

Abstract
A hybrid nanoparticle (NP) consisting of a pH sensitive lipid shell and a poly(lactic-co-glycolic) acid (PLGA) core was constructed. This hybrid NP has a mean size of 120.1 ± 8.8 nm and positively charged surface (zeta potential of 14.2 ± 1.4 mV). The lipid shell of the hybrid NP was quickly disintegrated in buffer with a pH of 5.5, which resembles the acidic environment of endosomes in dendritic cell (DC). Less than 20% of the antigen enclosed in pH-sensitive hybrid NP was released into human serum at physiological pH within 24 h, but more than 40% of the enclosed antigen was released within 8 h after pH was adjusted to 5.5. Fast uptake of the pH sensitive hybrid NP by DC was also observed. It was found that pH sensitive hybrid NP displayed faster degradation and antigen release compared to regular hybrid NPs after uptake by DC.
Keywords: PLGA nanoparticle, pH-sensitive lipid, Vaccine
1. Introduction
As the most professional antigen presenting cell (APC), DCs play a critical role in adaptive immune response [1]. Immature DCs are widely and sparsely distributed in the periphery tissues, including skin, nose and lungs, scanning surrounding areas for invading foreign materials [2]. Upon internalizing antigens, DCs will be activated, commence maturation and start to migrate into the nearest lymph node, where the processed antigen peptides associated with MHC molecules can be presented to naïve T cells, triggering the subsequent activation of naïve T cells [3]. The maturation of DCs is of particular importance to vaccine development, because rapid maturation may lead to rapid activation of naïve T cells, and the degree of maturation can also affect the magnitude of the immune response via the expression of MHC molecules, costimulatory molecules, and cytokines by DC [4].
Currently, NP based vaccine delivery systems are considered to hold great promise for enhancing the immunogenicity of conventional antigens due to their advantages of more pathogen like morphology, co-delivery of adjuvants, high antigen loading capacity, protection of antigens such as proteins, peptides, and DNAs from degradation during circulation, and sustained release of antigens [5–7]. Normally, NPs are designed to be stable enough to avoid premature release of the enclosed effector molecules. However, it is possible that such stability may overprotect antigens from enzymatic processing in endosome after uptake by DCs, resulting in slow and incomplete induction of immune response, thus compromising the efficacy of the vaccine. It is reasonable to believe that the timely release of antigens and adjuvants contained in NPs after internalization may facilitate the maturation of DCs and promote immune response. Therefore, it is desirable to develop a delivery system that could not only protect the antigen from premature degradation, but also enable the timely release of antigens and adjuvants in the endosomes of DCs.
In addition to the common advantages enjoyed by NPs as mentioned above, NPs made from poly(lactic-co-glycolic) acid (PLGA) or lipids have been extensively used as vaccine and drug delivery systems due to their biocompatibility and biodegradable characteristics [8–10]. Hybrid NPs composed of PLGA and lipids have been constructed to improve cancer drug delivery and imaging diagnosis [11,12]. Previous work in our lab (data not published) showed that liposome-polymeric hybrid NPs could potentially be applied as a vaccine delivery system due to their large antigen loading capacity, tunable physiochemical properties, and excellent stability in human plasma. However, previous researchers mainly focused on how to improve the stability of NPs for antigen delivery [13,14], while ignoring the possibility that “overprotection” may occur to delay the release of antigens in target cells [15].
In order to devise a NP for vaccine delivery that can minimize the loss of the enclosed antigens during circulation as well as enable timely release of the antigens in immune cells, a lipid-PLGA hybrid NP was constructed. The hybrid NP on one hand exhibited excellent stability in human plasma to enable extended circulation of NPs, and on the other hand allowed rapid release of the enclosed antigen in response to acidification in endosomes after uptake by DCs. These features should enable antigens delivered by this NP to induce rapid and complete immune response and thus to realize prolonged immunity. The hybrid nanoparticle consists of a shell made from pH sensitive lipids and an inner PLGA core. The physicochemical properties of the nano hybrid were evaluated in terms of size, surface charge, morphology, in vitro release profile of the enclosed antigen, its uptake by DCs as well as intracellular release of the enclosed antigen. All nanoparticles tested exhibited good stability under physiological pH. However, in contrast to non-pH sensitive hybrid NPs, the lipid shell of pH sensitive NPs could be readily disintegrated to allow rapid release of the enclosed antigens in response to a low pH challenge. All results to date suggest that the pH sensitive hybrid NPs are particularly suitable for addressing the “overprotection” and are capable of controlling the release of the enclosed antigens.
2. Experimental section
2.1. Materials
Lactel® 50:50 PLGA was purchased from Durect Corporation (Cupertino, CA). Fetal bovine serum (FBS), GM-CSF recombinant mouse protein, minimum essential medium (MEM) α, trypsin/EDTA, CellMask™ Orange Plasma membrane Stain, Alexa Fluor® 647 Hydrazide, tris(triethylammonium) salt were purchased from Life Technologies Corporation (Grand Island, NY). Lipids, including 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), cholesteryl hemisuccinate (CHEMS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000), and 1,2-diphytanoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD PE) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Keyhole limpet hemocyanin (KLH), poly (vinyl alcohol) (PVA, Mw 89,000-98,000), dichloromethane (DCM), rhodamine B (RB), paraformaldehyde, Triton™ X-100 were purchased from Sigma–Aldrich Inc. (Saint Louis, MO). 1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC) was purchased from Thermo Fisher Scientific Inc. (Rd, Rockford, IL). JAWSII (ATCC® CRL-11904™) immature dendritic cells were purchased from ATCC (Manassas, VA). All other chemicals were of analytical grade.
