

Abstract
Cathepsin B is a member of the papain family of cysteine proteases normally present in the lysosome, but it can translocate and function to degrade components of the extracellular matrix. It exhibits carboxyopeptidase, peptidyldipepidase, and endopeptidase activity. Aberrant overexpression of cathepsin B has been reported in invasive and metastatic cancers, including breast cancer, melanoma and colorectal cancer. It has been shown that oncogenic activation, such as the signaling of the ErbB pathways, can lead to cathepsin B overexpression. The degradation of the extracellular matrix is a key factor for cathepsin B to contribute to development and metastasis of tumors. An example of substrates for cathepsin B is E-cadherin, which is involved in adherens junctions, and the downregulation of E-cadherin in cancer is directly linked to invasion and metastasis. Recent studies also point to a role for cathepsin B in macrophages in the tumor microenvironment. The structure of cathepsin B is crystallographically solved, and several highly selective and potent inhibitors for cathepsin B have been developed. Yet it remains to be a challenge to demonstrate the clinical utility or benefit of any cathepsin B inhibitor. As cathepsin B is required for a cellular process called lysosomal membrane permeabilization (LMP), inhibition of cathepsin B would protect cancer cells from cell death induced by chemotherapeutic agents. It is expected that combining cathepsin B inhibitors with other approaches, such as nanoparticles, to direct the inhibition to the extracellular space may lead to better clinical approaches to treat cancers and metastasis.
1. Introduction
Cathepsin B belongs to the cathepsin family of lysosomal hydrolases. According to their active site amino acid, cathepsins can be divided into three sub-groups: cysteine (B, C, H, F, K, L, O, S, V, W and X/Z), aspartate (D and E) and serine (G) cathepsins (Rawlings et al., 2012). The lysosome relies on these protein hydrolases and other enzymes to carry out intracellular degradation before recycling cellular constituents. In addition to its localization in the lysosome, cathepsin B can be released from the cell and function to degrade components of the extracellular matrix (Sloane, 1990). Overexpression of cathepsin B has been observed in malignant tumors and has been found to be closely correlated with an array of cancers (invasive and metastatic). Aberrant regulation of cathepsin B can lead to amplified degradation of the extracellular matrix, and thereby attributes to the infiltrative nature of tumor cells. Cathepsin B is also found to participate in various intracellular processes such as autophagy and immune response.
2. Structure and Functions of Cathepsin B
The human cathepsin B gene is located on chromosome 8p22 and contains 12 exons (Fong et al., 1991). Cysteine cathepsins are synthesized as inactive precursors. For pro-cathepsin B, it has a N-terminal domain to cover the active site and binding sites (Figure 1A). Pro-cathepsins are normally activated in the acidic environment of lysosomes, where they are initially believed to function primarily as intracellular proteases that mediate proteolysis (Turk et al., 2001). The matured cathepsin B composes of a heavy chain of 25–26kDa and a light chain of 5kDa (Frlan and Gobec, 2006). Cathepsin B is different from other cathepsins with unique enzyme characteristics. Most cysteine cathepsins are endopeptidases, whereas cathepsin B has both endopeptidase and carboxyopeptidase activity (Turk et al., 2001).
Figure 1.

Crystal structures of pro-cathepsin B and cathepsin B. (A) The crystal structure of pro-cathepsin B (PDB:1MIR) (Cygler et al., 1996). The 62-residue pro-peptide region (magenta) is folded along the surface of mature cathepsin B (cygans) and covers the active site cleft. (B) The crystal structure of cathepsin B (PDB:1HUC) (Musil et al., 1991). The occluding loop (108–122) is shown in magenta on the surface of cathepsin B (orange). Two cysteine residues from the occluding loop form a di-sulfide bridge. Residues Cys29 and His199 form the catalytic dyad, and two histidines, His110 and His111, are positioned within the active site cleft. These functional residues are represented as green ball-and-stick model on the molecular surface model of cathepsin B. The S1, S2, S3 and S2′ active pocket are highlighted with bold labeling. The figure is prepared with PYMOL using reported structures in PDB.
Like other cysteine cathepsins, cathepsin B shares a conserved active site that is formed by cysteine (Cys29), and histidine (His199) residues (Figure 1B). The substrate binding cleft exists next to the active site, which is controlled by the occluding loop, an 18 residue long insertion. In addition, the occluding loop contains two His residues (His110 and His111) that can interact with the C-terminus carboxylic group of the substrate peptide and facilitate the access of substrate into the active site (Mohamed and Sloane, 2006). The interaction between the two His residues and the carboxylate group explains the carboxy dipeptidase activity of cathepsin B at an acidic pH (Mohamed and Sloane, 2006). The flexible nature of the occluding loop allows for cathepsin B to act as an endopeptidase as well when the occluding loop moves from the active site cleft and cleaves internal peptide bonds (Mort and Buttle, 1997). Cathepsin B, however, is less effective as an endopeptidase compared to other proteases in the papain family due to the large amount of energy needed to alter the conformation of the occluding loop (Cygler et al., 1996).
3. Overexpression of Cathepsin B in Cancers
Recent studies have revealed that cysteine cathepsins have profound functions beyond the protein turnover in normal cells and play roles in the development of heart, brain and skin (Reinheckel et al., 2001), bone resorption (Saftig et al., 1998) and antigen presentation (Shi et al., 1999).
Cathepsin B is produced constitutively, which classifies this protease as a housekeeping protein. However, it is highly upregulated in malignant tumors. Tumor cells are reported to first produce pro-cathepsin B, which is secreted but tethered to the cell surface in a Ca2+ dependent manner via the Annexin II tetramer complex (Mai et al., 2000). Once pro-cathepsin is converted to cathepsin B, it no longer binds to Annexin II and is released into the tumor microenvironment.
Overexpression of cathepsin B has been linked to breast, cervix, bladder, stomach, colon, ovary, bladder, lung, prostate, and thyroid cancers (Kuester et al., 2008). Levels of cathepsin B and C were significantly higher in cystic fluid of malignant ovarian tumors (Kolwijck et al., 2010). Many studies have demonstrated the association of elevated levels of cathepsin B with enhanced angiogenesis, invasion and metastasis. In colorectal carcinomas, elevated expression of cathepsin B in the tumor epithelial cells was associated with a significantly shorter survival of the patients (Campo et al., 1994). Expression of cathepsin B is usually the strongest in the advancing edge of tumors but is also detectable in stromal cells and normal epithelial cells adjacent to the tumors (Hirai et al., 1999).
