

Abstract
The umbrella and penknife models hypothesize that insecticidal Bacillus thuringiensis Cry toxins partition into the apical membrane of the insect midgut by insertion of only two α-helices from domain I of the protein, α-helices 4 and 5 in the case of the umbrella model and α-helices 5 and 6 in the case of the penknife model. Neither model envisages membrane partitioning by domains II and III. In this study, we present data suggesting that mutations in the domain II residue, F371, affect insertion of the whole toxin into Manduca sexta brush border membrane vesicles (BBMVs). Using steady state fluorescence measurements combined with a proteinase K protection assay, we show that mutants of F371 have lost their ability to insert into the BBMV, even though binding to cadherin is almost unaffected. The study also identifies a difference in partitioning of toxins into artificial lipid vesicles (SUVs) as opposed to native BBMVs. While the F371 mutations block insertion of domains I and II into BBMVs, they only block domain II insertion into SUVs. Bioassay and voltage clamping of midguts also confirm the fluorescence data that the noninserting mutants are nontoxic. Our study leads us to propose that, in contrast to previous models of individual free helices inserting into the membrane, the toxin enters into the membrane as a whole molecule or oligomers of the molecule, wherein the domain II residue F371 has a vital role to play in membrane insertion.
Insecticidal crystal proteins (Cry toxins) produced by the soil bacterium Bacillus thuringiensis are a large family of toxins that target a wide range of insects and nematodes but are harmless to mammals (1). The crystal toxins belonging to the Cry1A series target the insect order Lepidoptera. They are produced as inactive protoxins and are activated inside the alkaline lepidopteran gut by proteases. Crystal structures of the active toxin in solution (2, 3) have shown that the toxin has 3 structural domains. Domain I is an α-helical bundle made of 7 α-helices. Domain II is composed of antiparallel beta sheets, and domain III is a β-sandwich. The active form binds to one or more receptors on the brush border membrane vesicles (BBMV1) of the insect, including cadherins, alkaline phosphatases, and/or one or more forms of the aminopeptidases (4–6). The receptor bound toxin then is proposed to undergo several conformational changes such as aggregation and oligomer formation (7, 8) before inserting into the membrane.
Our understanding of the mechanism at the final step of toxin insertion into brush border membranes is incomplete. Initial models of insertion of toxin into the membrane, the umbrella and penknife models, suggest that only α-helix 4 and 5 of domain I, in the case of the umbrella model, and α-helices 5 and 6 in the case of the pen knife model, insert to form an ion channel (9–11). However, there is no conclusive evidence presented on the fate of domain II and domain III, which account for 60% of the bulk of the toxin, upon insertion. Studies using nonspecific proteases on Cry toxins that have been inserted into the BBMV show that almost the entire toxin of 60 kDa is protected inside the membrane (12–15); only α-helix 1 is lost from the active toxin in the membrane-bound state.
Site-directed mutagenesis studies involving replacement of single amino acids in domain II of Cry1Ab toxin (16, 17) show that mutations in phenylalanine 371 residue to several residues including cysteine, serine, alanine, valine, tyrosine, and phenylalanine do not affect the competition binding of the toxin to BBMV, but there was a significant loss of toxicity that is inversely correlated to the hydrophobicity of the replaced residues. This could have been due to the hydrophobic residue playing a role in tighter binding of the toxin to BBMV or due to the regions of domain II including loop 2 being inserted into the apical membrane of the gut. The present study presents evidence supporting the latter model of domain II inserting into the membrane.
EXPERIMENTAL PROCEDURES
Site-Directed Mutagenesis
The cell culture containing the B. thuringiensis δ-endotoxin gene for Cry1Ab (cry1Ab9033) was obtained from T. Yamamoto (Sandoz Agro Inc., Palo Alto, CA). Uracil-containing template of Cry1Ab was obtained as described (17). Primers for site-directed cysteine and alanine mutagenesis were obtained from Integrated DNA technologies, Inc. Site-directed mutagenesis was carried out using the MutaGene M13 In Vitro Mutagenesis kit as described in the manufacturer’s manual (BioRad). The presence of the expected mutations was confirmed by sequencing double-stranded DNA at the Plant Microbe Genomics Facility, Ohio State University, Columbus, Ohio.
