

Abstract
Objective
Kawasaki disease (KD) is the most common cause of acute vasculitis and acquired cardiac disease among US children. We have previously shown that both TLR2/MyD88 and IL-1β signaling are required for the Lactobacillus casei cell wall extract (LCWE)-induced KD vasculitis mouse model. The objectives of this study were to investigate the cellular origins of IL-1 production, the role of CD11c+ Dendritic Cells (DCs) and macrophages and the relative contribution of hematopoietic and stromal cells for IL-1 responsive cells, as well the MyD88 signaling in LCWE-induced KD mouse model of vasculitis.
Approach and Results
Using mouse knockout models as well as antibody depletion, we found that both IL-1α and IL-1β were required for LCWE-induced KD. Both DCs and macrophages were necessary and we found that MyD88 signaling was required in both hematopoietic and stromal cells. However, IL-1 response and signaling was critically required in non-endothelial stromal cells, but not hematopoietic cells.
Conclusions
Our results suggest that IL-1α and IL-1β as well as CD11c+ DCs and macrophages are essential for the development of KD vasculitis and coronary arteritis in this mouse model. Bone marrow chimera experiments suggest that MyD88 signaling is important in both hematopoietic and stromal cells, while IL-1 signaling and response is required only in stromal cells, but not in endothelial cells. Determining the role IL-1α and IL-1β and of specific cell types in the KD vasculitis mouse model may have important implications for the design of more targeted therapies and understanding of the molecular mechanisms of KD immunopathologies.
Keywords: Kawasaki disease, coronary artery vasculitis, dendritic cells, IL-1, MyD88
Introduction
Kawasaki disease (KD) is an acute febrile illness and systemic vasculitis1–5 that predominantly afflicts children <5 yrs of age. The etiology of KD remains unknown, although the current paradigm is that KD is triggered by an infectious agent (with a conventional Ag) that elicits an inflammatory response directed at cardiovascular tissues in genetically susceptible hosts6, 7. It often causes acute coronary as well as systemic arteritis with coronary artery aneurysms (CAA), and can lead to ischemic heart disease, myocardial infarction, and even death8–11. KD vasculitis, once thought of as an acute self-limiting disease, is now known to result in long term complications with ongoing vascular remodeling and myocardial fibrosis12. Indeed, long term cardiovascular complications among survivors of childhood KD are reported with increasing frequency.13–15 KD represents the leading cause of acquired heart disease among children in developed countries.9–11 While intravenous IgG (IVIG) treatment within the first 10 days of illness have reduced the cardiovascular complications of KD (CAA) from 25% down to 5%16, up to 20–25% of KD patients are IVIG-resistant and at even higher risk for developing CAA17. Therefore, discovery of more effective treatments for KD is one of the highest priorities in pediatric research18.
KD involves systemic inflammation with a distinct predilection for the coronary arteries. The resulting coronary arteritis in KD is characterized histologically by inflammatory cell infiltration and destruction of extracellular matrix, especially elastic tissue in vascular media, with resultant coronary artery aneurysm formation.19 The limited availability of tissue samples from patients with KD has significantly impeded our progress in understanding the etiology and pathogenesis of the disease, making the availability of a relevant animal model extremely valuable. Importantly, a well-described and widely used mouse model of KD vasculitis and coronary arteritis closely mimics the important histological as well as immune-pathological features of the cardiovascular lesions (i.e. coronary arteritis, aortitis, myocarditis, aneurysms, including abdominal aorta aneurysms (AAA) seen in KD)20–23. This mouse model (Lactobacillus casei-cell wall extract (LCWE)-induced KD vasculitis) also predicts efficacy of treatment in children with KD21, 23. While no animal model can fully mimic human disease, the LCWE-induced KD mouse model has been accepted by many in the Kawasaki research community as a reliable model to provide novel insights that can be tested in children.
Although both the trigger and the precise pathogenic mechanism of KD are unclear, there are strong indications that the pathology is immune mediated.24–27 We previously demonstrated that Caspase-1 activation and IL-1β are critically required for LCWE-induced KD vasculitis28, 29. We showed that an IL-1R antagonist (Anakinra) significantly blocked the coronary arteritis, aortitis and myocarditis associated with the LCWE-induced KD vasculitis model29. This experimental mouse study, together with several case reports of successful use of Anakinra in IVIG-unresponsive KD patients30, 31, lead to two recent clinical trials using this IL-1R antagonist for patients with IVIG-resistant KD. However, the exact mechanism by which IL-1 plays a role in KD pathogenesis is still unknown. IL-1α and IL-1β both bind to and activate the IL-1R, although their regulation and activity differ. Since the IL-1R antagonist blocks both of these cytokines, the role that IL-1α may play in this model is unknown. Most importantly, the host target cell(s) that respond to IL-1 in this experimental KD vasculitis model is also unknown.
We previously reported that DCs and macrophages are localized in the coronary lesions of LCWE-induced KD in mice24, similar to what has been reported in coronary lesions from KD patients.24, 32 While DCs are believed to mainly play a role as an antigen presenting cell, macrophages can provide wide ranging innate immune responses, including IL-1 secretion after inflammasome activation.
In this study we determined that CD11c+ DCs and macrophages are absolutely required for the LCWE-induced KD vasculitis model and these macrophages appear to be the cellular source of IL-1β production in the lesions. We also found that both IL-1α and IL-1β significantly contribute to LCWE-induced KD vasculitis model, and bone marrow chimera experiments show that IL-1 signaling is required only in stromal cells, but not in endothelial cells.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
CD11c+ macrophages in KD lesions have active Caspase-1 activity
IL-1 signaling and Caspase-1 are both required for LCWE-induced KD29. We previously reported that macrophages are present in LCWE-induced KD vasculitis and coronary arteritis lesions22. We now stained the coronary arteritis lesions from LCWE-induced KD mice with anti-F4/80 and anti-CD11c and observed the presence of F4/80 and CD11c+ double positive stained macrophages in the lesion site (Figure 1A). Inflammasome activity and IL-1β secretion is most commonly found in macrophages. Recent literature has indicated the involvement of CD11c+ macrophages in various proinflammatory disorders.33–35 However, which cells are producing IL-1, as well as which type of inflammasome is required is not known in this mouse model of KD. In order to determine which cells in the KD coronary lesions produce IL-1β, we assessed the expression of caspase-1 activity in heart sections of KD mice using FLICA, which identifies caspase-1 activity and can be used as a surrogate marker for IL-1β production. FLICA+ cells were readily visible in the aortitis and coronary arteritis lesions 7 days after LCWE injection but not in naive animals (Figure 1B). FLICA+ cells also expressed both F4/80 and CD11c, indicating that these cells were macrophages, and not DCs (Figure 1C). Thus it is likely that CD11c+ macrophages are the main source of IL-1β production in the LCWE-induced KD vasculitis lesions.
Figure 1. Caspase-1 activity is detected in CD11c and F4/80 positive cells in LCWE-induced vasculitis and coronary lesions.