By manipulating the cathepsin B expression levels with either overexpression or antisense oligos, Szpaderska et al. observed a correlation between cathepsin B levels and invasiveness of several tumor cell lines (Szpaderska and Frankfater, 2001). In a mouse model of pancreatic islet cell carcinogenesis, cathepsin B was upregulated together with several other cathepsins (cathepsin L, S, and C), and a significant association between increased levels of cathepsins B and L and tumor malignancy was observed (Gocheva et al., 2006). Conversely, inactivation of cathepsins B by mutation or knockout impaired tumor growth. Cathepsin B deficient mice demonstrated reduced tumor vascularity, increased tumor cell death, lowered cell proliferation and impaired tumor invasion. RNAi of cathepsin B was shown to reduce glioma cell invasion and angiogenesis both in vitro and in vivo (Gondi et al., 2004). In a study of IL-8 mediated endothelial cell migration, cathepsin B was found to be critical for IL-8 induced transactivation of EGFR (Schraufstatter et al., 2003).
We have detected significantly elevated serum cathepsin B levels in breast cancer patients (n=24) as compared with healthy controls (n=13) (Figure 2, p = 0.015). This observation is consistent with other reports on overexpression of cathepsin B in the cytosol of breast cancer tissue specimens and the correlation between higher cathepsin B content and risk of recurrence in breast cancer (Thomssen et al., 1995). It is not clear at this point if serum cathepsin B levels correlate with disease stages and metastasis status.
Figure 2.
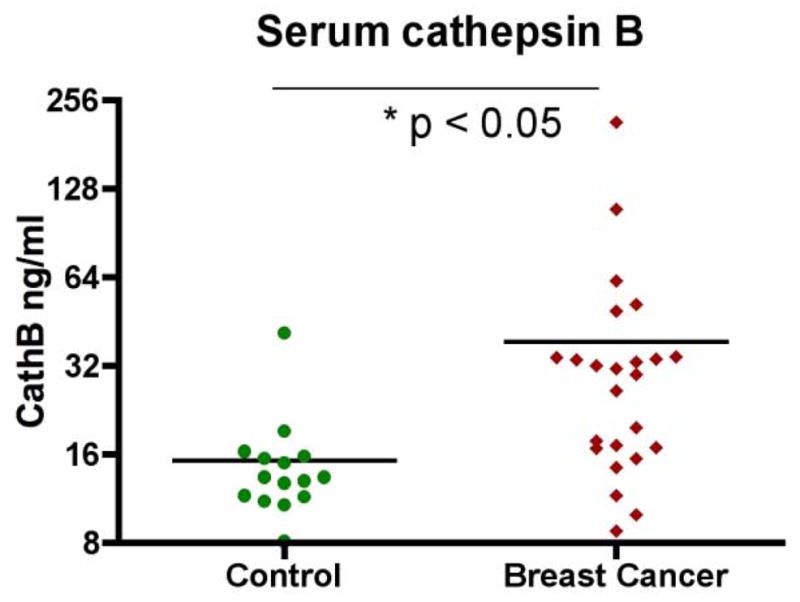
Serum levels of cathepsin B are elevated in breast cancer patients. Cathepsin B levels from both healthy volunteers (control) and breast cancer patients were determined by ELISA. The average levels of cathepsin B in breast cancer patients is significantly higher than that in the control (p < 0.05).
4. Activation of Cathepsin B in Malignant Cells
As studies have indicated that transformation of cells leads to changes in lysosomal activity, which include increases in lysosomal volume and protease activity, it is speculated that the expression of cathepsins such as cathepsin B is regulated by oncogenic pathways (Kirkegaard and Jaattela, 2009). Endogenous cathepsin inhibitors, such as stefin A (Chambers and Tuck, 1993), can also be regulated by oncogenes in an opposite way, but here we will only focus on the regulation of cathepsin B.
4.1. Activation of Cathepsin B by HER2 Signaling
Her2/neu has been implicated in the development of tumors and now the tyrosine kinase receptor has been validated as a clinical target for breast and stomach cancers (Zhang et al., 2007). Studies have shown that cathepsin B as well as cathepsin L are elevated in HER2 positive primary human breast cancer (Rafn et al., 2012). This outcome is closely related to the invasiveness of HER2-tumors. In the same study, it is also demonstrated that Cdc42-binding protein kinase beta, extracellular regulated kinase 2, p21-activated protein kinase 4, and protein kinase C alpha are essential mediators of HER2-induced cathepsin expression and breast cancer cell invasiveness. The HER2 signaling leads to the activation of myeloid zinc finger-1, which is a transcription factor that can bind to a HER2-responsive enhancer element in the first intron of the cathepsin B gene (CTSB)(Rafn et al., 2012).
HER2 and EGFR belong to the same family of receptor tyrosine kinases, and HER2 has been involved to form heterodimers with EGFR to mediate cell growth (Kokai et al., 1989). In CXCR2 positive microvascular endothelial cells (HMECs), EGF signaling is critical for IL-8-mediated cell migration (Schraufstatter et al., 2003). This process is determined to be highly dependent on the activation of cathepsin B, as inhibition of cathepsin B ablates the IL-8 effect on migration. These studies indicate that cathepsin B could be a good target to reverse oncogenic cellular changes that are induced by HER2/EGFR signaling.
4.2. Cathepsin B and Resistance to Chemotherapies
There is evidence that non-tumor cells in the tumor microenvironment can produce cathepsin B and contribute to invasion and metastasis. Indeed, in some highly invasive tumors, large numbers of myeloid cells (Gr-1+CD11b+) that secret cathepsin B are found at the leading edge of tumor margins (Shchors et al., 2013). In a mammary tumor model, treatment with chemotherapeutic agent paclitaxel (Taxol) increased macrophage infiltration into tumors (Shree et al., 2011). These tumor-associated macrophages (TMAs) expressed cathepsin B and protected against Taxol-induced tumor cell death in co-culture. These macrophages also protect tumor cells against death induced by other chemotherapeutics, specifically etoposide and doxorubicin. The tumor-protective activity of these macrophages was dependent on cathepsin, as it can be fully reversed by cathepsin inhibition (Shree et al., 2011).