2.2. Methods
2.2.1. Synthesis of keyhole limpet hemocyanin (KLH) containing PLGA (PK) NPs
PK NPs were prepared using a reported double emulsion solvent evaporation method with modifications [16]. Briefly, PLGA (20 mg) was dissolved in DCM (2 mL), followed by mixing with 50 mL of KLH (10 mg/mL) using vortex for 2 min. The resultant mixture was emulsified via sonication at 20% amplitude for 20 s using a sonic dismembrator (Model 500; Fisher Scientific, Pittsburg, PA). The primary emulsion was added drop-wise into 100 mL PVA (1% (w/v)), and continuously stirred for 10 min at 500 rpm. The above suspension was emulsified through sonication at 50% amplitude for 120 s. The secondary emulsion was stirred overnight to allow DCM to evaporate. Large particles were removed after settling the mixture in room temperature for 30 min. NPs in suspension were collected by centrifugation at 20,000 g, 4 °C for 30 min (Beckman Coulter Avanti J-251, Brea, CA). The pellet was washed 3 times using ultrapure water. The final suspension was freeze-dried (LABCONCO Freezone 4.5, Kansas City, MO), and NPs were stored at 2 °C for later use.
2.2.2. Assembly liposome-PK (LPK) hybrid NPs
A lipid film (10 mg) containing given lipids (as shown in Table 1) was hydrated with 5 mL, 55 °C pre-warmed hydration buffer (0.9% saline, 5% dextrose, and 10% sucrose). The resulting suspension was vigorously mixed using a vortex for 2 min, followed by incubation at 55 °C for 5 min and cooling to room temperature. PK NPs (10 mg) were added into the liposome suspension and pre-homogenized for 15 min using a Branson 2510 bath sonicator (Danbury, CT), followed by sonication on ice bath at 15% amplitude for 5 min (pulse on 20 s, pulse off 50 s) using a sonic dismembrator (Model 500; Fisher Scientific, Pittsburg, PA). The formed LPK NPs were collected by centrifugation at 20,000 g, 4 °C for 30 min, lyophilized, and stored at 2 °C.
Table 1.
Compositions and physicochemical properties of LPK hybrid NPs.
| LPK NPs | Lipid composition | Surface charge (mV ± SD) | Size (nm ± SD) | Polydispersity |
|---|---|---|---|---|
| LPK− | DOPC:cholesteral:DSPE-PEG2000 (w/w 8:0.5:1.5) | −11.7 ± 1.8 | 127.5 ± 5.2 | 0.18 ± 0.04 |
| LPK+ | DOTAP:cholesteral:DSPE-PEG2000 (w/w 8:0.5:1.5) | 24.7 ± 0.5 | 123.0 ± 7.4 | 0.20 ± 0.05 |
| LPKpH | DOPE:CHEMS:DSPE-PEG2000 (w/w 6.5:2.0:1.5) | 14.2 ± 1.4 | 120.1 ± 8.8 | 0.21 ± 0.02 |
2.2.3. Labeling KLH with RB or Alexa Fluor® 647 Hydrazide
The coupling of fluorescent dyes to KLH was done by a method described in a previous study [17]. 10 mg of EDC dissolved in 700 μL ultrapure water (pH 6.8) were mixed with 300 μL of 2 mg/mL RB. After incubation at 0 °C for 10 min, the product was mixed with 5 mg KLH (10 mg/mL) and stirred in darkness at room temperature for 12 h. For Alexa Fluor® 647 Hydrazide labeling, 5 mg of EDC in 800 μL ultrapure water (pH 6.8) was incubated with 5 mg KLH (10 mg/mL) at 0 °C for 10 min, followed by reaction with 100 μg Alexa Fluor® 647 Hydrazide in darkness at room temperature for 10 h. Fluorescently labeled KLH was purified using Microcon centrifugal filter units (50,000 MWCO, EMD Millipore, Billerica, MA), freeze-dried, and stored at 2 °C.
2.2.4. Characterizing physicochemical properties of NPs
1 mg of NPs was dispersed in 5 mL ultrapure water (pH 7.0) using a bath sonicator for 5 min. Each sample was diluted by 10 fold using ultrapure water, and the particle size (diameter, nm) and surface charge (zeta potential, mV) were measured using a Malvern Nano-ZS zetasizer (Malvern Instruments Ltd, Worcestershire, United Kingdom) at room temperature.
2.2.5. In vitro size stability of NPs under low pH
For these experiments, all the NPs were synthesized in the same batch. 1 mg of NPs was suspended in 5 mL, 10 mM phosphate buffered saline (PBS) with initial pH of 7.4, and then the pH was adjusted to 5.5 for both pH-sensitive NPs and regular hybrid NPs. The original particle size in pH 7.4 and that after being treated with pH 5.5 for 10 min were measured.
2.2.6. In vitro KLH release from NPs treated with low pH human plasma
5 mg of LPK NPs containing RB labeled KLH were suspended in 10 mL (5% v/v) human plasma (pH 7.4) and continuously stirred at room temperature. After 24 h incubation, the pH was lowered to 5.5 for each group and treated for another 48 h. The released KLH was separated from NPs via centrifugation at 20,000 g for 30 min at indicated time points. The NPs were re-suspended in human plasma and the released KLH in supernatant was measured using Synergy HT Multi-Mode Microplate Reader (BioTek Instruments, Inc., Winooski, VT), with excitation at 530 nm and emission at 590 nm. The percentage of released KLH at given time points was calculated using the following equation: % KLH released = Absor−bance at a given time point/Total absorbance × 100.
2.2.7. Imaging NPs using transmission electrical microscopy (TEM)
NP suspensions (1 mg/mL) before and after pH 5.5 treatment were dropped onto a 300-mesh Formvar-coated copper grid. After standing 10 min, the remaining suspension was carefully removed with wipes, and the samples were negatively stained using fresh 1% phosphotunstic acid for 60 s (phosphotungstic acid is electron dense and opaque for electrons, and it can easily adsorb onto nanoparticles. Therefore, it was used as the negative stain), and washed by ultrapure water twice. The dried samples were imaged on a JEOL JEM 1400 Transmission Electron Microscope (JEOL Ltd., Tokyo, Japan).