Transformation, Expression, and Protein Purification
Expression and purification of the Cry1Ab wild-type and mutant toxins were carried out as described elsewhere (18). Proteins, in protoxin form, were extracted from inclusion bodies by dissolving the crystals in 50 mM sodium carbonate at pH 10.5 at 37 °C with shaking for 2 h. The presence of the protein was assessed in SDS (8%)–PAGE gels. The total concentration of the proteins was estimated using Coomassie Protein Assay reagent (Pierce Biotechnologies, Inc.). Activated toxins were obtained by digesting the protoxin in 1/100 (trypsin/protoxin) at 37 °C for 30 min. The toxin was purified by anion exchange using Sepharose Q column (GE Healthcare) followed by size exclusion chromatography using Sephacryl S300 and Superdex 200 columns (GE Healthcare) in series. The elution solvent was 50 mM carbonate buffer at pH 10.5 at a flow rate of 1.0 mL/min and 2.0 mL/min for the 2 gel filtration columns.
Preparation of Small Unilamellar Vesicles
1-Palmitoyl-2-oleyl-sn-glycerol-3-phophatidylcholine, 1-palmitoyl-2-oleyl-sn-glycerol-3-phosphatidylethanolamine, and cholesterol (Avanti Polar lipids Inc.) were used in the ratio of 7:2:1 dissolved in chloroform and dried in a stream of nitrogen. These phospholipids are the predominant ones from studies that profiled the lipid environment in the insect midgut (19) and have been used in several earlier studies on insertion of Cry toxins into lipid bilayers or artificial vesicles to mimic the insect gut environment (20, 21). The resulting multilamelar vesicles were dried free of any residual chloroform. Vesicles were resuspended in 10 mM HEPES and 150 mM NaCl (pH 7.4) and sonicated in a Branson Sonifier water bath for 30 min to form small unilamellar vesicles (SUVs). Vesicles were subjected to light scattering to ensure a uniform size class among each batch of preparation (22).
Preparation of BBMV
M. sexta eggs (Carolina Biological Supplies, Inc.) were hatched and larvae were raised to the fifth instar on artificial diet (Bio Serv, Inc.). Dissection of the insect gut is as described elsewhere (23). BBMV were prepared by modified differential magnesium precipitation method (24). The final BBMV pellet was resuspended in binding buffer (10 mM HEPES and 150 mM NaCl at pH 7.4). Protein concentration was estimated using Coomassie Protein Assay reagent (Pierce Biotechnologies, Inc.)
Proteinase K Protection Assays
Pure toxin (10 μg) in 50 mM Na2CO3 (pH 10.5) was mixed with 100 μg of BBMV and incubated at 25 °C for 30 min. After incubation, 100 μg of proteinase K (Recombinant grade; Roche Biochemicals) was added to the mixture and incubated at 37 °C for 30 min. At the end of incubation with proteinase K, 1 mM PMSF was added to stop any further reaction. The mixture was centrifuged at 15,000g for 10 min. The pellet was washed in 10 mM HEPES and 150 mM NaCl (pH 7.4), and then treated with 1% n-octyl-β-D-glucopyranoside (Sigma) to dissolve the pellet and boiled for 3–5 min before loading on SDS–PAGE gels. Proteins were transferred from the gel onto a PVDF membrane, and Western blotting was performed using rabbit polyclonal anti-1A antibody (1:5000) and goat antirabbit HRP tagged antibody (BioRad). Blots were visualized using Immune-HRP substrate (BioRad).
Labeling of Purified Cysteine Mutant Toxins
Purified cysteine mutants were mixed with 10-fold molar excess of 6-acryloyl-2-dimethylaminonapthalene (Acrylodan) (Invitrogen Inc.) and incubated in the dark overnight. The labeled protein was purified off the free label using a desalting Sephadex G25 column (GE Healthcare). Purity of the labeled protein was checked on an 8% SDS–PAGE gel, and the degree of labeling was estimated using the molar extinction coefficient of acrylodan. Fifty micrograms of the labeled protein was mixed with 500 μg BBMV or 5 mg of SUV and incubated for 60 min. Bound forms of the labeled protein were separated from the unbound labeled proteins by either centrifuging the BBMV pellet down at 15,000g or by passing the SUVs through a Sephadex G100 column (GE Healthcare). The BBMV or SUV was treated with 500 μg of proteinase K and incubated for 30 min at 37 °C. One mM PMSF was added to stop the reaction. The reaction was then spun down at 15,000g to recover the BBMV or passed through Sephadex G 100 column (GE Healthcare) to recover the SUV.