In addition, it is reported that chemotherapeutic agents (e.g. gemcitabine and 5-fluorouracil) trigger lysosomal permeabilization in myeloid-derived suppressor cells (MDSCs) and the release of cathepsin B directly activates the Nlrp3 inflammasome to produce active caspase-1 (Bruchard et al., 2013). As a result, MDSCs secret IL-1β and limits the anti-tumor activity of these chemotherapies.
5. Substrates of Cathepsin B in the Extracellular Matrix
5.1. Cleavage Sites of Cathepsin B
Using a proteomic approach called “proteomic identification of protease cleavage sites” (PICS), Biniossek and colleagues have studied the cleavage sites of cathepsin B as an endopeptidase and compared the motif with that of cathepsins L and S (Biniossek et al., 2011) (Figure 3). The enzyme cleaves between the P1 and P1′ residues. According to the PICS study, cathepsin B prefers a glycine residue at the P3′ as well as the P1 position, and a phenylalanine at the P1′ position. It is noted that cathepsin B differs from cathepsin L and S by preferring an aromatic residue at the P1′ position.
Figure 3.
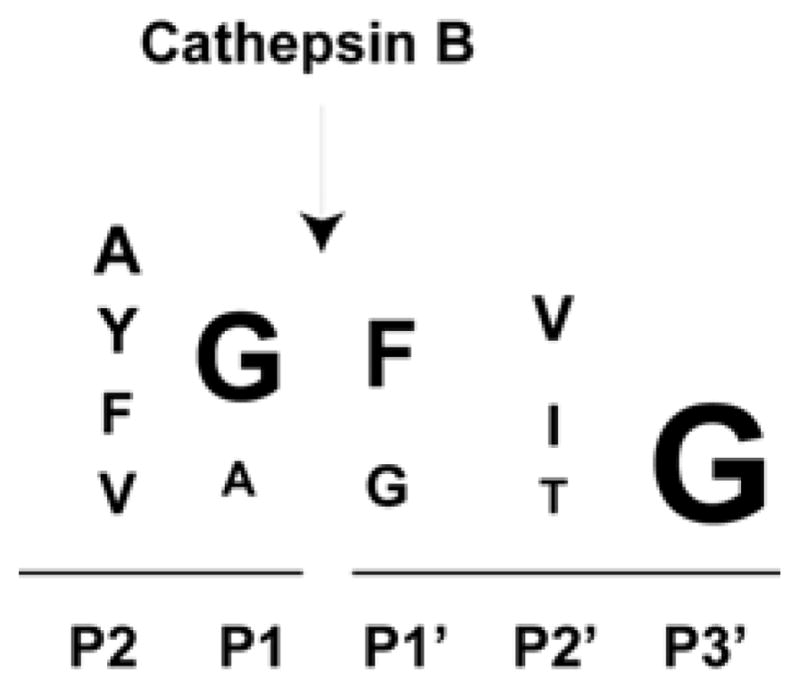
Motifs for cathepsin B substrates. The cathepsin B substrate peptide motifs based on the PICS study (Biniossek et al., 2011) are shown.
5.2. Active Roles of Cathepsin B in Tumor Microenvironment
Increased expression of proteinases or a decrease in levels of proteinase inhibitors have been thought to facilitate the ability of tumor cells to metastasize and invade tissues (Campo et al., 1994). In addition to degrading the basement membrane during extravasation and intravasation, proteinases can also detach cells from the primary tumor and allow the tumor cells to invade surrounding as well as remote tissues via vascular channels. Several studies have shown that cathepsin B staining is inversely correlated with basement membrane staining in lung, bladder, gastric, and colon carcinomas (Tan et al., 2013). This indicates that the confinement of tumor cells by the basement membrane could be broken by activation of cathepsin B and other proteases.
As stated above, overexpression and translocation of cathepsin B to the periphery of the tumor cell can degrade components of the extracellular matrix, mainly laminin, fibronectin, and collagen IV (Buck et al., 1992). In addition, studies have identified several critical substrates for cathepsin B that are particularly relevant to tumor invasion and metastasis.
E-cadherins as Cathepsin Substrates
The N-terminal extracellular domains of E-cadherin proteins on adjacent cells bind to each other in a homophilic manner to establish cell-cell junctions (Beavon, 2000). E-cadherin has been identified as an epithelial marker and its expression is greatly downregulated during the epithelial-mesenchymal transition, a critical process in embryogenesis, and in breast cancer stem cells (Mani et al., 2008). Thus, proteolysis of its extracellular domain represents an alternative to the well-documented mechanisms of mutational inactivation or transcriptional downregulation as a means of losing E-cadherin function. Several proteases have been shown to disable E-cadherin function by proteolysis: MMP3, MMP7, ADAM10 and plasmin (Lochter et al., 1997; Noe et al., 2001; Ryniers et al., 2002; Maretzky et al., 2005). Recently, Gocheva et al. identified E-cadherin as a target substrate of cathepsin B, L, and S, but not C. At the invasive tumor front of the islet tumor, cathepsin B localization correlated with downregulation of E-cadherin (Gocheva et al., 2006).
Pro-urokinase Plasminogen Activator (pro-uPA)
Cathepsin B and pro-uPA co-localize in the caveolae of tumor cells. Cathepsin B can activate pro-uPA to become uPA. Once activated, uPA converts plasminogen to plasmin, which subsequently activates MMPs. Downregulation of caveolin-1, the main structure protein of caveolae, leads to reduced secretion of both procathepsin B and pro-uPA (Cavallo-Medved et al., 2005). Meanwhile, downregulation of caveolin-1 also reduces degradation of the extracellular matrix protein collagen IV and the invasion of these cells through Matrigel.
Tissue inhibitors of Metalloproteinases (TIMP)
As indicated by their names, TIMPs are inhibitors of matrix metalloproteinases (MMPs) and can prevent degradations of extracellular matrix. Cathepsin B has been shown to deplete the tissue inhibitors of matrix metalloproteinases TIMP-1 and TIMP-2 (Kostoulas et al., 1999) and thus will lead to higher MMP activity to promote cell invasion. In addition, TIMPs are also angiogenesis inhibitors, and cathepsin B and MMPs are found at sites of neovascularization in cancer as well as in osteoarthritis (Kostoulas et al., 1999). By degrading TIMPs, cathepsin B raises the activity of the MMPs, even in the absence of over-production of these enzymes, and stimulates angiogenesis. This will prepare blood vessels for invasion in cancer development and metastasis. However, the exact mechanism of each TIMP is not clearly defined since a reverse correlation of TIMP-1/TIMP-2 with MMPs was not observed in several reports (Naka et al., 2008; Sameni et al., 2008). In a prostate cell line, increased secretion of TIMP-3, but not TIMP-1 and TIMP-2, was observed when β1-integrin was knocked down and extracellular matrix degradation was decreased (Sameni et al., 2008).