WT mice were i.p. injected with LCWE (250 ug) and heart tissues were collected 1 week after the injection. (A) Images were taken at the aortic root at the level of the 2nd branch of the coronary artery from aorta. F4/80 and CD11c single or double positive cells were detected in the coronary artery and aortitis lesions of LCWE-injected WT mice. (B) The heart sections of naïve and LCWE-injected mice were stained with H&E or FLICA (Green), specific for active caspase-1, and DAPI (Blue). (C) The serial heart sections of LCWE-injected mice were co-stained with FLICA (Green), and antibodies against CD11c (Red) or F4/80 (Blue). White arrows indicate the CD11c, F4/80 and FLICA positive cells. The scale bar is 250μm.
NLRP3 is required for LCWE-induced KD vasculitis and coronary lesions
We previously reported that the NLRP3 inflammasome is required for Caspase-1 activity and IL-1β production in bone marrow derived macrophages in-vitro29. However, while we showed that IL-1 signaling and Caspase-1 are absolutely required in vivo for the LCWE-induced KD vasculitis29, we did not directly investigate the role of NLRP3 in KD lesion induction. We therefore injected Nlrp3−/− and WT mice with LCWE and harvested the hearts 14 days later. We observed that Nlrp3−/− mice were protected and developed significantly reduced vasculitis lesions and myocarditis compared with WT mice (Figure 2A–C). These data confirmed the involvement of the NLRP3 inflammasome in LCWE-induced KD vasculitis. We also assessed the role of the AIM2 inflammasome, and did not find a role for AIM2 inflammasome in this model as Aim2−/− mice were not protected and develop severe KD-vasculitis with similar intensity to WT mice (data not shown).
Figure 2. NLRP3 and CD11c+ cells are required for LCWE-induced vasculitis.

(A–C) C57BL6/J WT or Nlrp3−/− mice were i.p. injected with LCWE and their hearts harvested 14 days after injection. (A) H&E staining, (B) Heart vessel inflammation score, and (C) Myocardium inflammation. (D-H) CD11c+ DTR transgenic mice were i.p. injected with 8 ng/g body weight of diphtheria toxin (DTx) for depletion of CD11c+ cells on day -1 and day 1. LCWE was administrated on day 0. Control mice were injected with PBS instead of toxin or LCWE. The hearts were harvested on day 7 and analyzed by H&E staining. (D) Representative H&E images of DTx treated mice, (E) Heart vessel inflammation score, (F) incidence of KD lesions, (G) and myocardial inflammation score were evaluated as described in Methods. (H) Splenocytes were re-stimulated on day 14 from LCWE injection and IFN-γ in the supernatants was analyzed by ELISA. Data shown are mean±SE and were compared by the normalized unpaired Student t test with Mann-Whitney post test (B, C, and H), by the normalized One way ANOVA with Tukey’s post hoc test (E and G), and Fisher exact test for incidence of KD lesions (F). A probability value of P≤0.05 was considered statistically significant. The scale bar is 250μm.
CD11c+ dendritic cells and macrophages are required for the development of LCWE-induced KD vasculitis and coronary arteritis
In addition to CD11c+ macrophages, there were also many CD11c single positive cells (Fig. 1A), indicating a large number of DCs in the lesion as we have previously reported22. To investigate the requirement of DCs or macrophages in the LCWE-induced KD model, we treated the CD11c-DTR transgenic mice with diphtheria toxin (DTx) on days −1 and 1 relative to LCWE injection (day 0) to deplete CD11c+ cells. CD11c is a cell surface marker for DCs and some peripheral macrophages and these mice express the human Diphtheria toxin receptor under the CD11c promoter. The animals were sacrificed on day 7 and their hearts were harvested. CD11c positive cells were depleted after DTx injection as confirmed by flow cytometry analysis (data not shown). Mice depleted of CD11c+ cells by DTx developed significantly less KD vasculitis and coronary arteritis lesions compared to PBS injected control mice, and DTx itself had no effect on naïve CD11c-DTR mice (Figure 2D). CD11c+ cells depletion also resulted in a significant reduction in incidence as well as vascular inflammation and myocardial inflammation severity scores when compared to controls (Figure 2E–G). Furthermore, IFN-γ production by splenocytes was significantly reduced after LCWE restimulation, while it was unaffected after anti-CD3 stimulation (Fig. 2H). We next treated mice with clodronate liposomes to more specifically deplete macrophages during LCWE-induced KD34, although some DCs can also be targeted this way. Similar to the DTR model, we also found that macrophages were required for LCWE-induced KD vasculitis and coronary lesions (Supplemental Figure IA–E). Taken together, these data demonstrate that CD11c+ DCs and or macrophages play a critical role in LCWE-induced KD vasculitis, coronary arteritis and myocarditis.
Both IL-1α and IL-1β are required for LCWE-induced vasculitis
Our data revealed that macrophages are likely critically required for LCWE-induced KD in mice. We also found that CD11c+ macrophages had inflammasome activity at the lesions, thus making them the likely source of IL-1β. We previously reported that Il1r1−/− mice were completely protected and that IL-1Ra treatment can prevent LCWE-induced KD vasculitis and coronary arteritis.29 However, both IL-1α and IL-1β can bind to and activate the same IL-1 receptor and IL-1Ra treatment would block both cytokines. To test the role of IL-1α and IL-1β in LCWE-induced KD vasculitis and coronary arteritis, we injected Il1a−/− and Il1b−/− mice with LCWE and examined their hearts at day 14. We observed that both Il1a−/− and Il1b−/− mice were protected from KD vasculitis and showed significantly diminished vascular inflammatory lesions, coronary arteritis and myocarditis compared with WT mice (Figure 3A–D). These results demonstrate that both IL-1α and IL-1β play critical roles in LCWE-induced KD vasculitis and myocarditis.
Figure 3. Critical Role of IL-1α and IL-1β in LCWE-induced vasculitis and coronary arteritis.

C57BL/6J, Il1a−/− and Il1b−/− mice were i.p injected with 250 μg of LCWE and their heart was harvested and analyzed by H&E staining 14 days later. C57BL/6J mice were i.p administrated with 80 μg of anti-IL-1α mAb and/or 200 μg of anti-IL-1β mAb at day -1, 2, and 5 from 250 μg of LCWE injection. Their hearts were harvested on day 7 for H&E staining. (A and E) Representative histology, (B and F) Heart vessel inflammation score, (C and G) incidence of KD lesions and (D and H) myocardial inflammation score were evaluated as described in Methods. Data shown are mean ± SE and were compared by the normalized One way ANOVA with Tukey’s post hoc test (B and D) and Fisher exact test for incidence (C). A probability value of P≤0.05 was considered statistically significant. The scale bar is 250 μm.
Recent reports have suggested that Il1a−/− mice may have diminished NLRP3 inflammasome activation and IL-1β secretion.36 We therefore wished to confirm the data obtained with IL1 KO mice by depleting the specific cytokines with anti-IL-1α and anti-IL-1β monoclonal antibodies alone or in combination in LCWE-induced KD vasculitis. Monoclonal anti-IL-1α and/or anti-IL-1β mAb were injected on days -1, 2, 5 from LCWE injection. Mice treated with anti-IL-1α or anti-IL-1β mAb were protected as they developed significantly less KD vasculitis, coronary arteritis, as well as myocarditis compared with mice injected with isotype control antibody (Figure 3E–H). Interestingly, while mice that received either anti-IL-1α or anti-IL-1β mAb still displayed some small residual vasculitic lesions, when both antibodies were given together, the mice were completely protected (Figure 3E–H), similar to IL-1R antagonist (Anakinra)-treated mice29 These results indicate that either anti-IL-1α or IL-1β mAb alone can significantly protect against LCWE-induced KD vasculitis and myocarditis, but the protection is not complete unless the two mAbs are given together or an IL-1R antagonist such as Anakinra, which blocks both IL-1α and IL-1β, is used.