Fluorescence Measurements
Steady state fluorescence measurements were carried out on Fluoromax 3 fluorimeter (JY Horiba Instruments). The labeled proteins were excited at 360 nm, and emission intensity was measured from 390–600 nm. The labeled protein in solution, bound to BBMV and proteinase K treated, were measured simultaneously to avoid any instrumentation error. Spectra were corrected for background from buffer and/or the vesicles. Each experiment was performed three times. Emission spectra were plotted using relative fluorescence of the mutants in buffer to that in the membrane before and after proteinase K treatment. The fluorescence values on the Y axis represent relative arbitrary units and are not scaled to absolute values of intensity of fluorescence.
Surface Plasmon Resonance Analysis
Surface plasmon resonance experiments on BIAcore 3000 were performed for kinetic analysis. BIAcore’s carboxymethylated dextran matrix (CM5) sensor chip was used. The analysis temperature was set to 25 °C. Around 15,000 RU of anti-MBP IgG was immobilized on the surface of flow cell 2 on the CM5 sensor chip using an EDC/NHS-mediated amine coupling procedure. A freshly prepared solution of 50 mM NHS (N-hydroxysuccinimide) and 0.2 M EDC [1-ethyl-3-(3-dimethylaminopropyl) carbodiimide] was injected for 7 min to activate the flow cell. Anti-MBP IgG was reconstituted in 10 mM NaCOOH at pH 5.0 and injected at a flow rate of 10 μL/min. Excess activated ester groups on the surface were deactivated using a 7 min injection of 1 M ethanolamine-HCl at pH 8.5. Flow cell 1 was activated with 50 mM NHS and 0.2 M EDC and blocked with 1 M ethanolamine-HCl at pH 8.5 without immobilization of anti-MBP IgG, serving as the reference surface.
Maltose binding protein–receptor fusion protein, MBP-CAD-D was constructed as a subclone of the M. sexta CAD gene provided to us by the Adang laboratory in pMECA vector (25) including CAD regions 11 and 12. MBP-CAD-D at 50 μg/mL was injected at a flow rate of 5 μL/min to interact with IgG on the chip. Over a 3 min period of time, approximately 100 RU of MBP-receptor was captured. The (anti-MBP IgG)-(MBP-CAD-D) surface was allowed to stabilized for 1 min before wild type toxin at various concentrations was injected. Buffer only was included as a blank. The flow rate for toxin injections was at 30 μL/min. The association phase was 3 min, and the dissociation phase was 10 min. Regeneration was achieved by two 30-s injections of 10 mM glycine at pH 1.8 at 100 μL/min. The control flow cell that was activated and blocked without immobilization of the antibody had both MBP-receptor fusion protein and toxin flowing through in each cycle. Running buffer in all experiments was HBS-P buffer (0.01 M HEPES at pH 7.4, 0.15 M NaCl, and 0.005% surfactant). MBP-CAD-D fusion protein was purified in the same buffer, while toxin was prepared in sodium carbonate buffer and filter-dialyzed into the HBS-P buffer.
Toxicity Bioassays
Toxicity levels were determined on first instar M. sexta larvae. The median lethal concentration (LC50) was estimated by diet surface contamination assays. First instar larvae were confined in 24-well sterile dishes (Falcon) containing solidified artificial diet surface-contaminated with activated toxin. Five to six toxin concentrations were prepared for each assay. Twenty M. sexta larvae were used for each concentration. Mortalities were scored after 5 days. The LC50 for each toxin was calculated by Probit analysis using SoftTox (WindowChem Software, Inc.).