6. Cathepsin B Inhibitors
Current cathepsin inhibitors are mostly active site inhibitors derived from epoxysuccinyl (Murata et al., 1991), vinyl sulfone or nitrile based compounds (Vasiljeva et al., 2007; Mirkovic et al., 2011). For cathepsin B, both epoxysuccinyl and nitrile inhibitors with potent activity have been identified, while peptide vinyl sulfone inhibitors in general have poor activity and selectivity. E-64 is an irreversible epoxysuccinyl-based inhibitor with broad-spectrum inhibitory activity to cathepsin and other proteases such as calpains. JPM-OEt is structurally related to E-64 but with more specific activity to cathepsins. This pan-cathepsin inhibitor has been shown to enhance chemotherapy regimens in a multistage cancer mouse model (Bell-McGuinn et al., 2007). CA-074, a specific inhibitor for cathepsin B, is also derived from the E-64 scaffold (Towatari et al., 1991). A methyl ester derivative of CA-074, CA074Me, was further developed to improve cell permeability. However, CA074Me has compromised cathepsin B selectivity and shows good inhibition against cathepsin L (Montaser et al., 2002; Mihalik et al., 2004).
The complex crystal structure of human cathepsin B with a dipeptidyl nitrile inhibitor (compound 3) was first obtained (Greenspan et al., 2001). Compound 3 had an IC50 of 45nM for cathepsin B and was an improvement after molecular modeling from an initial inhibitor, Compound 19, which had a weak IC50 of 62 μM. The high-resolution 1.9 Å crystal structure demonstrated that the nitrile inhibitor binds the enzyme at Cys29 in the active site by forming a reversible thioimidate ester intermediate. An analysis of the co-crystal structure led to the addition of a carboxylated aromatic ring (Compound 39) to form a salt-bridge with His110 in the S2′ pocket of the enzyme, which improved the IC50 to 1.8 nM. Removing a bulky ring from the inhibitor close to Tyr75 in the S3 pocket of the enzyme (Compound 10) barely had any effect on inhibitory activity against cathepsin B but produced ~100 fold selectivity over other cathepsins (Greenspan et al., 2001).
Epoxysuccinyl inhibitors have also been co-crystalized with cathepsin B and the crystallographic studies confirm the binding of inhibitors to the active site. This family of inhibitors, such as E64c (Yamamoto et al., 2002), CA030 (Turk et al., 1995), CA074 (Yamamoto et al., 2000), and NS134 (Stern et al., 2004), are irreversible inhibitors derived from the natural compound E64. These inhibitors form a covalent bond with the SH group of Cys29 in cathepsin B. Comparison of the potency of these epoxysuccinyl inhibitors also reveals that binding to both S1′ and S2′ sites in the enzyme is required for a potent inhibition (IC50: 20–30 nM) (Watanabe et al., 2006). CA074 (Figure 4) has a very high selectivity for cathepsin B over other enzymes since its free carboxylic acid can interact with the two histidine residues in the occluding loop structure.
Figure 4.
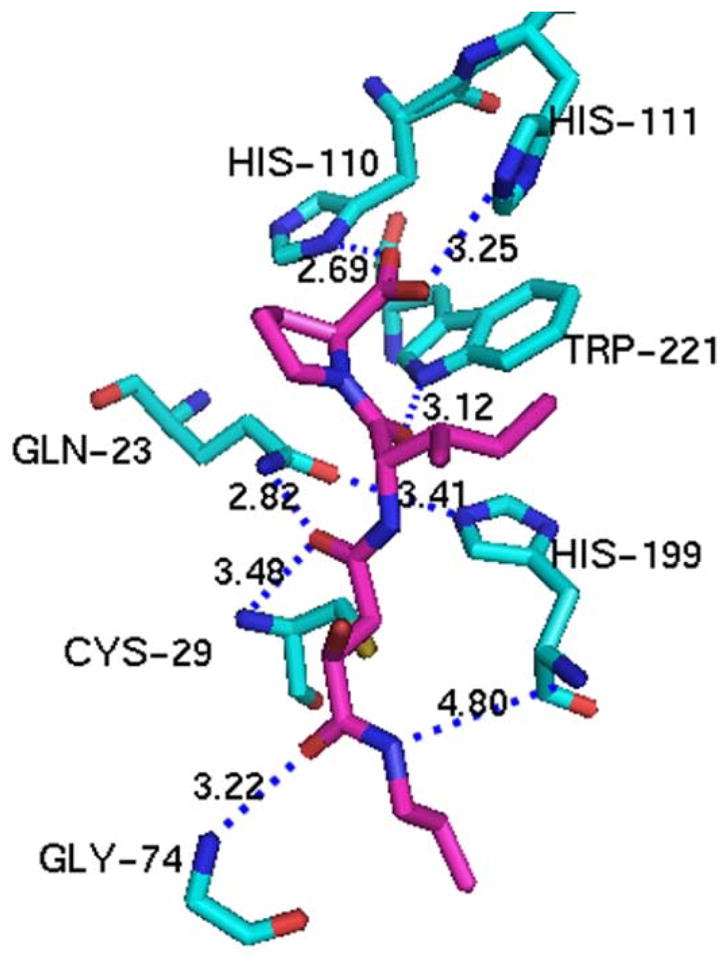
Interaction of CA-074 with cathepsin B. The crystal structure of the CA-074 and cathepsin B complex (PDB: 1QDQ) has revealed the mode of binding of CA-074 (magenta) to the active site (cygans) of cathepsin B. The side chain of interacting residues are labeled using the stick model. Interatomic distances (less than 3.5 Å) between the functional groups of CA-074 and cathepsin B active sites are shown as dashed blue lines. Double hydrogen bonding between the two carboxyl oxygen atoms of CA-074 and the nitrogen atoms of His110 and His111 of the occluding loop are important for cathepsin B-specific inhibition of CA-074. The figure was prepared with PYMOL based on published structures in PDB (Yamamoto et al., 2000).