MyD88 in CD11c+ cells is not sufficient for the development of LCWE-induced KD vasculitis
We previously reported that MyD88 signaling is important in the development of LCWE-induced KD vasculitis, as MyD88-deficient mice were completely protected.29 MyD88 signaling is required for most TLR signaling, as well as IL-1R1 signaling and we have already shown that LCWE-induced KD model requires both TLR2 and IL-1R1 signaling.28, 29 Since our data in Figure 1 suggested that both DCs and macrophages may be critically required, we reasoned that MyD88 signaling in DCs and or macrophages may be important but we wished to investigate if that would be sufficient to induce the vasculitis lesions in this experimental model. To test this, we injected MyD88-deficient mice that express transgenic MyD88 only in CD11c+ cells (Cd11c-Myd88-TG/Myd88−/−)37 with LCWE. We observed that MyD88 expression alone in CD11c+ cells was not sufficient to restore LCWE-induced KD vasculitis, as both full Myd88−/− and Cd11c-Myd88-TG/Myd88−/− mice were protected from KD vasculitis development (Figure 4A–D). These observations suggest that while CD11c+ DCs and macrophages are required for LCWE-induced KD vasculitis, MyD88 signaling is also required in cells other than or in addition to CD11c+ DCs, most likely in IL-1 responsive stromal cells.
Figure 4. MyD88 in CD11c+ cells is not sufficient for LCWE-induced KD vasculitis.
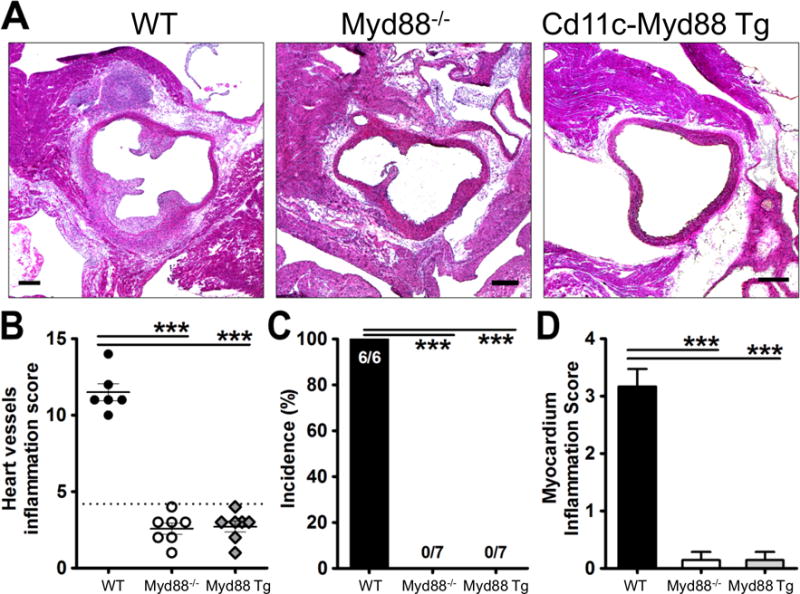
C57BL/6J, Myd88−/− and CD11c-Myd88 Tg mice were i.p injected with 250 μg of LCWE and their heart was harvested 14 days later. (A) Hearts were embedded in OCT compound and analyzed by H&E staining. (B) Heart vessel inflammation score, (C) Incidence of KD lesions, and (D) myocardial inflammation score were assessed as described in Methods. Data shown are mean±SE and were compared by the normalized One-way ANOVA with Tukey’s post hoc test (B and D) and Fisher exact test for incidence (C). A probability value of P≤0.05 was considered statistically significant. The scale bar is 250μm (A).
MyD88 signaling is required in both hematopoietic and stromal cells for the development of LCWE-induced KD vasculitis mouse model
In previous studies we found that LCWE signals via TLR2 and MyD88, that Myd88−/− bone marrow derived macrophage (BMDM) do not express IL-6 and TNF-α in vitro in response to LCWE28, and that IL-1 signaling is critically important in LCWE-induced KD vasculitis.29 To further define in which cellular compartment MyD88 is important for LCWE-induced KD vasculitis development, we next generated Myd88−/− bone marrow chimeric mice. Chimerism was typically greater than 90% (Supplemental Figure II). Irradiated WT mice reconstituted with WT BM transplantation (control mice) developed KD vasculitis lesions as expected (Figure 5A–D), while irradiated WT mice reconstituted with Myd88−/− BM were completely protected form developing KD vasculitis (Figure 5A–D). Unexpectedly, irradiated recipient Myd88−/− mice transplanted with WT BM were also completely protected, indicating a critical requirement for MyD88 in not only hematopoietic cells, but also in stromal cells. These data would also explain why the CD11c-MyD88-TG mice were unable to develop LCWE-induced KD vasculitis and coronary arteritis (Figure 4). Since LCWE-signals via TLR2/MyD88 pathway28 and IL-1 signaling also require MyD88 for signaling, MyD88 would be required both for initial IL-1 production following the LCWE injection as well as subsequent IL-1R signaling in the target cells.
Figure 5. MyD88 is important in both hematopoietic and stromal cells in LCWE-induced KD coronary vasculitis.
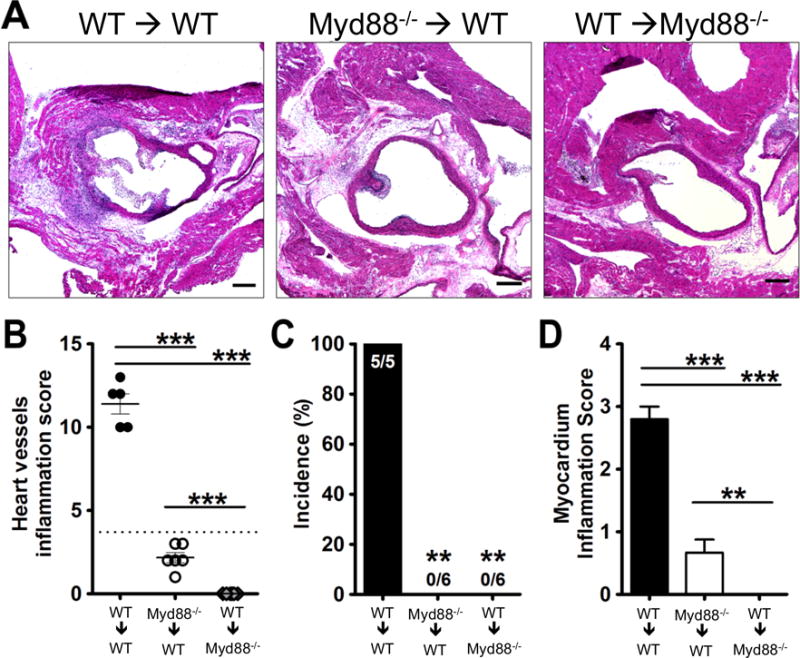
C57BL/6J or Myd88−/− were irradiated (7.5Gy) followed by bone marrow transplantation to create chimeric mice (WT→WT, Myd88−/−→WT, WT→Myd88−/−). Following 8 weeks of recovery, LCWE was injected and two weeks later the mice were sacrificed and their hearts analyzed by H&E staining. (A) Representative histology, (B) Heart vessel inflammation score, (C) Incidence of KD lesions, and (D) myocardial inflammation score were evaluated as described in Methods. Data shown are mean±SE and were compared by the normalized One-way ANOVA with Tukey’s post hoc test (B and D) and Fisher exact test for incidence (C). A probability value of P≤0.05 was considered statistically significant. The scale bar is 250μm.