Secondary Structural Studies by Circular Dichroism
Purified proteins (labeled or unlabeled with acrylodan) were used for CD analysis at 2 uM concentration in 50 mM Na2CO3 buffer at pH 10.5. Five hundred microliter samples were used in a 1 cm path length quartz cuvette (Starna Cells Inc.). The spectra were collected in an AVIV CD2 spectropolarimeter at 25 °C. Ellipticity was measured as a function of wavelength from 300 to 200 nm in 1 nm increments and plotted on SigmaPlot 2000 (Jandel Scientific Co.). Each CD spectra shown is the average of 10 scans.
Voltage Clamp Analysis
Voltage clamp analysis was performed as described earlier (23). After stabilization of the midguts in the buffer (26), 100 ng of each toxin was added into the lumen side of the chamber. The volume of the lumen chamber is 3.75 mL. The inhibition of short circuit current (Isc) was measured with a DVC-1000 Vage/current clamp (World Precision Instruments, Sarasota, FL) connected to a MacLab-4 (AD Instruments, Mountain View, CA). Data analysis was performed with SigmaPlot 2000 (Jandel Scientific Co.). Recorded data was normalized to the percentage of Isc remaining. Each experiment was repeated at least 3 times.
RESULTS
Production of Stable Toxins
The desired cysteine and alanine mutations were obtained as described in Experimental Procedures and were verified using DNA sequencing. The wild type and mutant proteins were expressed in E. coli DH5α as 130 kDa protoxin molecules. All four proteins used in this study were digested with trypsin to yield 65 kDa toxin molecules. The protoxin and toxin molecule sizes were verified on an 8% SDS–PAGE gel (data not shown). The trypsin activated mutant toxins displayed the same stability as the wild type activated toxin. All activated toxins were purified through an ion exchange column and 2 gel filtration columns to remove all smaller fragments and higher order molecules generated in the process of solubilization and/or activation. All toxins showed a 65 kDa band on 8% SDS–PAGE gels post-purification (data not shown). The secondary structures of the purified mutants were compared to that of the wild type Cry1Ab using CD spectrometry.
Toxicity Bioassays
The biological activity of each toxin was measured using the surface contamination method against M. sexta larvae and the results reported as LC50 (concentration required to kill 50% of the larva tested) as shown in Table 1.
Table 1.
Bioassay Measurements of Cry1Ab and Its Mutants on 1st Instar Larvae of M. sexta (Tobacco Hornworms) Using the Surface Contamination Methoda
| sample | LC50 M. sexta ng/cm2 |
|---|---|
| Cry1Ab | 20.00 [7.5–31.7] |
| 1AbV171C | 40.3 [26.6–53.4] |
| 1AbF371C | >2000 |
| 1AbV171C/F371A | >2000 |
| acrylodan labeled 1Ab V171C | 36.6 [24.6–48.2] |
| acrylodan labeled 1Ab F371C | >2000 |
| acrylodan labeled 1Ab V171C/F371A | >2000 |
Eight larvae were used per concentration of toxin. The results were measured after 5 days of incubation and calculated as LC50 using Probit analysis (Softtox).
Surface Plasmon Resonance Analysis
The binding of the mutant toxins to the toxin binding cadherin repeats 11 and 12 in the BT-R1 sequence was compared to that of Cry1Ab wild type toxin. Table 2 shows that compared to the wild type (KD ~18 nM), the 1AbF371C (KD ~ 48 nM) and the double mutant 1AbF371A/V171C (KD ~ 66 nM) have only a minor decrease in binding.
Table 2.
Binding Measurements of the Toxin 1Abwt and Its Mutants to Cadherin Repeats 11 and 12 Using Surface Plasmon Resonance Analysis (Biacore)a
| sample | ka (1/Ms) | kd (1/s) | KD (M) | χ2 | |
|---|---|---|---|---|---|
| 1Abwt: | exp 1 | 6.51E+04 | 1.17E-03 | 1.79E-08 | 1.08 |
| exp 2 | 6.42E+04 | 1.21E-03 | 1.89E-08 | 0.859 | |
| 1AbV171C: | exp 1 | 7.64E+04 | 1.84E-03 | 2.41E-08 | 6.02 |
| exp 2 | 7.04E+04 | 1.55E-03 | 2.21E-08 | 1.1 | |
| 1AbF371C: | exp 1 | 3.66E+04 | 1.79E-03 | 4.89E-08 | 5.38 |
| exp 2 | 3.83E+04 | 1.83E-03 | 4.78E-08 | 6.32 | |
| 1AbF371A/V171C: | exp 1 | 1.83E+04 | 1.25E-03 | 6.80E-08 | 6.58 |
| exp 2 | 1.83E+04 | 1.17E-03 | 6.41E-08 | 2.15 |
The results are expressed as KD values (ratio of the kd:ka) and are expressed in molar units. Sets of measurements for each sample were performed twice, as shown.