2.2.8. Imaging LPK NPs using confocal laser scanning microscope (LSM)
Fluorescently labeled LPKpH NPs were prepared exactly using the methods mentioned above, except that KLH was labeled with Alexa Fluor® 647 Hydrazide and 0.5 mg NBD PE was supplemented into 10 mg of the existing pH sensitive lipids to label lipid layer. A Zeiss LSM 510 Laser Scanning Microscope (Carl Zeiss, German) was used to image these freshly labeled NPs. Lipids layer with NBD PE was viewed using 488 nm laser and PLGA core with Alexa Fluor® 647 was viewed using 633 nm laser.
2.2.9. Fourier transform infrared (FT-IR) spectroscopy analysis of NPs
The spectrum of freeze-dried pH sensitive liposome, PLGA NPs, and the hybrid NPs were recorded on a Thermo Nicolet 6700 FT-IR spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). The spectrum was taken from 4000 to 400 cm−1.
2.2.10. Flow cytometry measurement of endocytosis of NPs by DCs
JAWSII (ATCC® CRL-11904™) immature dendritic cells from ATCC were cultured with alpha minimum essential medium (80%v) including ribonucleosides, deoxyribonucleosides, 4 mM L-glutamine, 1 mM sodium pyruvate and 5 ng/mL murine GM-CSF, and fetal bovine serum (20%v) at 37 °C, 5% CO2 in 12 well plates (CORNING, Tewksbury, MA). NPs were assembled according to the above mentioned method, except 0.25 mg NBD PE was added into the existing lipids. 200 μg of NPs were added into each well containing 2 × 106 cells, and incubated for 5 h. After incubation, the medium was immediately removed and cells were washed 5 times with ultrapure water. Cells were detached from culture plate using trypsin/EDTA solution and centrifuged at 200 g for 10 min, then cell pellets were re-suspended in 10 mM PBS (pH 7.4). Cell samples were immediately analyzed by flow cytometry (BD FACSAriaI, BD, Franklin Lakes, NJ).
2.2.11. Imaging endocytosis of NPs by DCs using LSM
Cells were cultured in a 4 well chamber slide (Thermo Fisher Scientific Inc., Rd, Rockford, IL) using the same method described above. 0.1 mg NPs were incubated with 2 × 105 cells for 8 h at 37 °C, 5% CO2. After incubation, the medium was immediately removed and cells were washed with 5 times with ultrapure water. Freshly prepared 4% (w/v) paraformaldehyde (500 μL) was added into each well, and cells were fixed for 15 min. This was followed by washing 3 times with PBS (10 mM, pH 7.4). Fixed cells were permeabilized using 500 μL of 0.1% (v/v) Triton™ X-100 for 15 min at room temperature, and washed 3 times using PBS (10 mM, pH 7.4). Cells were stained using 500 μL of freshly diluted 1X CellMask™ Orange Plasma membrane Stain for 15 min, and washed 3 times using PBS (10 mM, pH 7.4). Cell samples were covered with a glass cover and sealed by nail polish. Images were acquired using a Zeiss LSM 510 Laser Scanning Microscope (Carl Zeiss, Germany).
2.2.12. Imaging distribution of NPs in DCs using TEM
Petri dishes containing 4 × 106 immature DCs were supplemented with 500 μg of the three types of NPs. After 5 h incubation, the medium containing un-internalized NPs was removed and cells were washed with 5 times with ultrapure water. Cell samples were prepared for TEM using the following procedure: Cells were washed 2 times in 0.1 M Na-Cacodylate for 15 min each, and then post-fixed in 1% OsO4 in 0.1 M Na-Cacodylate for 1 h. OsO4 was discarded, and the samples were washed two times for 10 min each in 0.1 M Na-Cacodylate. Cell samples were dehydrated in solutions containing increasing ethanol concentration as follows: 15%, 30%, 50%, 70%, 95%, and 100% (15 min in each ethanol solution). Dehydration was completed by submerging cell samples in propylene oxide for 15 min. Cells were infiltrated with a 50:50 solution of propylene oxide:Poly/Bed 812 for 6–24 h, then embedded using freshly prepared 100% Poly/Bed 812 in flat embedding molds, and placed in a 60 °C oven for at least 48 h to cure. Images were acquired using a JEOL JEM 1400 Transmission Electron Microscope (JEOL Ltd., Tokyo, Japan).
2.2.13. Statistical analysis
All experiments were performed in at least triplicate. Data were expressed as mean ± standard deviation (SD). Significant tests were conducted using one-way ANOVA following Tukey test (JMP pro 10). Differences were considered significant at P-values that were less than or equal to 0.05.
3. Results and discussion
Lipid-PLGA hybrid NPs have been widely accepted as outstanding systems for delivery of cancer chemotherapeutic agents [18]. In a previous study, we found that lipid-PLGA hybrid NPs may also be used as vaccine delivery systems due to their high antigen loading capacity, ability in co-delivering both antigen and adjuvants, and desired stability for long term storage [19]. However, to be an excellent vaccine delivery system, the above mentioned features of lipid-PLGA NPs are not enough. A desirable delivery system needs to meet at least three criteria: 1) antigen-containing NPs can maintain high stability during circulation, reducing the loss of payloads; 2) NPs are able to readily disassemble to release the payloads upon internalization by the target cells, maximizing their efficacy; 3) NPs can be easily taken up by antigen presenting cells (APCs).
The main findings of this study are that 1) a lipid-PLGA hybrid NP with a pH-sensitive lipid layer can be constructed via sonication mediated fusion, 2) this pH-sensitive hybrid NP exhibited high sensitivity to low pH challenge, 3) DCs could effectively internalize pH-sensitive hybrid NPs, and 4) antigen was released more rapidly from pH-sensitive hybrid NPs than non pH-sensitive hybrid NPs after internalization by DCs.