IL-1 signaling is required in stromal cells but not hematopoietic cells for LCWE-induced KD vasculitis
We previously reported that Il1r1−/− mice are completely protected and that IL-1Ra treatment can prevent LCWE-induced KD vasculitis and coronary lesions.29 Here, we observed that MyD88 was required in both stromal and hematopoietic cells, consistent with requirement for MyD88 downstream of either TLR2 or IL-1R1 signaling. We therefore sought to determine next the specific cellular compartment where IL-1R1 signaling is required. We generated bone marrow chimeric mice between WT and Il1r1−/− mice to determine the IL-1 responsive cellular compartments. Irradiated WT mice reconstituted with WT BM transplantation (control mice) developed KD vasculitis lesions as expected (Figure 6A–D), but irradiated WT mice reconstituted with Il1r1−/− BM also developed normal KD vasculitis and coronary arteritis after LCWE injection (Figure 6A–D). However, irradiated Il1r1−/− recipient mice that received WT BM did not develop LCWE-induced vasculitis and coronary lesions (Figure 6A–D). These data suggest that stromal IL-1 signaling is indispensable to LCWE-induced vasculitis and coronary arteritis and that IL-1 signaling in hematopoietic cells is not required for the development of this cardiovascular pathology.
Figure 6. IL-1 signaling is required in stromal cells for LCWE-induced coronary vasculitis.
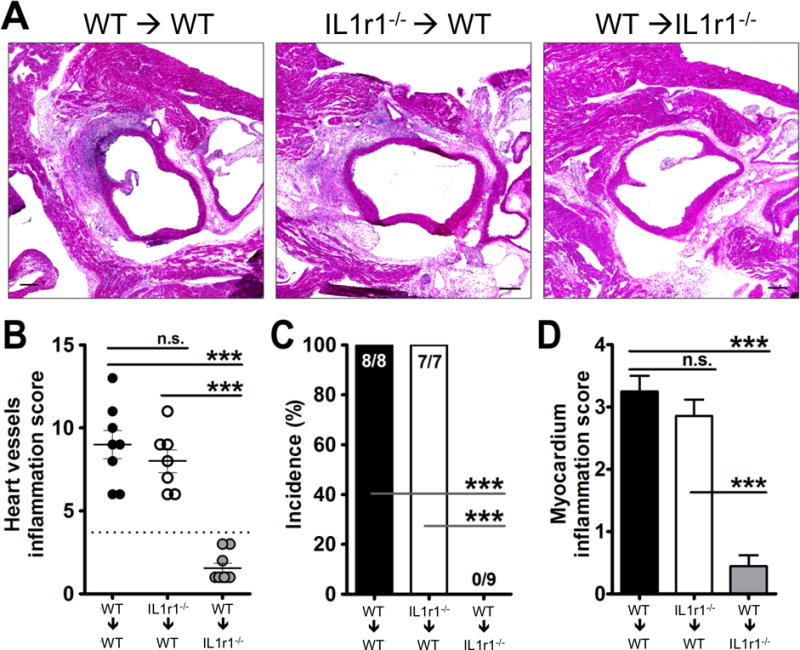
CD45.1 WT (Ly5.1) or Il1r1−/− were irradiated (7.5Gy) followed by bone marrow transplantation to create chimeric mice (WT→WT, Il1r1−/−→WT, and WT→Il1r1−/−). 8 weeks after irradiation and transplantation the mice were i.p injected with LCWE. Two weeks later the mice were sacrificed and the hearts were analyzed by H&E. (A) Representative histology, (B) Heart vessel inflammation score, (C) incidence of KD lesions, and (D) and myocardial inflammation score were evaluated as described in Methods. Data shown are mean±SE and were compared by the normalized One way ANOVA with Tukey’s post hoc test (B and D) and Fisher exact test for incidence (C). A probability value of P≤0.05 was considered statistically significant. The scale bar is 250μm.
Endothelial MyD88 is not required in the development of LCWE-induced KD vasculitis
To begin to dissect which stromal cell is the IL-1/MyD88 responsive cell type i.e. vascular endothelial cells versus vascular smooth muscle cells (VSMC) or others, we first investigated the role of EC. Since IL-1 signaling requires MyD88, and we observed that MyD88 was also required in stromal cells, we sought to identify if MyD88 specifically in EC was required for LCWE-induced KD vasculitis. To this effect, we generated endothelial cell-specific MyD88-deficient mice. We crossed the Myd88fl/fl mice to Tek-Cre mice to create endothelial MyD88 conditional knockout mice (ECMyd88−/−)38. TEK receptor tyrosine kinase is expressed almost exclusively in ECs39. These mice have normal MyD88 function except in their vascular ECs where exon 3 of MyD88 is removed by the cre recombinase, thus making them unresponsive to TLR/MyD88 signaling, as well as IL-1α/β signaling (Supplemental Figure III).40 We found that the endothelial MyD88 conditional knockout mice (ECMyd88−/−) were not protected and developed KD vasculitis with no differences compared to WT mice (Fig. 7A–C). Control Myd88fl/fl mice (no cre) also developed lesions as expected. These results indicate that endothelial MyD88 signaling is not required for LCWE-induces KD vasculitis in mice, suggesting that the stromal cell responsive to IL-1/MyD88 is not endothelial cells, but some other stromal cell type, such as VSMC.
Figure 7. Endothelial MyD88 is not involved in pathogenesis of LCWE induced KD vasculitis and coronary arteritis.

C57BL6/J WT, Myd88−/−, and ECMyD88−/− conditional knock-out mice or their control mice (Myd88fl/fl mice) were i.p. injected with 250 μg LCWE and their hearts were harvested on day 14 for H&E staining. (A) Representative histology, (B) Heart vessel inflammation score, and (C) incidence of KD lesions was evaluated as described in Methods. Data shown are mean±SE and were compared by the normalized One-way ANOVA with Tukey’s post hoc test (B) and Fisher exact test for incidence (C). A probability value of P≤0.05 was considered statistically significant. The scale bar is 250μm.
Discussion
KD is the leading cause of acquired heart disease in children in the United States and the developed world.9–11 However, while the underlying etiology and mechanisms leading to vessel inflammation, coronary artery lesions, and aneurysms that are the hallmarks of KD remain largely unknown, various studies have characterized which immune cells infiltrate into the cardiovascular lesions seen in KD24–27.