7. Cathepsin B as a Target for Cancer Therapies
Although a variety of cathepsin B inhibitors have been developed and investigated for the inhibition of tumor invasion to treat different cancers, none has yet succeeded in demonstrating clinical evidence to treat cancers. However, what has been learned from the use of inhibitors to block cathepsin B function is still valuable and will provide insights to the development of clinically useful agents.
As cathepsin B has been confirmed to play a vital role in invasion and metastasis, specific cathepsin B inhibitors will be more valuable for late stage cancers. In an immunocompetent model of breast cancer with spontaneous bone metastasis, intraperitoneal administration of the highly selective cathepsin B inhibitor CA-074, but not the broad spectrum cysteine cathepsin inhibitor JPM-OEt, reduced metastasis in tumor-bearing animals (Withana et al., 2012).
Cathepsin B inhibitors may also help potentiate current chemotherapies. Since cathepsin B was reported to protect tumors against cell death induced by chemotherapeutic agents such as Taxol, combination of Taxol and cathepsin inhibitors in vivo significantly enhanced efficacy against primary and metastatic tumors (Shree et al., 2011).
For cathespin B inhibitors, there have been two issues that hamper the advancement of clinical application. One is the specificity of the compound. The other is the complex role of cathepsin B in cancer treatment. As we mentioned above, advances have been made for the first issue after the atomic resolution of cathepsin B crystal structures were solved in complex with its inhibitors. For the second issue, recent studies have shed light on this problem and are leading towards more promising ways to use inhibitors in cancer treatment.
It turns out cathepsin B is required for a cellular process called lysosomal membrane permeabilization (LMP). LMP could be induced by a variety of reagents including TNF and chemotherapeutic agents and will lead to apoptosis as well as necrosis of cells. In cathepsin B deficient hepatocytes, TNF fails to induce LMP (Groth-Pedersen and Jaattela, 2013). Pharmacological and genetic inhibition of cathepsin B are also able to rescue WEHI-S murine fibrosarcoma cells from apoptosis triggered by TNF or TRAIL (Foghsgaard et al., 2001). These studies demonstrate that cathepsin B is essential for TNF-induced LMP. In addition, in non-small lung cancer cells, blocking of cathepsin B activity can also prevent the appearance of multinucleated cells, which is an early characteristic of cell death induced by microtubule-disturbing agents. Furthermore, cathepsin was also shown to protect cancer cells against paclitaxel induced cell death (Broker et al., 2004).
Since cathepsin B plays a critical role in LMP (Kirkegaard and Jaattela, 2009) induced cell death, complete inhibition of cysteine cathepsin activity in cancer treatment may accidentally interfere with drug-induced lysosomal cell death and result in poorer responses to therapies. A better cathepsin B targeting strategy is to limit the inhibitor effect to the extracellular space or prevent lysosomal exocytosis.
7.1. Nanoparticle or Liposome Inhibitors for Cathepsin B
Mikhaylov and colleagues reported a cathepsin B inhibitor conjugated to a highly biocompatible liposomal nanocarrier (Mikhaylov et al., 2014). This inhibitor, termed LNC-NS-629, is designed to target extracellular cathepsin B from tumor and stromal cells in the tumor microenvironment. The lipidated inhibitor NS-629 can form a covalent bond with the catalytic Cys29 of cathepsin B, thereby this nanoparticle is also an irreversible inhibitor. Treating in vivo mammary tumor model, LNC-NS-629 shows clear strong staining on tumor cells and stromal cells. The staining in the macrophages was much stronger, possibly due to the capacity of phagocytosis in this type of cells. The staining on tumor cells was dominantly pericellular, confirming the selective targeting of extracellular cathepsin B (Mikhaylov et al., 2014). Most importantly, LNC-NS-629 was able to function as a drug delivery system. When doxorubicin was encapsulated in LNC-NS-629, its potency against tumor cells was improved 22-fold (Mikhaylov et al., 2014).
8. Pro-drugs that Utilize Cathepsin B Activity
Doxorubicin is a popular and effective anticancer drug used to treat vast numbers of solid tumors. In its salt form, doxorubicin can disseminate into tissues and intracellular compartments through either passive diffusion or active transport (Zhong, et al., 2012). Doxorubicin must be administered at low doses due to its indiscriminative toxic effects on cells, which include but are not limited to bone marrow toxicity, cardiotoxicity, nephrotoxicity, and hepatotoxicity. Pro-drugs, derivatives of drugs that are inactive except when metabolized at the site of action, are therefore synthesized to reduce the toxicity of the drug.
Several cathepsin B cleavable pro-drugs have been developed (Zhong et al., 2013). These pro-drugs contain the tetrapeptide linker that is cleavable by cathepsin B: Gly-Phe-Leu-Gly (see Figure 3). Two pro-drugs, PK1 and PK2, have advanced to phase II clinical trials (Seymour et al., 2009).
PK1
PK1 comprises doxorubicin covalently bound to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer by the cathepsin B cleavable linker. In phase I trial, PK1 showed better maximum tolerated dose (MTD) of 320 mg/m2 (doxorubicin equivalent), which is 4–5 fold higher than that of free doxorubicin (Vasey et al., 1999). In a phase II trial in patients with breast, non-small cell lung and colorectal cancers, 6 out of 62 patients showed partial responses (PR) (Seymour et al., 2009). Compared with doxorubicin, PK1 demonstrated reduced non-specific organ toxicity in both phase I and phase II trials.
PK2
PK2 is also a HPMA copolymer with the cathepsin B cleavable linker but contains both doxorubicin and galactosamine (Julyan et al., 1999). The inclusion of galactosamine is designed to target the asialoglycoprotein receptor (ASGPR) that is selectively expressed in hepatocytes and hepatoma cell lines (Zhong et al., 2013). In a clinical trial with a total of 31 primary or metastatic liver cancer patients, 3 patients showed partial responses, with one in continuing partial remission 47 months after therapy (Seymour et al., 2002). However, study of tissue distribution revealed that more PK2 was retained in the normal liver than the tumor tissue (16.9% vs 3.3% of administrated dose).
Currently, neither PK1 nor PK2 is under an active development program. Clearly, the accumulation of PK2 in normal liver tissue is a serious concern. For PK1, although the pro-drug might be less toxic to normal tissues as indicated in clinical trial, the therapeutic efficacy over free doxorubicin is not obvious. As revealed in two animal models, PK1 requires co-administrated cathepsin B to release doxorubicin (Satchi et al., 2001), and PK1 alone is inferior to free doxorubicin in the xenografted tumor growth control.