Proteinase K Protection Assays
Protection assays were carried out on toxin bound BBMV to determine if each of our toxins inserted into the BBMV. Western blot analysis show that while the wild type and the toxic mutant 1AbV171C were protected from 10-fold excess of proteinase K even after 30 min of incubation at 37 °C, seen as a 60 kDa band on the gel, the domain II mutant 1AbF371C and the double mutant 1AbV171C/F371A were completely digested by the nonspecific protease in the same conditions (Figure 1). The results suggest that the change in residue F371 to cysteine has an affect on the insertion process. We have observed that F371A has the same affect (not shown).
Figure 1.
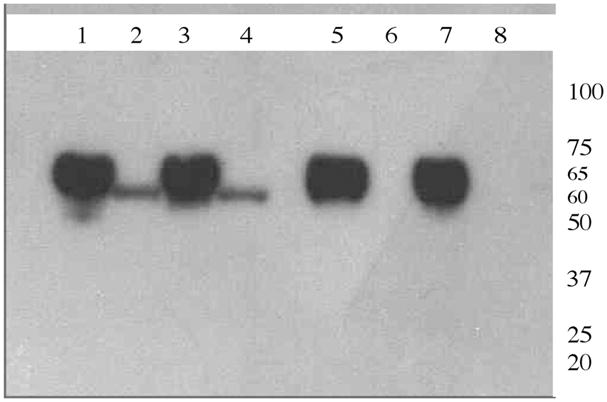
Proteinase K protection assay of Cry1Ab wt and its mutants. Reaction was run on 4–20% SDS–PAGE gels, and the membranes were blotted using anti-1A polyclonal antibody and HRP tagged anti rabbit secondary antibody. Lane1: Pure Cry1Abwt (10 μg). Lane 2: proteinase K treated BBMV bound to Cry1Ab wt. Lane 3: Pure 1AbV171C (10 μg). Lane 4: proteinase K treated BBMV bound to 1AbV171C. Lane 5: Pure 1AbF371C (10 μg). Lane 6: proteinase K treated BBMV bound to 1AbF371C. Lane 7: Pure 1AbF371A/V171C (10 μg). Lane 8: proteinase K treated BBMV bound to 1AbF371A/V171C.
Circular Dichroism Analysis
The cysteine mutants in the activated form were labeled with acrylodan, and the secondary structure of the labeled mutants were checked using circular dichroism spectrophotometry. CD spectra of the cysteine mutants showed that the labeling had not affected the global structure of the mutant 1AbF371C, and the double mutant 1AbF371A/V171C when compared to the wild-type (Figure 2B and C). The CD spectrum of 1AbV171C differs from the wild-type, but the labeled and unlabeled spectra are similar (Figure 2A).
Figure 2.
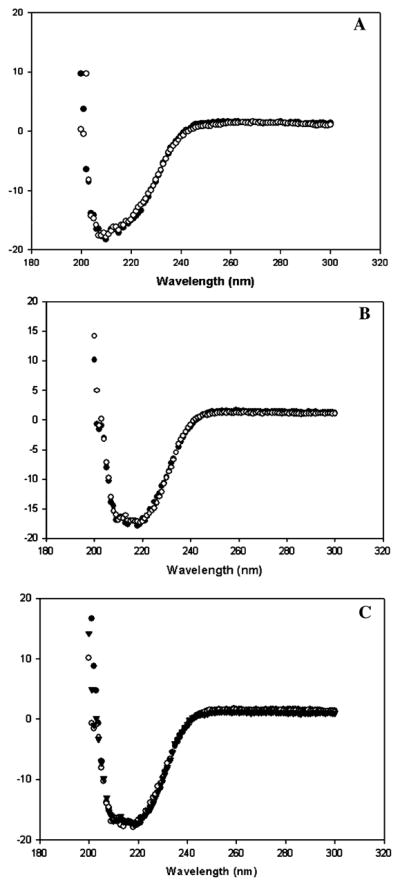
Circular dichroism spectra of Cry1Ab mutants V171C (A), 1AbF371C (B), and 1AbF371A/V171C (Figure 2C) before (○) and after (●) labeling with acrylodan expressed in mol · deg−1 · cm−1 units.