IL-1β plays a critical role in auto-inflammatory diseases as well as chronic inflammatory diseases such as atherosclerosis, diabetes41–46, and more recently was also linked to KD vasculitis29. Serum level of IL-1β is significantly increased in KD patients compare to age matched healthy controls47. IL-1 related genes are upregulated in KD peripheral blood during acute phase of illness48. Previous studies have shown that IVIG influences the production and release of IL-1β in KD patients49, 50 and genetic studies showed that IL-1β promoter polymorphisms with increased IL-1β production is associated with IVIG resistance41. In addition, IL-1α, often considered an alarmin, acting early during inflammatory responses and linked to many immunopathologies51, also plays a critical role in acute and chronic inflammation, and recent studies suggest that IL-1α may even regulate IL-1β secretion42, 52–54. In our study, while both IL-1α and IL-1β are required for LCWE-induced vascultis, it is possible that IL-1α and IL-1β may play a role sequentialy in inflammaton, early and late respectively, similar to what other recent studies have reported36, 55–57. While specific function blocking mAbs of either IL-1α or IL-1β alone did lead to a significant reduction in KD vasculitis, blocking both cytokines at the same time proved to be more effective in completely preventing any KD lesion formation. Thus therapies that block both cytokines, such as anti IL-1R antagonists (Anakinra) may prove to be more efficacious than each individual specific mAbs.
While IVIG reduces the rate of coronary artery abnormalities (CAA), morbidity and mortality associated with KD, up to 20–25% of patients are resistant to IVIG and have a higher risk of CAA, and discovery of novel more effective treatment for KD is a priority in pediatric research18. Progress for more effective and targeted treatments have been hindered due to a lack of a specific etiologic agent and an incomplete understanding of the molecular mechanisms mediating the cardiovascular pathology of KD. Additionally, the severely limited availability of human tissue samples has significantly impeded any progress in our understanding of the etiology and pathology of KD, making the availability of a relevant animal model of KD vasculitis extremely valuable. Indeed, recent progress in KD pathogenesis was made with the advent of human genetic studies (GWAS) combined with relevant mouse models of KD, and these two exciting areas of research recently have converged upon the importance of IL-1 pathway in KD pathogenesis4, 29. We have shown that IL-1β is critical in LCWE-induced KD vasculitis, coronary arteritis and myocarditis, and that an IL-1R antagonist (Anakinra) can effectively block these KD induced cardiovascular pathologies29.
Recent GWAS studies have discovered that several genetic polymorphisms (SNPs) are associated with increased risk for KD and CAA58, 59. Among these SNPs that are associated with increased risk for KD as well as risk for CAA, one that has attracted a major interest is found in the inositol 1,4,5,-triphosphate 3 kinase (ITPKC) gene4, 60–62. ITPK3 acts as a negative regulator of intracellular Ca2+ influx61, 62. Ca2+ influx plays a critical in NLRP3 inflammasome activation63, and the specific ITPKC SNPs associated with KD risk lead to sustained elevation of intracellular Ca2+, and therefore increased NLRP3 inflammasome activation and IL-1α, IL-1β production, providing a possible mechanistic link between these SNPs and KD64. The convergence of emerging genetic data with our finding that IL-1 signaling plays a crucial role in this experimental mouse model of KD29, now provide a very strong rational for further investigating the role of IL-1R antagonist therapies in KD patients. Following these studies, several case reports were published describing successful use and outcomes with Anakinra, in IVIG-non-responder KD patients30, 31. Importantly, all of these new data have led to two Phase II clinical trials for Anakinra in IVIG-resistant KD patients (NCT02179853 in UCSD and Univ. of Paris).
Using BM chimera experiments, we found that IL-1 signaling is required in non-hematopoietic cells (i.e. stromal cells), but not endothelial cells (EC) in the LCWE-induced KD mouse model. The likely candidates among the stromal cells are smooth muscle cells (SMC), fibroblasts or pericytes, the former being the most likely candidate. It is intriguing that, IL-1 signaling drives proliferation of smooth muscle cells (SMC) and myofibroblast formation65–67, a pathologic hallmark of subacute arteriopathy seen in KD (both patients and the animal model). Indeed, SMC-derived myofibroblast actively proliferate in an uncontrolled fashion in KD arterial wall leading to luminal myofibroblast proliferation (LMP), progressing to life-threating coronary artery stenosis and infarction, features that are present both in the KD patients and in the LCWE-induced KD mouse model12, 68. Although therapies are available to reduce risk of thrombosis in diseases of the coronary arteries, no therapies are available to reduce or prevent the LMP and coronary stenosis. Therefore, the pathophysiology of LMP must be better understood in order to develop novel targets to prevent or treat this detrimental ongoing vascular remodeling. Additionally, IL-1 induced SMC proliferation is driven by matrix metalloproteinases, including MMP3 and MMP965, 66, 69, both of which are implicated in human KD5, 48, 67, 70–72. Thus it is very intriguing that our data point towards non-endothelial cells stromal cells, such as VSMC as the key IL-1 responsive cells. The specific confirmation of this must await the generation of VSMC-specific IL-1R deficient mice (ongoing studies).
One possible caveat to our bone marrow chimera studies is the recent finding that many resident tissue macrophages originate from yolk-sac derived myeloid precursors73. While in some tissues the authors found that the macrophages can be replaced over time by bone marrow derived cells, no data exists regarding the heart. However, as our WT to WT chimeras developed LCWE-induced KD vasculitis normally, we do not think that they play a critical role in this model and can be functionally substituted by bone marrow derived cells.
We previously observed that both DCs and macrophages are present in close contact to CD8 T cells in the LCWE-induced KD coronary arteritis lesions in mice22, similar to what was described in human KD lesions24. Interestingly, we now observed that the majority of these macrophages were CD11c+, indicating a more specialized kind of macrophage34, 74, 75. Indeed, only CD11c+ macrophages were also FLICA positive (i.e. caspase 1 activity), suggesting that they may be the primary producers of IL-1β locally in the lesions. These cells are likely critical for the development of KD inflammation observed as mice depleted of CD11c+ cells were nearly completely protected from developing KD lesions. Additionally, mice depleted of phagocytic cells by clodronate liposomes were also completely protected. Recently, in another model of vascultis, CD11c+ macrophages were also found to be important for the induction of coronary arteritis.34
In our study FLICA activity was detected in CD11c+ macrophages 1 week after LCWE injection and it is possible that other cell types may have inflammasome activity earlier and or later in the progression of pathology. In contrast to our study, Chen et al found Caspase-1 activity localized to the coronary endothelium, and not in macrophages after LCWE injection.76 Indeed, other studies have found that inflammasome activation can occur in endothelial cells.77–79 However, in Chen et al’s study, the coronary lesions generated after LCWE injection were dramatically smaller than in our study, thus making comparison between the two studies difficult.76 Additionally, they find evidence for active caspase-1 in the coronary ECs even under naive conditions, which is unusual considering that inflammasome activation is tightly regulated.80
Our data strongly suggest the role of both IL-1α and IL-1β in LCWE-induced KD vasculitis model. These findings have important implications for the design of clinical studies to investigate the role of IL-1 in KD patients. The role of IL-1 was reported using this experimental mouse model, and these data has now led to clinical trials in children with KD with the IL-1R antagonist. Our new studies have placed IL-1R1 signaling as a critical step is KD cardiovascular lesion development, without which pathology is prevented. As anti-IL-1 therapeutics already exist for several chronic inflammatory diseases, IL-1 offers an attractive target for prevention and treatment of cardiovascular lesions seen in KD. Clinical trials to investigate the efficacy of anti-IL-1 modalities to prevent and treat KD vasculitis and aneurysm development should include agents that inhibit both IL-1α and IL-1β.