9. Perspectives
In summary, due to the extensive study of cathepsin B functions and progress in the atomic understanding of cathepsin B structure, a variety of inhibitors have been developed to combat this enzyme in cancer. Since cathepsin B has some overlapping functions with other members of cathepsins, the ideal selectivity for cathepsin B inhibitors needs to be carefully defined to be broad enough to sufficiently block similar enzymes while sparing others to minimize unintended toxicity. A novel approach termed activity-based protein profiling (ABPP) was developed to screen compound libraries for activity to inhibit multiple cathepsins in defined tissues simultaneously (Sadaghiani et al., 2007). This approach has highlighted a particular compound, ASM7, which has good activity against both cathepsin B, H and X (IC50: 40–70 nM), and could be useful to reevaluate available inhibitors for required specificity. In addition, combining cathepsin B inhibitors with other approaches, such as nanoparticles, may lead to better clinical approaches to treat cancers and metastasis.
Acknowledgments
This work was supported by grants from the Breast Cancer Research Foundation and the National Institutes of Health (R01CA089481, R01CA149425, R01CA157766, and R43CA171417).
References
- Beavon IR. The E-cadherin-catenin complex in tumour metastasis: structure, function and regulation. Eur J Cancer. 2000;36(13 Spec):1607–1620. doi: 10.1016/s0959-8049(00)00158-1. [DOI] [PubMed] [Google Scholar]
- Bell-McGuinn KM, Garfall AL, Bogyo M, Hanahan D, Joyce JA. Inhibition of cysteine cathepsin protease activity enhances chemotherapy regimens by decreasing tumor growth and invasiveness in a mouse model of multistage cancer. Cancer Res. 2007;67(15):7378–7385. doi: 10.1158/0008-5472.CAN-07-0602. [DOI] [PubMed] [Google Scholar]
- Biniossek ML, Nagler DK, Becker-Pauly C, Schilling O. Proteomic identification of protease cleavage sites characterizes prime and non-prime specificity of cysteine cathepsins B, L, and S. Journal of proteome research. 2011;10(12):5363–5373. doi: 10.1021/pr200621z. [DOI] [PubMed] [Google Scholar]
- Broker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004;64(1):27–30. doi: 10.1158/0008-5472.can-03-3060. [DOI] [PubMed] [Google Scholar]
- Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, Kanellopoulos J, Martin F, Rebe C, Apetoh L, Ghiringhelli F. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nature medicine. 2013;19(1):57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. The Biochemical journal. 1992;282 (Pt 1):273–278. doi: 10.1042/bj2820273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campo E, Munoz J, Miquel R, Palacin A, Cardesa A, Sloane BF, Emmert-Buck MR. Cathepsin B expression in colorectal carcinomas correlates with tumor progression and shortened patient survival. The American journal of pathology. 1994;145(2):301–309. [PMC free article] [PubMed] [Google Scholar]
- Cavallo-Medved D, Mai J, Dosescu J, Sameni M, Sloane BF. Caveolin-1 mediates the expression and localization of cathepsin B, pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J Cell Sci. 2005;118(Pt 7):1493–1503. doi: 10.1242/jcs.02278. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Tuck AB. Ras-responsive genes and tumor metastasis. Critical reviews in oncogenesis. 1993;4(2):95–114. [PubMed] [Google Scholar]
- Cygler M, Sivaraman J, Grochulski P, Coulombe R, Storer AC, Mort JS. Structure of rat procathepsin B: model for inhibition of cysteine protease activity by the proregion. Structure. 1996;4(4):405–416. doi: 10.1016/s0969-2126(96)00046-9. [DOI] [PubMed] [Google Scholar]
- Foghsgaard L, Wissing D, Mauch D, Lademann U, Bastholm L, Boes M, Elling F, Leist M, Jaattela M. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. The Journal of cell biology. 2001;153(5):999–1010. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong D, Chan MM, Hsieh WT. Gene mapping of human cathepsins and cystatins. Biomedica biochimica acta. 1991;50(4–6):595–598. [PubMed] [Google Scholar]
- Frlan R, Gobec S. Inhibitors of cathepsin B. Current medicinal chemistry. 2006;13(19):2309–2327. doi: 10.2174/092986706777935122. [DOI] [PubMed] [Google Scholar]
- Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, Hanahan D, Joyce JA. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20(5):543–556. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. RNAi-mediated inhibition of cathepsin B and uPAR leads to decreased cell invasion, angiogenesis and tumor growth in gliomas. Oncogene. 2004;23(52):8486–8496. doi: 10.1038/sj.onc.1207879. [DOI] [PubMed] [Google Scholar]
- Greenspan PD, Clark KL, Tommasi RA, Cowen SD, McQuire LW, Farley DL, van Duzer JH, Goldberg RL, Zhou H, Du Z, Fitt JJ, Coppa DE, Fang Z, Macchia W, Zhu L, Capparelli MP, Goldstein R, Wigg AM, Doughty JR, Bohacek RS, Knap AK. Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. Journal of medicinal chemistry. 2001;44(26):4524–4534. doi: 10.1021/jm010206q. [DOI] [PubMed] [Google Scholar]
- Groth-Pedersen L, Jaattela M. Combating apoptosis and multidrug resistant cancers by targeting lysosomes. Cancer Lett. 2013;332(2):265–274. doi: 10.1016/j.canlet.2010.05.021. [DOI] [PubMed] [Google Scholar]
- Hirai K, Yokoyama M, Asano G, Tanaka S. Expression of cathepsin B and cystatin C in human colorectal cancer. Human pathology. 1999;30(6):680–686. doi: 10.1016/s0046-8177(99)90094-1. [DOI] [PubMed] [Google Scholar]
- Julyan PJ, Seymour LW, Ferry DR, Daryani S, Boivin CM, Doran J, David M, Anderson D, Christodoulou C, Young AM, Hesslewood S, Kerr DJ. Preliminary clinical study of the distribution of HPMA copolymers bearing doxorubicin and galactosamine. Journal of controlled release: official journal of the Controlled Release Society. 1999;57(3):281–290. doi: 10.1016/s0168-3659(98)00124-2. [DOI] [PubMed] [Google Scholar]