3.1. Characterization of the physicochemical properties of LPK hybrid NPs
The hydrodynamic size and ploydispersity of various NPs were characterized using dynamic light scattering (DLS), and the results are shown in Table 1. The average sizes for negatively charged LPK (LPK−) NP, positively charged LPK (LPK+) NP, and pH sensitive LPK (LPKpH) NP are 127.5 ± 5.2 nm, 123.0 ± 7.4 nm, and 120.1 ± 8.8 nm (mean ± SD), respectively. Surface charges, measured in term of zeta potential, were −11.7 ± 1.8 mV, 24.7 ± 0.5 mV, and 14.2 ± 1.4 mV, respectively, which evidently depend on the lipid composition of NPs. The polydisperisty of NPs ranges between 0.18 ± 0.04 and 0.21 ± 0.02 for the three different types of particles. The hybridization of lipids (labeled with NBD PE, green) and PLGA NPs containing KLH (labeled with Alexa Fluor® 647 Hydrazide, red) was verified via confocal microscopy. As shown in Fig. 1, all particles that were labeled with red (left panel) were concomitantly stained with green fluorescence (middle panel), indicating a lipid shell was successfully formed outside PK NPs. Moreover, no disassociated liposomes in the merged image were detected, suggesting that liposomes could be readily coated onto PK NPs. The size and narrow size distribution of NPs displayed in the confocal images were consistent with the size and small polydispersity of NPs measured by DLS. In order to further confirm that pH sensitive lipids were coated onto PLGA NPs, the FTIR spectrums of pH-sensitive liposomes, PLGA NP, and pH sensitive liposome-PLGA (LPpH) hybrid NP were taken. As shown in Fig. 2, a peak of LPpH NP was observed at 1081 nm, which includes contributions by the peaks at 1062 nm and 1089 nm of the liposome and PLGA NP, respectively. Compared to the other two NPs, an increased peak is shown at 1228 nm for LPpH NPs, due to the contributions of the peaks at 1220 nm and 1234 nm of the liposome and PLGA NPs, respectively. A combined peak also occurs at 1739 nm with LPpH NPs. In addition, a unique peak belonging to liposome at 2852 nm is shown in hybrid NPs. Similarly, a peak at 3297 nm, which only occurs with PLGA NPs, was also detected in hybrid NPs. Considering all the evidence together, the results from FTIR analysis prove that the pH sensitive liposome was successfully hybridized with PLGA NPs.
Fig. 1.
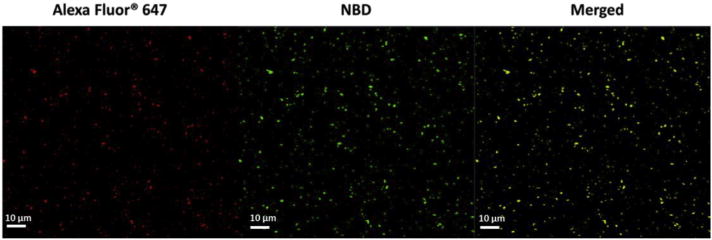
Confocal LSM images of LPKpH NPs, in which KLH was labeled with Alexa Fluor® 647, and lipids were labeled with NBD.
Fig. 2.
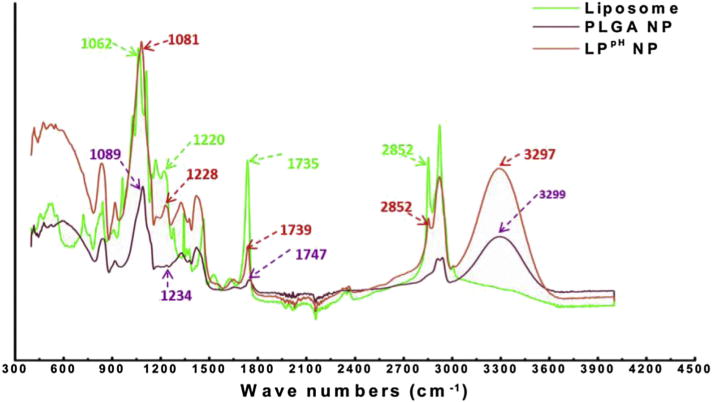
FTIR spectra of the pH sensitive liposome, PLGA NP, and LPpH hybrid NP. A unique peak belonging to liposome at 2852 nm is shown in LPpH hybrid NP. Similarly, a peak at 3297 nm, which only occurs with PLGA NPs, is also shown in hybrid NPs.
The morphology of PK NPs and LPK NPs was studied using TEM (Fig. 3). PK NP is schematically illustrated in Fig. 3A, and its TEM image is displayed in Fig. 3C. PK NP is a spherical particle with a diameter around 100 nm. In contrast, LPK NP has a lipid shell outside PK NP, as illustrated in Fig. 3B, and its TEM image (Fig. 3D) confirms that there is a lipid layer coating the white spherical PK NP.
Fig. 3.
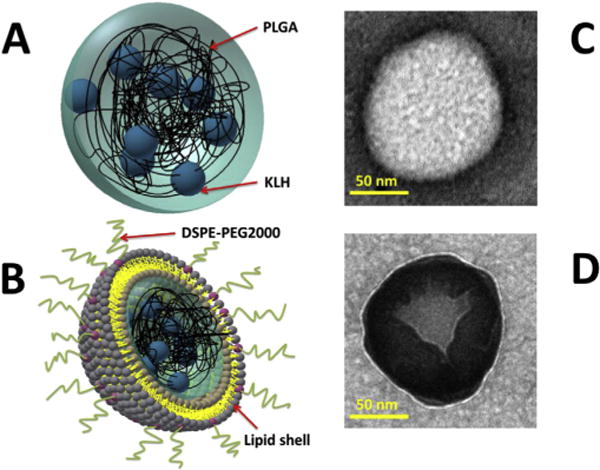
Schematic illustration of PK NP (A) and LPK NP (B). TEM images of PK NP (C) and LPK NP (D). PK NP is a spherical particle (white) with a diameter around 100 nm. In contrast, LPK NP has a lipid shell (grey) outside PK NP core (white).