Supplementary Material
Significance.
This study highlights the critical requirement of IL-1α and IL-1β signaling, as well as CD11c+ DCs and macrophages in LCWE-induced vasculitis and coronary artery arteritis mouse model. IL-1 signaling is required only in stromal cells, most likely in VSMCs but not in endothelial cells.
These results further emphasize the importance of IL-1 and its downstream effects as they relate to the development of cardiovascular pathologies of KD and further strengthen the rational and need to design anti-IL-1 therapies for KD patients.
Acknowledgments
The authors would like to thank Wenxuan Zhang for technical support.
Funding Sources: Supported by grants from the National Institutes of Health AI072726 and AI070162 to M.A, American Heart Association 12POST9750014 to Y.L., and American Heart Association 15POST22810013 to D.W.
Non-standard Abbreviations and Acronyms
- LCWE
Lactobacillus casei cell wall extract
- EC
endothelial cell
- KD
Kawasaki Disease
Footnotes
Disclosures
None.
References
- 1.Kawasaki T, Kosaki F, Okawa S, Shigematsu I, Yanagawa H. A new infantile acute febrile mucocutaneous lymph node syndrome (mlns) prevailing in japan. Pediatrics. 1974;54:271–276. [PubMed] [Google Scholar]
- 2.Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, Crawford SE, Russo PA, Hammadeh R, Takahashi K, Orenstein JM. Rna-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute kawasaki disease. PLoS One. 2008;3:e1582. doi: 10.1371/journal.pone.0001582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowley AH, Shulman ST, Garcia FL, Guzman-Cottrill JA, Miura M, Lee HL, Baker SC. Cloning the arterial iga antibody response during acute kawasaki disease. J Immunol. 2005;175:8386–8391. doi: 10.4049/jimmunol.175.12.8386. [DOI] [PubMed] [Google Scholar]
- 4.Onouchi Y, Ozaki K, Burns JC, et al. A genome-wide association study identifies three new risk loci for kawasaki disease. Nature Genetics. 2012 doi: 10.1038/ng.2220. [DOI] [PubMed] [Google Scholar]
- 5.Shimizu C, Matsubara T, Onouchi Y, et al. Matrix metalloproteinase haplotypes associated with coronary artery aneurysm formation in patients with kawasaki disease. J Hum Genet. 2010;55:779–784. doi: 10.1038/jhg.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of kawasaki disease-cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. 2008;6:394–401. doi: 10.1038/nrmicro1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shulman ST, Rowley AH. Kawasaki disease: Insights into pathogenesis and approaches to treatment. Nat Rev Rheumatol. 2015 doi: 10.1038/nrrheum.2015.54. [DOI] [PubMed] [Google Scholar]
- 8.Holman R, Belay E, Christensen K, Folkema A, Steiner C, Schonberger L. Hospitalizations for kawasaki syndrome among children in the united states, 1997–2007. Pediatr Infect Dis J. 2010;29:483–488. doi: 10.1097/INF.0b013e3181cf8705. [DOI] [PubMed] [Google Scholar]
- 9.Burns JC. Kawasaki disease update. Indian J Pediatr. 2009;76:71–76. doi: 10.1007/s12098-009-0031-3. [DOI] [PubMed] [Google Scholar]
- 10.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of kawasaki disease: A statement for health professionals from the committee on rheumatic fever, endocarditis and kawasaki disease, council on cardiovascular disease in the young, american heart association. Circulation. 2004;110:2747–2771. doi: 10.1161/01.CIR.0000145143.19711.78. [DOI] [PubMed] [Google Scholar]
- 11.Burns JC, Glodé MP. Kawasaki syndrome. Lancet. 2004;364:533–544. doi: 10.1016/S0140-6736(04)16814-1. [DOI] [PubMed] [Google Scholar]
- 12.Orenstein JM, Shulman ST, Fox LM, Baker SC, Takahashi M, Bhatti TR, Russo PA, Mierau GW, de Chadarévian JP, Perlman EJ, Trevenen C, Rotta AT, Kalelkar MB, Rowley AH. Three linked vasculopathic processes characterize kawasaki disease: A light and transmission electron microscopic study. PLoS ONE. 2012;7:e38998. doi: 10.1371/journal.pone.0038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta-Malhotra M, Gruber D, Abraham SS, et al. Atherosclerosis in survivors of kawasaki disease. J Pediatr. 2009;155:572–577. doi: 10.1016/j.jpeds.2009.04.054. [DOI] [PubMed] [Google Scholar]
- 14.Fukazawa R. Long-term prognosis of kawasaki disease: Increased cardiovascular risk? Curr Opin Pediatr. 2010;22:587–592. doi: 10.1097/MOP.0b013e32833e12f7. [DOI] [PubMed] [Google Scholar]
- 15.Paredes N, Mondal T, Brandão LR, Chan AK. Management of myocardial infarction in children with kawasaki disease. Blood Coagul Fibrinolysis. 2010;21:620–631. doi: 10.1097/MBC.0b013e32833d6ec2. [DOI] [PubMed] [Google Scholar]
- 16.Burns JC, Capparelli EV, Brown JA, Newburger JW, Glode MP. Intravenous gamma-globulin treatment and retreatment in kawasaki disease. Us/canadian kawasaki syndrome study group. The Pediatric Infectious Disease Journal. 1998;17:1144–1148. doi: 10.1097/00006454-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E, Eichenfield JR, Martin DD, Newburger JW, Burns JC. Resistance to intravenous immunoglobulin in children with kawasaki disease. The Journal of pediatrics. 2008;153:117–121. doi: 10.1016/j.jpeds.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rowley AH. Finding the cause of kawasaki disease: A pediatric infectious diseases research priority. Journal of Infectious Diseases. 2006;194:1635–1637. doi: 10.1086/509514. [DOI] [PubMed] [Google Scholar]
- 19.Kato H, Sugimura T, Akagi T, Sato N, Hashino K, Maeno Y, Kazue T, Eto G, Yamakawa R. Long-term consequences of kawasaki disease. A 10- to 21-year follow-up study of 594 patients. Circulation. 1996;94:1379–1385. doi: 10.1161/01.cir.94.6.1379. [DOI] [PubMed] [Google Scholar]
- 20.Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group b lactobacillus casei cell walls in aqueous suspension. Arthritis & Rheumatism. 1985;28:652–659. doi: 10.1002/art.1780280609. [DOI] [PubMed] [Google Scholar]
- 21.Myones BL, Bathoria JM, Lehman TJ, Shulman ST. Human ivig inhibits lactobacillus casei-inducible coronary arteritis in a murine model. Proceedings of the V International Kawasaki Disease Symposium. 1995:252–256. [Google Scholar]
- 22.Schulte DJ, Yilmaz A, Shimada K, Fishbein MC, Lowe EL, Chen S, Wong M, Doherty TM, Lehman T, Crother TR, Sorrentino R, Arditi M. Involvement of innate and adaptive immunity in a murine model of coronary arteritis mimicking kawasaki disease. J Immunol. 2009;183:5311–5318. doi: 10.4049/jimmunol.0901395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehman TJA, Sherry B, Gietl DM, Nguyen HT, Cerami A. Suppression of lactobacillus casei cell wall-induced coronary arteritis in mice by antibody to murine tumor necrosis factor. Proceedings of the Third International Conference on Kawasaki Disease. 1988:203–206. [Google Scholar]
