR cardiac amyloidosis patients and 36 AL primary cardiac amyloidosis patients, evidence of a plasma cell dyscrasia was observed in all AL primary cardiac amyloidosis patients, but only in 24% of senile ATTR cardiac amyloidosis patients[2]. This finding suggested that it is unlikely to have AL primary cardiac amyloidosis without evidence of a plasma cell dyscrasia, but it is possible to have senile ATTR cardiac amyloidosis in the presence of a plasma cell dyscrasia.
Magnetic resonance imaging (MRI) is being increasingly used for the diagnosis of cardiac amyloidosis. MRI is generally considered not specific enough to differentiate various subtypes of amyloid. Recently, it was suggested that transmural and right ventricular late gadolinium enhancement (LGE) patterns on cardiac magnetic resonance (CMR) imaging were more characteristic of senile ATTR cardiac amyloidosis [5]. NT-pro-BNP was elevated in cardiac amyloidosis[6]. Recently, Pinney et al found that NT pro-BNP and age at diagnosis were both associated with risk of senile ATTR cardiac amyloidosis. They developed a diagnosis algorithm incorporating the level of NT pro-BNP and age at diagnosis for distinguishing senile ATTR cardiac amyloidosis from AL primary cardiac amyloidosis[2]. They also found that patients ≤70 years old with an NT pro-BNP < 183 pmol/L and patients >70 years old with an NT pro-BNP <1420 pmol/L were more likely to have senile ATTR cardiac amyloidosis. The positive predictive value using this combination of age and NT pro-NBP was 90% and negative predictive value was 85%. We did not have the level of NT pro-NBP for our Case 1, 2, and 3. For Case 4, this model of prediction did not work. The patient was 69 years old and had a significantly elevated NT pro-BNP (2800 pg/ml, that is, 326 pmol/L), and should be less likely to be senile ATTR cardiac amyloidosis. Again, our cases further underscored the importance of cardiac biopsy for definitive diagnosis and accurate subtyping of cardiac amyloidosis.
Biopsy followed by Congo red staining and immunohistochemistry used to be considered the gold standard for diagnosing and typing amyloid. Recently, Satoskar et al retrieved 229 native endomyocardial biopsies[7]. There were eight cases that showed strong transthyretin staining by immunohistochemistry. Mass spectrometry was performed in five out of these eight biopsies and all five cases revealed AL type amyloidosis. These data suggested a strong false positive immunostaining for transthyretin in cardiac amyloid. We have not routinely used immunohistochemistry staining for the diagnosis or subtyping of cardiac amyloidosis. All of our samples were sent to Mayo Clinic for LS/MS spectrometry subtyping.
For AL primary cardiac amyloidosis, the patients can be treated with supportive care, systemic chemotherapy, and autologous hematopoietic stem cell transplant. Currently there is no effective FDA-approved treatment for senile ATTR cardiac amyloidosis. Heart transplant was reported in extremely rare cases[8]. Several novel agents such as diflunisal and Fx-1006A to stabilize the native state of transthyretic tetramer are in phase II/III clinical trial[9]. More recently, antitransthyretin small interfering RNA was packaged in lipid nanoparticles and administered to 32 patients with transthyretin amyloidosis patients. The treatment led to a reduction of transthyretin level up to 67% at 28 days[10]. Several clinical trials are actively recruiting patients, and it is expected that new drugs are on the horizon. Currently, the mainstay of management for senile ATTR cardiac amyloidosis is to optimize supportive cardiac care including diuresis.
In summary, our cases demonstrated the importance of cardiac biopsy for the accurate diagnosis of cardiac amyloidosis. Correctly identifying the type of amyloid is vital and dictates treatment and prognosis.
ACKNOWLEDGEMENT
This work was supported by MUSC Hollings Cancer Center Startup Fund, Hollings Cancer Center ACS IRG, ASCO Conquer Cancer Foundation Career Development Award, NIH 1K08HL 103780-01A1, and NIH 3P30CA138313-01S3. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other funding agents.
Footnotes
CONSENT
Consents were obtained from the patients for publishing these data.
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHORS’ CONTRIBUTION
Ms. Roof participated in the data collection and the writing of the manuscript. Dr. Lazarchick is the hematopathologist. Drs. Coker and Kang participated in the care of the patients. All the authors wrote and approved the manuscript.
REFERENCES
- 1.Kyle RA, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362–1369. doi: 10.1056/NEJMoa054494. [DOI] [PubMed] [Google Scholar]
- 2.Pinney JH, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):e000098. doi: 10.1161/JAHA.113.000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dispenzieri A, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol. 2004;22(18):3751–3757. doi: 10.1200/JCO.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 4.Dispenzieri A, et al. Prognostication of survival using cardiac troponins and Nterminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2004;104(6):1881–1887. doi: 10.1182/blood-2004-01-0390. [DOI] [PubMed] [Google Scholar]
- 5.Dungu JN, et al. CMR-based differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):133–142. doi: 10.1016/j.jcmg.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 6.Damy T, et al. Role of natriuretic peptide to predict cardiac abnormalities in patients with hereditary transthyretin amyloidosis. Amyloid. 2013;20(4):212–220. doi: 10.3109/13506129.2013.825240. [DOI] [PubMed] [Google Scholar]
- 7.Satoskar AA, et al. Strong transthyretin immunostaining: potential pitfall in cardiac amyloid typing. Am J Surg Pathol. 2011;35(11):1685–1690. doi: 10.1097/PAS.0b013e3182263d74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pepys MB. Amyloidosis. Annu Rev Med. 2006;57:223–241. doi: 10.1146/annurev.med.57.121304.131243. [DOI] [PubMed] [Google Scholar]
- 9.Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14(30):3219–3230. doi: 10.2174/138161208786404155. [DOI] [PubMed] [Google Scholar]
- 10.Coelho T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]