3.2. Stability of NPs in response to a low pH treatment
The stability of NPs was evaluated by measuring the change of their size. The size distributions for each particle before and after the low pH treatment are shown in Fig. 4. Neither LPK− NPs (Fig. 4A) nor LPK+ NPs (Fig. 4B) underwent significant size changes after being treated with pH 5.5 PBS buffer for 10 min. The average sizes with and without the low pH treatment were 123.6 ± 1.8 nm and 122.6 ± 2.0 nm, 122.9 ± 2.0 nm and 120.7 ± 3.7 nm for LPK− NPs (Fig. 4A) and LPK+ NPs (Fig. 4B), respectively. In contrast, significant size changes under conditions of low pH were detected for pH sensitive LPK NPs. The average size of LPKpH NPs increased from 118.4 ± 2.3 nm to 146.6 ± 1.5 nm after 10 min of the low pH treatment (Fig. 4C). Evidently, the size distribution curve of LPKpH NPs shifted to the right after a low pH challenge, which was consistent with the increase in average size of LPKpH NPs. The stability of NPs was also studied using TEM, and the morphologies of NPs in both pH 7.4 and pH 5.5 PBS buffer were compared (Fig. 5). The three NPs share strong resemblance in their morphology in pH 7.4 PBS buffer: there is an apparent lipid shell (grey layer) coating the PK NPs (white core). The size distribution, which centers around 100 nm, is also in agreement with those obtained by DLS and confocal microscopy. Not surprisingly, non-pH sensitive NPs remained intact after being suspended in pH 5.5 PBS buffer for 10 min (Fig. 5A and B), but pH sensitive NPs disassembled in the low pH environment (Fig. 5C). Formerly regularly shaped lipid shells can be seen to disintegrate into thread-like lipid aggregates (blue arrow), and PK NPs (red square) were released and distributed around the debris of lipid shells.
Fig. 4.
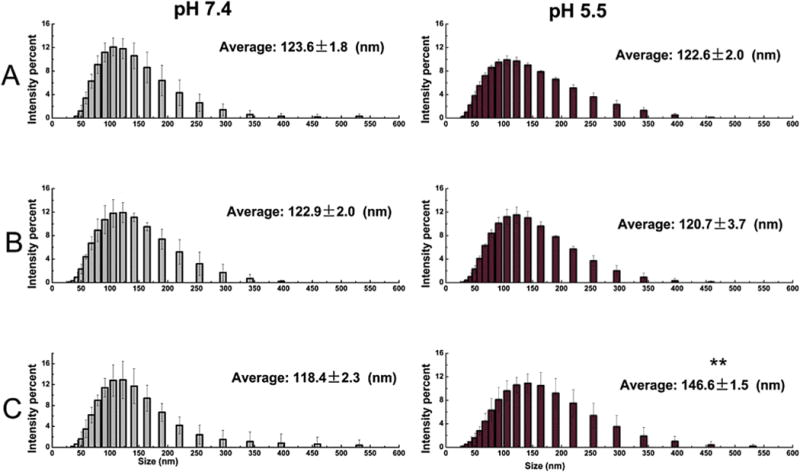
The stability (size) of the three LPK NPs in response to a pH 5.5 treatment for 10 min (A) LPK− NPs, (B) LPK+ NPs, (C) LPKpH NPs. ** indicates that significant size increase occurred after LPKpH NPs were treated with pH 5.5 PBS buffer (P-value < 0.05).
Fig. 5.
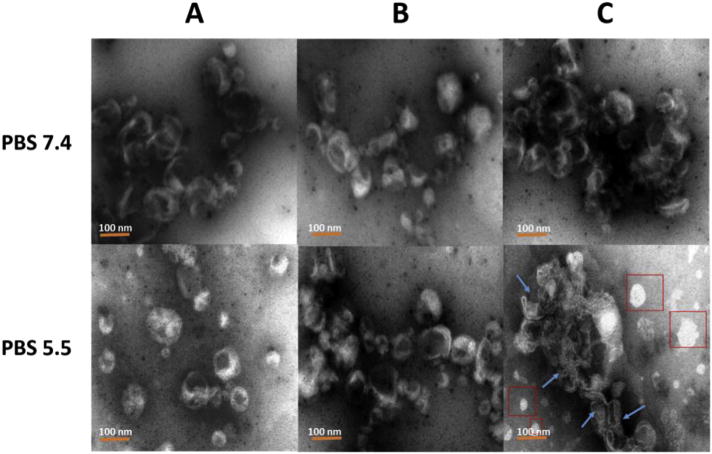
TEM images of the three LPK NPs in both pH 7.4 and pH 5.5 PBS buffer. (A) LPK− NPs, (B) LPK+ NPs, (C) LPKpH NPs. In contrast to other two NPs, LPKpH NP underwent significant morphology change after low pH treatment. The blue arrows show degraded lipid layer, and red boxes show released PK NPs. The scale bars represent 100 nm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The rapid disintegration of LPKpH NPs in acidic environment probably resulted from poor stability of DOPE in the lipid layer under low pH condition [20]. Lipid layer of the hybrid NP can protect antigens and adjuvants from premature release during circulation. However, it may also hinder the release of antigen and adjuvants after uptake by DCs, resulting in slow or even incomplete maturation of DCs. Therefore, the rapid disassociation of lipid layer from PLGA core in response to low pH challenge is of great significance. It may allow rapid release of antigens and adjuvants, providing DCs strong stimuli for their rapid and complete maturation.
3.3. In vitro release of KLH from LPK NPs
In this study, KLH was used as a model antigen for evaluating the capacity of controlled release of antigen by hybrid NPs. To mimic the conditions in vivo, NPs underwent two phases of pH treatment, in which NPs were treated with pH 7.4 human serum for the first 24 h and pH 5.5 human serum for an additional 48 h. In the first 24 h, all NPs exhibited a similarly sustained and slow KLH release profile, of which less than 20% of the KLH was released for all NPs, reflecting the high stability of LPK hybrid NPs under pH 7.4. Such a slow release at pH 7.4 can minimize antigen loss during circulation and allow sufficient amount of antigen to reach immune cells to induce strong immune response. However, significantly different KLH release profiles of the LPK NPs were observed after the pH was lowered to 5.5 at the 24 h time point. In the first 8 h after the pH change, 40% of the KLH was released from LPKpH NPs, while only 11% and 14% of KLH were released from LPK+ NPs and LPK− NPs, respectively. The accelerated KLH release from LPKpH NPs is in agreement with the results from NP stability study in Figs. 4 and 5. In response to a low pH environment, the hybrid structure of LPKpH NPs could disintegrate rapidly, resulting in accelerated antigen release. This accelerated antigen release under low pH in human serum suggests that antigen might also be rapidly released from NPs in the acidic environment of DC endosomes, which might lead to rapid development of a strong immune response.