- Kirkegaard T, Jaattela M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta. 2009;1793(4):746–754. doi: 10.1016/j.bbamcr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Kokai Y, Myers JN, Wada T, Brown VI, LeVea CM, Davis JG, Dobashi K, Greene MI. Synergistic interaction of p185c-neu and the EGF receptor leads to transformation of rodent fibroblasts. Cell. 1989;58(2):287–292. doi: 10.1016/0092-8674(89)90843-x. [DOI] [PubMed] [Google Scholar]
- Kolwijck E, Massuger LF, Thomas CM, Span PN, Krasovec M, Kos J, Sweep FC. Cathepsins B, L and cystatin C in cyst fluid of ovarian tumors. J Cancer Res Clin Oncol. 2010;136(5):771–778. doi: 10.1007/s00432-009-0716-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostoulas G, Lang A, Nagase H, Baici A. Stimulation of angiogenesis through cathepsin B inactivation of the tissue inhibitors of matrix metalloproteinases. FEBS Lett. 1999;455(3):286–290. doi: 10.1016/s0014-5793(99)00897-2. [DOI] [PubMed] [Google Scholar]
- Kuester D, Lippert H, Roessner A, Krueger S. The cathepsin family and their role in colorectal cancer. Pathology, research and practice. 2008;204(7):491–500. doi: 10.1016/j.prp.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139(7):1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai J, Finley RL, Jr, Waisman DM, Sloane BF. Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. The Journal of biological chemistry. 2000;275(17):12806–12812. doi: 10.1074/jbc.275.17.12806. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, de Strooper B, Hartmann D, Saftig P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc Natl Acad Sci U S A. 2005;102(26):9182–9187. doi: 10.1073/pnas.0500918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalik R, Imre G, Petak I, Szende B, Kopper L. Cathepsin B-independent abrogation of cell death by CA-074-OMe upstream of lysosomal breakdown. Cell Death Differ. 2004;11(12):1357–1360. doi: 10.1038/sj.cdd.4401493. [DOI] [PubMed] [Google Scholar]
- Mikhaylov G, Klimpel D, Schaschke N, Mikac U, Vizovisek M, Fonovic M, Turk V, Turk B, Vasiljeva O. Angewandte Chemie. 2014. Selective Targeting of Tumor and Stromal Cells By a Nanocarrier System Displaying Lipidated Cathepsin B Inhibitor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkovic B, Renko M, Turk S, Sosic I, Jevnikar Z, Obermajer N, Turk D, Gobec S, Kos J. Novel mechanism of cathepsin B inhibition by antibiotic nitroxoline and related compounds. Chem Med Chem. 2011;6(8):1351–1356. doi: 10.1002/cmdc.201100098. [DOI] [PubMed] [Google Scholar]
- Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nature reviews Cancer. 2006;6(10):764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Montaser M, Lalmanach G, Mach L. CA-074, but not its methyl ester CA-074Me, is a selective inhibitor of cathepsin B within living cells. Biol Chem. 2002;383(7–8):1305–1308. doi: 10.1515/BC.2002.147. [DOI] [PubMed] [Google Scholar]
- Mort JS, Buttle DJ. Cathepsin B. The international journal of biochemistry & cell biology. 1997;29(5):715–720. doi: 10.1016/s1357-2725(96)00152-5. [DOI] [PubMed] [Google Scholar]
- Murata M, Miyashita S, Yokoo C, Tamai M, Hanada K, Hatayama K, Towatari T, Nikawa T, Katunuma N. Novel epoxysuccinyl peptides. Selective inhibitors of cathepsin B, in vitro. FEBS Lett. 1991;280(2):307–310. doi: 10.1016/0014-5793(91)80318-w. [DOI] [PubMed] [Google Scholar]
- Musil D, Zucic D, Turk D, Engh RA, Mayr I, Huber R, Popovic T, Turk V, Towatari T, Katunuma N, et al. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: the structural basis for its specificity. The EMBO journal. 1991;10(9):2321–2330. doi: 10.1002/j.1460-2075.1991.tb07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka T, Kuester D, Boltze C, Schulz TO, Samii A, Herold C, Ostertag H, Roessner A. Expression of matrix metalloproteinases-1, -2, and -9; tissue inhibitors of matrix metalloproteinases-1 and -2; cathepsin B, urokinase plasminogen activator; and plasminogen activator inhibitor, type I in skull base chordoma. Human pathology. 2008;39(2):217–223. doi: 10.1016/j.humpath.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Noe V, Fingleton B, Jacobs K, Crawford HC, Vermeulen S, Steelant W, Bruyneel E, Matrisian LM, Mareel M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J Cell Sci. 2001;114(Pt 1):111–118. doi: 10.1242/jcs.114.1.111. [DOI] [PubMed] [Google Scholar]
- Rafn B, Nielsen CF, Andersen SH, Szyniarowski P, Corcelle-Termeau E, Valo E, Fehrenbacher N, Olsen CJ, Daugaard M, Egebjerg C, Bottzauw T, Kohonen P, Nylandsted J, Hautaniemi S, Moreira J, Jaattela M, Kallunki T. ErbB2-driven breast cancer cell invasion depends on a complex signaling network activating myeloid zinc finger-1-dependent cathepsin B expression. Mol Cell. 2012;45(6):764–776. doi: 10.1016/j.molcel.2012.01.029. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2012;40(Database issue):D343–350. doi: 10.1093/nar/gkr987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinheckel T, Deussing J, Roth W, Peters C. Towards specific functions of lysosomal cysteine peptidases: phenotypes of mice deficient for cathepsin B or cathepsin L. Biol Chem. 2001;382(5):735–741. doi: 10.1515/BC.2001.089. [DOI] [PubMed] [Google Scholar]
- Ryniers F, Stove C, Goethals M, Brackenier L, Noe V, Bracke M, Vandekerckhove J, Mareel M, Bruyneel E. Plasmin produces an E-cadherin fragment that stimulates cancer cell invasion. Biol Chem. 2002;383(1):159–165. doi: 10.1515/BC.2002.016. [DOI] [PubMed] [Google Scholar]
- Sadaghiani AM, Verhelst SH, Gocheva V, Hill K, Majerova E, Stinson S, Joyce JA, Bogyo M. Design, synthesis, and evaluation of in vivo potency and selectivity of epoxysuccinyl-based inhibitors of papain-family cysteine proteases. Chem Biol. 2007;14(5):499–511. doi: 10.1016/j.chembiol.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz JD, Schu P, von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A. 1998;95(23):13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sameni M, Dosescu J, Yamada KM, Sloane BF, Cavallo-Medved D. Functional live-cell imaging demonstrates that beta1-integrin promotes type IV collagen degradation by breast and prostate cancer cells. Molecular imaging. 2008;7(5):199–213. [PMC free article] [PubMed] [Google Scholar]