- 24.Yilmaz A, Rowley A, Schulte DJ, Doherty TM, Schröder NWJ, Fishbein MC, Kalelkar M, Cicha I, Schubert K, Daniel WG, Garlichs CD, Arditi M. Activated myeloid dendritic cells accumulate and co-localize with cd3+ t cells in coronary artery lesions in patients with kawasaki disease. Experimental and Molecular Pathology. 2007;83:93–103. doi: 10.1016/j.yexmp.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi K, Oharaseki T, Yokouchi Y, Naoe S, Saji T. Kawasaki disease: Basic and pathological findings. Clin Exp Nephrol. 2012 doi: 10.1007/s10157-012-0734-z. [DOI] [PubMed] [Google Scholar]
- 26.Brown TJ, Crawford SE, Cornwall ML, Garcia F, Shulman ST, Rowley AH. Cd8 t lymphocytes and macrophages infiltrate coronary artery aneurysms in acute kawasaki disease. J Infect Dis. 2001;184:940–943. doi: 10.1086/323155. [DOI] [PubMed] [Google Scholar]
- 27.Rowley AH, Shulman ST, Mask CA, Finn LS, Terai M, Baker SC, Galliani CA, Takahashi K, Naoe S, Kalelkar MB, Crawford SE. Iga plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute kawasaki disease. J Infect Dis. 2000;182:1183–1191. doi: 10.1086/315832. [DOI] [PubMed] [Google Scholar]
- 28.Rosenkranz ME, Schulte DJ, Agle LMA, Wong MH, Zhang W, Ivashkiv L, Doherty TM, Fishbein MC, Lehman TJA, Michelsen KS, Arditi M. Tlr2 and myd88 contribute to lactobacillus casei extract-induced focal coronary arteritis in a mouse model of kawasaki disease. Circulation. 2005;112:2966–2973. doi: 10.1161/CIRCULATIONAHA.105.537530. [DOI] [PubMed] [Google Scholar]
- 29.Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, Fishbein MC, Lehman TJ, Arditi M. Interleukin-1β is crucial for the induction of coronary artery inflammation in a mouse model of kawasaki disease. Circulation. 2012;125:1542–1550. doi: 10.1161/CIRCULATIONAHA.111.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shafferman A, Birmingham JD, Cron RQ. High dose anakinra for treatment of severe neonatal kawasaki disease: A case report. Pediatr Rheumatol Online J. 2014;12:26. doi: 10.1186/1546-0096-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing kawasaki disease rescued by il-1 receptor blockade and extracorporeal membrane oxygenation. Annals of the Rheumatic Diseases. 2012 doi: 10.1136/annrheumdis-2012-201658. [DOI] [PubMed] [Google Scholar]
- 32.Rowley AH, Shulman ST, Mask CA, Finn LS, Terai M, Baker SC, Galliani CA, Takahashi K, Naoe S, Kalelkar MB, Crawford SE. Iga plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute kawasaki disease. Journal of Infectious Diseases. 2000;182:1183–1191. doi: 10.1086/315832. [DOI] [PubMed] [Google Scholar]
- 33.Uchida K, Satoh M, Inoue G, Onuma K, Miyagi M, Iwabuchi K, Takaso M. Cd11c+ macrophages and levels of tnf-alpha and mmp-3 are increased in synovial and adipose tissues of osteoarthritic mice with hyperlipidemia. Clin Exp Immunol. 2015 doi: 10.1111/cei.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Motomura Y, Kanno S, Asano K, Tanaka M, Hasegawa Y, Katagiri H, Saito T, Hara H, Nishio H, Hara T, Yamasaki S. Identification of pathogenic cardiac cd11c+ macrophages in nod1-mediated acute coronary arteritis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015 doi: 10.1161/ATVBAHA.114.304846. ATVBAHA.114.304846. [DOI] [PubMed] [Google Scholar]
- 35.Persson EK, Scott CL, Mowat AM, Agace WW. Dendritic cell subsets in the intestinal lamina propria: Ontogeny and function. Eur J Immunol. 2013;43:3098–3107. doi: 10.1002/eji.201343740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Pasare C, Medzhitov R. Control of b-cell responses by toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 38.Takakura N, Huang XL, Naruse T, Hamaguchi I, Dumont DJ, Yancopoulos GD, Suda T. Critical role of the tie2 endothelial cell receptor in the development of definitive hematopoiesis. Immunity. 1998;9:677–686. doi: 10.1016/s1074-7613(00)80665-2. [DOI] [PubMed] [Google Scholar]
- 39.Dumont DJ, Yamaguchi TP, Conlon RA, Rossant J, Breitman ML. Tek, a novel tyrosine kinase gene located on mouse chromosome 4, is expressed in endothelial cells and their presumptive precursors. Oncogene. 1992;7:1471–1480. [PubMed] [Google Scholar]
- 40.Dagvadorj J, Shimada K, Chen S, Jones HD, Tumurkhuu G, Zhang W, Wawrowsky KA, Crother TR, Arditi M. Lipopolysaccharide induces alveolar macrophage necrosis via cd14 and the p2×7 receptor leading to interleukin-1α release. Immunity. 2015;42:640–653. doi: 10.1016/j.immuni.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weng K-P, Hsieh K-S, Ho T-Y, Huang S-H, Lai C-R, Chiu Y-T, Huang S-C, Lin C-C, Hwang Y-T, Ger LP. Il-1b polymorphism in association with initial intravenous immunoglobulin treatment failure in taiwanese children with kawasaki disease. Circulation journal: official journal of the Japanese Circulation Society. 2010 doi: 10.1253/circj.cj-09-0664. [DOI] [PubMed] [Google Scholar]
- 42.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dinarello CA. A clinical perspective of il-1β as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 44.Van Tassell BW, Toldo S, Mezzaroma E, Abbate A. Targeting interleukin-1 in heart disease. Circulation. 2013;128:1910–1923. doi: 10.1161/CIRCULATIONAHA.113.003199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhaskar V, Yin J, Mirza AM, Phan D, Vanegas S, Issafras H, Michelson K, Hunter JJ, Kantak SS. Monoclonal antibodies targeting il-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein e-deficient mice. Atherosclerosis. 2011;216:313–320. doi: 10.1016/j.atherosclerosis.2011.02.026. [DOI] [PubMed] [Google Scholar]
- 47.Maury CP, Salo E, Pelkonen P. Circulating interleukin-1 beta in patients with kawasaki disease. N Engl J Med. 1988;319:1670–1671. doi: 10.1056/NEJM198812223192515. [DOI] [PubMed] [Google Scholar]
- 48.Senzaki H. The pathophysiology of coronary artery aneurysms in kawasaki disease: Role of matrix metalloproteinases. Arch Dis Child. 2006;91:847–851. doi: 10.1136/adc.2005.087437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leung DY, Cotran RS, Kurt-Jones E, Burns JC, Newburger JW, Pober JS. Endothelial cell activation and high interleukin-1 secretion in the pathogenesis of acute kawasaki disease. Lancet. 1989;2:1298–1302. doi: 10.1016/s0140-6736(89)91910-7. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki H, Uemura S, Tone S, Iizuka T, Koike M, Hirayama K, Maeda J. Effects of immunoglobulin and gamma-interferon on the production of tumour necrosis factor-alpha and interleukin-1 beta by peripheral blood monocytes in the acute phase of kawasaki disease. Eur J Pediatr. 1996;155:291–296. doi: 10.1007/BF02002715. [DOI] [PubMed] [Google Scholar]