The true percentage of KLH released from non-pH sensitive NPs should be smaller than the values shown in Fig. 6, because the KLH release rates were still noticeably high even at 72 h. The sharp increase in KLH release of LPKpH NPs was likely due to the removal of the protection provided by the lipid shell after low pH treatment. Between 32 h and 72 h, sustained release of KLH continued for all NPs, except the release rates were considerably higher than those during the first 24 h. When approaching 72 h, LPKpH NPs displayed a nearly flat release curve, indicating LPKpH NPs had almost depleted its payload. The other two NPs showed a considerably higher release rates, suggesting that a large amount of KLH was still retained in the NPs. It is known that PLGA is subject to continuous bulk erosion in aqueous solution [11]. The stored antigen in PLGA core may consistently be released during PLGA degradation. However, due to the existence of the lipid layer, the released antigen may be retained in the inter-space between the PLGA core and the lipid layer. Te removal of the lipid layer caused burst release of antigen.
Fig. 6.
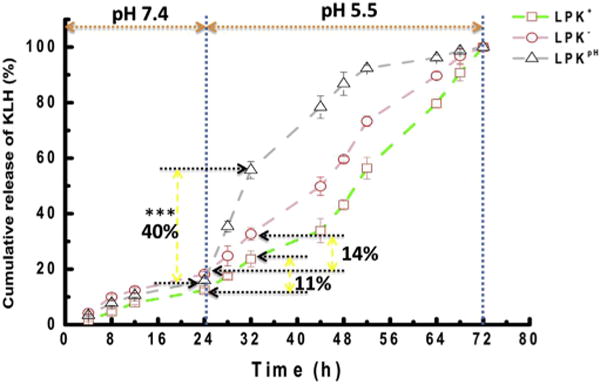
KLH release profile of LPK NPs in human serum with two phases of pH treatment, in which from 0 to 24 h, pH was 7.4, and from 24 to 72 h, pH was 5.5. *** indicates that the percentage of KLH released from LPKpH between 24 h and 32 h was significantly higher than that from either LPK− NPs or LPK+ NPs (P-value < 0.01).
It is also important to note that LPKpH NPs displayed outstanding stability as the other two NPs did in human serum at pH 7.4. This stability might be attributed to the protection offered by the long PEG chains placed on the surface of the particles and reduced diffusion of enzymes from the human serum into the inner core of NPs due to the barrier provided by the lipid shell [21,22]. As a matter of fact, PEG molecules have been routinely used in biodegradable NPs to improve stability of NPs [23]. Reducing the release of the enclosed antigens or adjuvant during circulation is of great value, because it can not only reduce the loss of effector molecules that target immune cells, but also minimize the potential systemic toxicity caused by adjuvants [24].
3.4. The uptake of LPK NPs by DCs
The uptake rates of the three NPs by DCs were compared using flow cytometry. 200 μg of NPs labeled with NBD were incubated with 2 × 106 DCs for 5 h, and the fluorescence intensity in the cells was measured. The results (Fig. 7) show that DCs internalized more LPK+ NPs than either LPKpH or LPK− NPs. The mean fluorescence intensities of the internalized NPs were 9164, 7448, 5631, respectively. To some degree, the uptake rate of the NPs by DCs appears to be related to their surface charge, implying that positive surface charge might facilitate NP uptake. This finding was consistent with the results from a previous study [19]. Since cell membrane is negatively charged, the positive surface charge of NPs may improve cellular uptake via electrostatic interactions between immune cells and NPs. It was also shown that positive charges on the surface of NPs could promote the DCs maturation and enhance vaccine induced immune response [25]. In addition to the positive surface charges, the presence of fusion-promoting DOPE molecules in the lipid shell may also contribute to the faster uptake of LPKpH NPs than LPK− NPs [26]. The fast uptake of NPs based vaccine is meaningful in at least three aspects: 1) it may allow fast development of immune response, 2) it may enable immune system to use vaccine components more efficiently, and 3) it reduces the potential systemic toxicity from adjuvants that may be released from NPs after long term circulation.
Fig. 7.
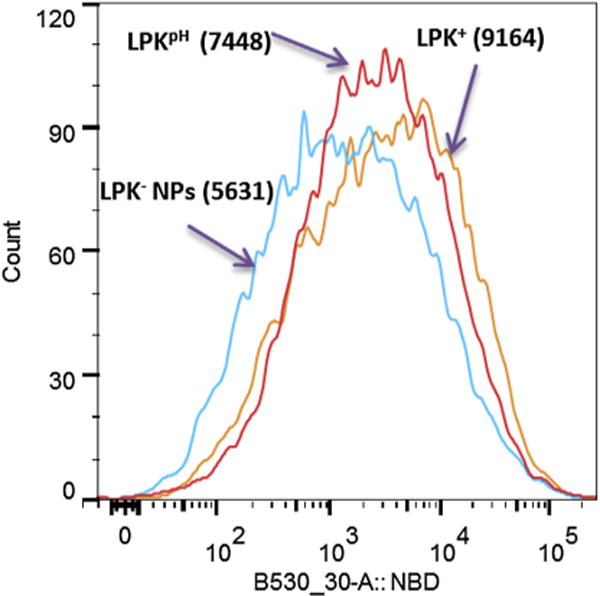
The intensity of NBD fluorescence in DCs, which internalized the three LPK NPs, respectively. LPK+ NPs exhibited the fastest uptake rate by DCs, while LPKpH NP could be more readily taken up by DCs in comparison to LPK− NPs. The numbers in parentheses represent the mean NBD intensity.