- Satchi R, Connors TA, Duncan R. PDEPT: polymer-directed enzyme prodrug therapy. I. HPMA copolymer-cathepsin B and PK1 as a model combination. British journal of cancer. 2001;85(7):1070–1076. doi: 10.1054/bjoc.2001.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraufstatter IU, Trieu K, Zhao M, Rose DM, Terkeltaub RA, Burger M. IL-8-mediated cell migration in endothelial cells depends on cathepsin B activity and transactivation of the epidermal growth factor receptor. Journal of immunology. 2003;171(12):6714–6722. doi: 10.4049/jimmunol.171.12.6714. [DOI] [PubMed] [Google Scholar]
- Schraufstatter IU, Trieu K, Zhao M, Rose DM, Terkeltaub RA, Burger M. IL-8-mediated cell migration in endothelial cells depends on cathepsin B activity and transactivation of the epidermal growth factor receptor. J Immunol. 2003;171(12):6714–6722. doi: 10.4049/jimmunol.171.12.6714. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, Doran J, Young AM, Burtles S, Kerr DJ. Hepatic drug targeting: phase I evaluation of polymer-bound doxorubicin. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2002;20(6):1668–1676. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, Boivin C, Hesslewood S, Twelves C, Blackie R, Schatzlein A, Jodrell D, Bissett D, Calvert H, Lind M, Robbins A, Burtles S, Duncan R, Cassidy J. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. International journal of oncology. 2009;34(6):1629–1636. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- Shchors K, Nozawa H, Xu J, Rostker F, Swigart-Brown L, Evan G, Hanahan D. Increased invasiveness of MMP-9-deficient tumors in two mouse models of neuroendocrine tumorigenesis. Oncogene. 2013;32(4):502–513. doi: 10.1038/onc.2012.60. [DOI] [PubMed] [Google Scholar]
- Shi GP, Villadangos JA, Dranoff G, Small C, Gu L, Haley KJ, Riese R, Ploegh HL, Chapman HA. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity. 1999;10(2):197–206. doi: 10.1016/s1074-7613(00)80020-5. [DOI] [PubMed] [Google Scholar]
- Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, Bell-McGuinn KM, Zabor EC, Brogi E, Joyce JA. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes & development. 2011;25(23):2465–2479. doi: 10.1101/gad.180331.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane BF. Cathepsin B and cystatins: evidence for a role in cancer progression. Seminars in cancer biology. 1990;1(2):137–152. [PubMed] [Google Scholar]
- Stern I, Schaschke N, Moroder L, Turk D. Crystal structure of NS-134 in complex with bovine cathepsin B: a two-headed epoxysuccinyl inhibitor extends along the entire active-site cleft. The Biochemical journal. 2004;381(Pt 2):511–517. doi: 10.1042/BJ20040237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szpaderska AM, Frankfater A. An intracellular form of cathepsin B contributes to invasiveness in cancer. Cancer Res. 2001;61(8):3493–3500. [PubMed] [Google Scholar]
- Tan GJ, Peng ZK, Lu JP, Tang FQ. Cathepsins mediate tumor metastasis. World journal of biological chemistry. 2013;4(4):91–101. doi: 10.4331/wjbc.v4.i4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomssen C, Schmitt M, Goretzki L, Oppelt P, Pache L, Dettmar P, Janicke F, Graeff H. Prognostic value of the cysteine proteases cathepsins B and cathepsin L in human breast cancer. Clin Cancer Res. 1995;1(7):741–746. [PubMed] [Google Scholar]
- Towatari T, Nikawa T, Murata M, Yokoo C, Tamai M, Hanada K, Katunuma N. Novel epoxysuccinyl peptides. A selective inhibitor of cathepsin B, in vivo. FEBS Lett. 1991;280(2):311–315. doi: 10.1016/0014-5793(91)80319-x. [DOI] [PubMed] [Google Scholar]
- Turk D, Podobnik M, Popovic T, Katunuma N, Bode W, Huber R, Turk V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-A resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry. 1995;34(14):4791–4797. doi: 10.1021/bi00014a037. [DOI] [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. Embo J. 2001;20(17):4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, Thomson AH, Murray LS, Hilditch TE, Murray T, Burtles S, Fraier D, Frigerio E, Cassidy J. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates. Cancer Research Campaign Phase I/II Committee. Clinical cancer research: an official journal of the American Association for Cancer Research. 1999;5(1):83–94. [PubMed] [Google Scholar]
- Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr Pharm Des. 2007;13(4):387–403. doi: 10.2174/138161207780162962. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Yamamoto A, Tomoo K, Matsumoto K, Murata M, Kitamura K, Ishida T. Quantitative evaluation of each catalytic subsite of cathepsin B for inhibitory activity based on inhibitory activity-binding mode relationship of epoxysuccinyl inhibitors by X-ray crystal structure analyses of complexes. Journal of molecular biology. 2006;362(5):979–993. doi: 10.1016/j.jmb.2006.07.070. [DOI] [PubMed] [Google Scholar]
- Withana NP, Blum G, Sameni M, Slaney C, Anbalagan A, Olive MB, Bidwell BN, Edgington L, Wang L, Moin K, Sloane BF, Anderson RL, Bogyo MS, Parker BS. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer research. 2012;72(5):1199–1209. doi: 10.1158/0008-5472.CAN-11-2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tomoo K, Hara T, Murata M, Kitamura K, Ishida T. Substrate specificity of bovine cathepsin B and its inhibition by CA074, based on crystal structure refinement of the complex. J Biochem. 2000;127(4):635–643. doi: 10.1093/oxfordjournals.jbchem.a022651. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Tomoo K, Matsugi K, Hara T, In Y, Murata M, Kitamura K, Ishida T. Structural basis for development of cathepsin B-specific noncovalent-type inhibitor: crystal structure of cathepsin B-E64c complex. Biochimica et biophysica acta. 2002;1597(2):244–251. doi: 10.1016/s0167-4838(02)00284-4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, Greene MI. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117(8):2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong YJ, Shao LH, Li Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review) Int J Oncol. 2013;42(2):373–383. doi: 10.3892/ijo.2012.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]