- 51.Guo H, Callaway JB, Ting JP. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, Kopf M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent il-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol. 2013;14:1045–1053. doi: 10.1038/ni.2704. [DOI] [PubMed] [Google Scholar]
- 53.Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, Kündig TM. Inflammasome activation and il-1β target il-1α for secretion as opposed to surface expression. Proc Natl Acad Sci U S A. 2011;108:18055–18060. doi: 10.1073/pnas.1109176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamari Y, Shaish A, Shemesh S, Vax E, Grosskopf I, Dotan S, White M, Voronov E, Dinarello CA, Apte RN, Harats D. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-e-deficient mice lacking bone marrow-derived interleukin-1α. Biochem Biophys Res Commun. 2011;405:197–203. doi: 10.1016/j.bbrc.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 55.Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. Il-1α and il-1β recruit different myeloid cells and promote different stages of sterile inflammation. Journal of immunology (Baltimore, Md: 1950) 2011;187:4835–4843. doi: 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- 56.Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, Rider P, Kaplanov I, Aychek T, Dinarello CA, Apte RN, Voronov E. Non-redundant properties of il-1α and il-1β during acute colon inflammation in mice. Gut. 2013 doi: 10.1136/gutjnl-2012-303329. [DOI] [PubMed] [Google Scholar]
- 57.Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burgner D, Davila S, Breunis WB, Ng SB, Li Y, Bonnard C, Ling L, Wright VJ, Thalamuthu A, Odam M, Shimizu C, Burns JC, Levin M, Kuijpers TW, Hibberd ML, International Kawasaki Disease Genetics C A genome-wide association study identifies novel and functionally related susceptibility loci for kawasaki disease. PLoS Genet. 2009;5:e1000319. doi: 10.1371/journal.pgen.1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang C-J, Kuo H-C, Chang J-S, Lee J-K, et al. Replication and meta-analysis of gwas identified susceptibility loci in kawasaki disease confirm the importance of b lymphoid tyrosine kinase (blk) in disease susceptibility. PLoS ONE. 2013;8:e72037. doi: 10.1371/journal.pone.0072037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuo H-C, Hsu Y-W, Lo M-H, Huang Y-H, Chien S-C, Chang W-C. Single-nucleotide polymorphism rs7251246 in itpkc is associated with susceptibility and coronary artery lesions in kawasaki disease. PLoS ONE. 2014;9:e91118. doi: 10.1371/journal.pone.0091118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Onouchi Y, Gunji T, Burns JC, et al. Itpkc functional polymorphism associated with kawasaki disease susceptibility and formation of coronary artery aneurysms. Nature Genetics. 2008;40:35–42. doi: 10.1038/ng.2007.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lou J, Xu S, Zou L, Zhong R, Zhang T, Sun Y, Lu X, Liu L, Li C, Wang L, Xiong G, Wang W, Gong F, Wu J. A functional polymorphism, rs28493229, in itpkc and risk of kawasaki disease: An integrated meta-analysis. Mol Biol Rep. 2012 doi: 10.1007/s11033-012-2022-0. [DOI] [PubMed] [Google Scholar]
- 63.Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the nlrp3 inflammasome. Trends in Immunology. 2014 doi: 10.1016/j.it.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alphonse MP, Duong TT, Shimizu C, Hoang LT, McCrindle BW, Franco A, Schurmans S, Philpott DJ, Hibberd ML, Burns JC, Kuijpers TW, Yeung RS. Abstract o.21: Inositol 1,4,5 triphosphate 3- kinase c regulates nlrp3 inflammasome activation in kawasaki disease. Circulation. 2015(131):AO21–AO21. [Google Scholar]
- 65.Bonin PD, Fici GJ, Singh JP. Interleukin-1 promotes proliferation of vascular smooth muscle cells in coordination with pdgf or a monocyte derived growth factor. Exp Cell Res. 1989;181:475–482. doi: 10.1016/0014-4827(89)90104-3. [DOI] [PubMed] [Google Scholar]
- 66.Sasu S, Beasley D. Essential roles of ikappab kinases alpha and beta in serum- and il-1-induced human vsmc proliferation. Am J Physiol Heart Circ Physiol. 2000;278:H1823–1831. doi: 10.1152/ajpheart.2000.278.6.H1823. [DOI] [PubMed] [Google Scholar]
- 67.Alexander MR, Moehle CW, Johnson JL, Yang Z, Lee JK, Jackson CL, Owens GK. Genetic inactivation of il-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. The Journal of Clinical Investigation. 2012;122:70–79. doi: 10.1172/JCI43713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arditi M. Animal models for understanding kawasaki disease. International Kawasaki Disease Symposium. 2015 [Google Scholar]
- 69.Johnson JL, Dwivedi A, Somerville M, George SJ, Newby AC. Matrix metalloproteinase (mmp)-3 activates mmp-9 mediated vascular smooth muscle cell migration and neointima formation in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:e35–44. doi: 10.1161/ATVBAHA.111.225623. [DOI] [PubMed] [Google Scholar]
- 70.Chua PK, Melish ME, Yu Q, Yanagihara R, Yamamoto KS, Nerurkar VR. Elevated levels of matrix metalloproteinase 9 and tissue inhibitor of metalloproteinase 1 during the acute phase of kawasaki disease. Clin Diagn Lab Immunol. 2003;10:308–314. doi: 10.1128/CDLI.10.2.308-314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hong YM, Jin H-S, Park IS, Hong S-J. Association of the matrix metalloproteinase-3 (−439c/g) promoter polymorphism with kawasaki disease in korean children. Heart Vessels. 2008;23:341–347. doi: 10.1007/s00380-008-1041-1. [DOI] [PubMed] [Google Scholar]
- 72.Takeshita S, Tokutomi T, Kawase H, Nakatani K, Tsujimoto H, Kawamura Y, Sekine I. Elevated serum levels of matrix metalloproteinase-9 (mmp-9) in kawasaki disease. Clinical & Experimental Immunology. 2001;125:340–344. doi: 10.1046/j.1365-2249.2001.01608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald H-R. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nature Immunology. 2014;16:36–44. doi: 10.1038/ni.3052. [DOI] [PubMed] [Google Scholar]
- 76.Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, Li PL. Endothelial nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochimica et biophysica acta. 2014 doi: 10.1016/j.bbamcr.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abe J-I, Berk BC. Athero-prone flow activation of the srebp2-nlrp3 inflammasome mediates focal atherosclerosis. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xiao H, Lu M, Lin TY, et al. Srebp2 activation of nlrp3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.113.002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xiang M, Shi X, Li Y, Xu J, Yin L, Xiao G, Scott MJ, Billiar TR, Wilson MA, Fan J. Hemorrhagic shock activation of nlrp3 inflammasome in lung endothelial cells. The Journal of Immunology. 2011 doi: 10.4049/jimmunol.1102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.