Dendritic cell is the most professional antigen presenting cell that plays a core role in eliciting immune response [27]. The fate of NPs in DCs after internalization is of particular interest to vaccine researchers, because NP degradation and trafficking may have profound impact on the types and magnitude of immune responses [28,29]. An intense and sustained stimulation of DCs by the antigens and adjuvants released from NPs may lead to a strong and prolonged adaptive immune response. The internalization of NPs by DCs was also measured using confocal microscopy. KLH was labeled with Alexa Fluor® 647, the lipid shell was labeled with NBD, and cells were labeled with CellMask™ Orange Plasma Membrane Stain. As shown in Fig. 8, all three NPs were internalized by DCs, indicated by the presence of both red and green fluorescence in the cells. However, the state of NPs and their distribution in cells differed from one to another. The green fluorescence in LPK− NPs (Fig. 8A) was obviously weaker than that of the either LPK+ NPs (Fig. 8B) or LPKpH (Fig. 8C), suggesting that less LPK− NPs was internalized by DCs compared to the other two NPs. This is consistent with the results obtained from flow cytometry. As shown in Alexa Fluor® 647 panels, most of LPK+ and LPK− NPs existed as distinct and intact particles, while the red fluorescence in LPKpH NPs spread over the whole cell. These observations suggest that most of the non-pH sensitive NPs remained stable in cells, but pH sensitive NPs underwent significant degradation, resulting in the release of KLH into the entire cell. As discussed above, the rapid antigen release may come from the antigen accumulated in the inter-space between PLGA core and lipid layer. Such a burst release of antigen may result in more processed antigenic peptides. These antigenic peptides could subsequently associate with MHC molecules, form peptide-MHC (pMHC) complexes, and be transported to the membrane surface of DC for T cell activation [30]. To prime naive T cells, there has to be a sufficient number of interactions between T cell receptors and pMHC complexes. Higher density of pMHC complexes may thus increase the likelihood of such interactions [31]. Therefore, burst release of antigen from pH-sensitive hybrid NPs in DCs may promote the magnitude of immune response.
Fig. 8.
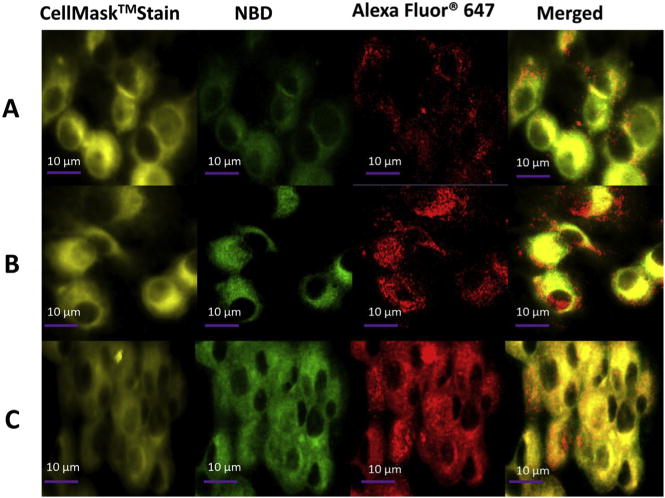
Confocal LSM images of DCs after LPK NP uptake, in which KLH was labeled with Alexa Fluor® 647, and lipids were labeled with NBD. Dendritic cells were treated with (A) LPK− NPs, (B) LPK+ NPs, and (C) LPKpH NPs, respectively. The intracellular release of KLH from LPKpH NPs was faster than those in other two NPs.
The endocytosis of NPs by DCs was further examined with TEM (Fig. 9). No endosome vesicles were formed in the control group, to which no NPs were added (Fig. 9A). On the other hand, numerous endosomes (blue arrow) were observed in DCs treated with the three NPs (Figs. 9 B–D). Consistent with previous results, DCs internalized less LPK− NPs (Fig. 9B) than either LPK+ (Fig. 9C) or LPKpH NPs (Fig. 9D). In particular, two huge rod-like endosomes containing considerable numbers of LPK+ NPs were present in DCs, implying that positively charged NPs might induce the formation of larger endosomes for NPs uptake. Another interesting and important phenomenon was that the NPs in endosomes of DCs treated with either of the non-pH sensitive NPs retained a regular spherical shape, while the morphology of the NPs in the pH sensitive group could not be defined, indicating that LPKpH NPs might have been degraded by acidification in endosomes.
Fig. 9.
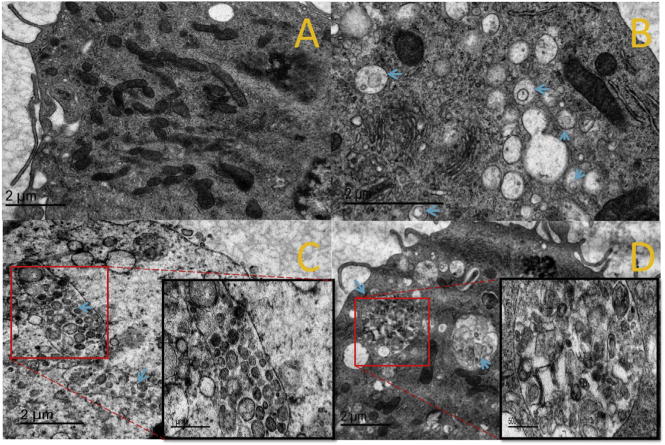
TEM images of DCs after LPK NPs internalization. (A) Control group, (B) LPK− NPs, (C) LPK+ NPs, (D) LPKpH NPs. Blue arrows show the position of endosomes, which were more numerous in groups treated with NPs compared to the control group, and LPKpH NPs underwent significant degradation in endosomes. Images in black boxes are zoomed-in pictures of endosomes. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The results of the present experiment suggest that shortly after being internalized by DCs, the lipid shell of LPKpH would be removed, leading to a burst release of KLH followed by sustained and prolonged release. Such a two-phase antigen release in DCs may initially enable a strong immune response and provide prolonged stimulation to the immune system.
4. Conclusion
In this study, we have shown that a pH-sensitive liposome-polymeric hybrid NP can be successfully manufactured via sonication assisted hybridization of a pH-sensitive lipid layer and a PLGA core. The hybrid NP had a size of 120.1 ± 8.8 nm and a surface charge of 14.2 ± 1.4 mV. Largely due to its nano-range size, positive surface charges, and hydrophobic lipid shell, it can be readily internalized by DC.
In buffer at physiological pH, pH-sensitive hybrid NP displayed slow antigen release as regular hybrid NPs, indicating that the pH-sensitive NP may minimize antigen loss during circulation. However, under acidic environment, the hybrid structure of the pH-sensitive hybrid NP was readily disassembled, and the enclosed antigen was more rapidly released. After internalization by DC, pH-sensitive NP released antigen faster than regular hybrid NPs. The rapid release of the enclosed antigens in DC may enable the hybrid NPs to serve as a potential delivery system of antigens and adjuvants for rapid and complete activation of the immune system.
Acknowledgments
This work was financially supported by National Institutes of Health, more specifically, National Institute on Drug Abuse (R21DA030083 and U01DA036850).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
Yun Hu participated in experimental design and execution and drafting of the manuscript. Zongmin Zhao participated in part of the experiments. Chenming Zhang participated in experiment design, coordinated the experiments, and conducted manuscript revision. Marion Ehrich participated in experiment design. Kristel Fuhrman participated in experiments related to dendritic cell culture. All authors read and approved the final manuscript.
References
- 1.Caminschi I, Shortman K. Trends Immunol. 2012;33:71–77. doi: 10.1016/j.it.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Shortman K, Lahoud MH, Caminschi I. Exp Mol Med. 2009;41:61–66. doi: 10.3858/emm.2009.41.2.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Adv Drug Deliv Rev. 2011;63:943–955. doi: 10.1016/j.addr.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau J, Steinman RM. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 5.Park YM, Lee SJ, Kim YS, Lee MH, Cha GS, Jung ID, Kang TH, Han HD. Immune Netw. 2013;13:177–183. doi: 10.4110/in.2013.13.5.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith DM, Simon JK, Baker JR., Jr Nat Rev Immunol. 2013;13:592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Date AA, Destache CJ. Biomaterials. 2013;34:6202–6228. doi: 10.1016/j.biomaterials.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin-Banderas L, Duran-Lobato M, Munoz-Rubio I, Alvarez-Fuentes J, Fernandez-Arevalo M, Holgado MA. Mini Rev Med Chem. 2013;13:58–69. [PubMed] [Google Scholar]
- 9.Gregory AE, Titball R, Williamson D. Front Cell Infect Microbiol. 2013;3:13. doi: 10.3389/fcimb.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joshi VB, Geary SM, Salem AK. AAPS J. 2013;15:85–94. doi: 10.1208/s12248-012-9418-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC. Biomaterials. 2009;30:1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 12.Valencia PM, Basto PA, Zhang L, Rhee M, Langer R, Farokhzad OC, Karnik R. ACS Nano. 2010;4:1671–1679. doi: 10.1021/nn901433u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galindo-Rodriguez SA, Allemann E, Fessi H, Doelker E. Crit Rev Ther Drug Carr Syst. 2005;22:419–464. doi: 10.1615/critrevtherdrugcarriersyst.v22.i5.10. [DOI] [PubMed] [Google Scholar]
- 14.Prasad S, Cody V, Saucier-Sawyer JK, Fadel TR, Edelson RL, Birchall MA, Hanlon DJ. Pharm Res. 2012;29:2565–2577. doi: 10.1007/s11095-012-0787-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelosi A, Shepherd R, Walmsley AM. Biotechnol Adv. 2012;30:440–448. doi: 10.1016/j.biotechadv.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 16.Yang YY, Chia HH, Chung TS. J Control Release. 2000;69:81–96. doi: 10.1016/s0168-3659(00)00291-1. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Zheng H, Huang W, Zhang C. Hum Vaccin Immunother. 2014;10:64–72. doi: 10.4161/hv.26635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. ACS Nano. 2008;2:1696–1702. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, Ehrich M, Fuhrman K, Zhang C. Nanoscale Res Lett. 2014;9:434. doi: 10.1186/1556-276X-9-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hafez IM, Ansell S, Cullis PR. Biophys J. 2000;79:1438–1446. doi: 10.1016/S0006-3495(00)76395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gindy ME, Ji S, Hoye TR, Panagiotopoulos AZ, Prud’homme RK. Biomacromolecules. 2008;9:2705–2711. doi: 10.1021/bm8002013. [DOI] [PubMed] [Google Scholar]
- 22.Martins S, Sarmento B, Ferreira DC, Souto EB. Int J Nanomed. 2007;2:595–607. [PMC free article] [PubMed] [Google Scholar]
- 23.Li SD, Huang L. J Control Release. 2010;145:178–181. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montomoli E, Piccirella S, Khadang B, Mennitto E, Camerini R, De Rosa A. Expert Rev Vaccin. 2011;10:1053–1061. doi: 10.1586/erv.11.48. [DOI] [PubMed] [Google Scholar]
- 25.Ma Y, Zhuang Y, Xie X, Wang C, Wang F, Zhou D, Zeng J, Cai L. Nanoscale. 2011;3:2307–2314. doi: 10.1039/c1nr10166h. [DOI] [PubMed] [Google Scholar]
- 26.Huth US, Schubert R, Peschka-Suss R. J Control Release. 2006;110:490–504. doi: 10.1016/j.jconrel.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Liu YJ, Kanzler H, Soumelis V, Gilliet M. Nat Immunol. 2001;2:585–589. doi: 10.1038/89726. [DOI] [PubMed] [Google Scholar]
- 28.Cubas R, Zhang S, Kwon S, Sevick-Muraca EM, Li M, Chen C, Yao Q. J Immunother. 2009;32:118–128. doi: 10.1097/CJI.0b013e31818f13c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallace A, West K, Rothman AL, Ennis FA, Lu S, Wang S. Hum Vaccin Immunother. 2013;9:2095–2102. doi: 10.4161/hv.26009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanzavecchia A, Sallusto F. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 31.Anikeeva N, Gakamsky D, Scholler J, Sykulev Y. PLoS One. 2012;7:e41466. doi: 10.1371/journal.pone.0041466. [DOI] [PMC free article] [PubMed] [Google Scholar]