Fluorescence Measurements
The fluorescence emission of acrylodan is highly sensitive to the environment of the fluorophore, with the fluorescence maxima in a hydrophilic environment of ≥480 nm and <460 nm in a hydrophobic environment (27, 28). Our studies with the label show that while the excited-state emission of the acrylodan bound to the protein is very low in solution there is a dramatic blue shift in fluorescence emission when the labeled protein is in either artificial SUVs or in BBMVs. The dipole moment of the label being highly sensitive to the environment, the emission of each labeled mutant is different in the free form itself depending on the location of the mutation (27). While a blue shift is the predominant indication of the change in the environment, the intensity of the emission is also a representation of the environment of the label (28).
The acrylodan-labeled toxic mutant protein, 1AbV171C shows a blue shift in maximal emission wavelength from 500 ± 10 nm in free solution to 462 ± nm in BBMV and to about the same value (462 ± 11 nm) in BBMV treated with proteinase K (Figure 3A). Table 3 records the maximal wavelengths for each mutant bound to either SUV or BBMV before and after proteinase K treatment. That the shift was not from hydrophobic effects of the receptors outside the vesicles was ensured by the emission spectra from the proteinase K treated BBMV or SUVs bearing the labeled toxin since receptors are not present in the case of SUVs and are removed from BBMV by proteinase K. The intensity of emission of the spectra both before and after proteinase K treatment was similar for this mutant suggesting that the particular region of the toxin was embedded into the vesicles. The acrylodan-labeled domain II mutant, 1AbF371C mixed with vesicles (both BBMVs and SUVs) showed blue shift in the spectra but upon proteinase K treatment showed a significant drop in fluorescence intensity (Figure 3C and 3F).
Figure 3.

Steady state fluorescence spectra of Cry1Ab mutants labeled with acrylodan. The samples were excited at 360 nm and the resultant emission recorded from 390 to 650 nm. The relative fluorescence (y-axis) is expressed in arbitrary units. Correction of the spectra was made against either a buffer blank for the free protein in solution or against SUV or BBMV for the protein bound to the respective vesicles. (−) represents the spectra of the purified labeled protein in solution. (· · ·) represents the spectra of the pure labeled protein bound to SUV or BBMV before proteinase K treatment. (---) represents the spectra of labeled protein bound to BBMV or SUV after proteinase K treatment. The y-axis represents the relative fluorescence intensity of each sample under one condition (buffer) to another (in membrane before and after proteinase K) in arbitrary units and may not be interpreted as an absolute value of intensity of fluorescence. A: Cry1AbV171C treated with BBMV. B: Cry1AbF371A/V171C treated with BBMV. C: Cry1AbF371C treated with BBMV. D: Cry1AbV171C treated with SUV. E: Cry1AbF371A/V171C treated with SUV. F: Cry1AbF371C treated with SUV.
Table 3.
| (A) | |||
|---|---|---|---|
| maximal emission wavelength of acrylodan labeled protein (nm) |
|||
| in carbonate buffer | bound to BBMV | bound to BBMV + PK | |
| 1AbV171C (Figure 3A) | 500 ± 10 | 462 ± 9 | 462 ± 11 |
| 1AbV171C/F371A (Figure 3B) | 486 ± 0.5 | 465 ± 15 | 458 ± 5 |
| 1AbF371C (Figure 3C) | 484 ± 1 | 482 ± 2 | 497 ± 2 |
| (B) | |||
| maximal emission wavelength of acrylodan labeled protein (nm) |
|||
| in carbonate buffer | bound to SUV | bound to SUV + PK | |
| 1AbV171C (Figure 3D) | 493 ± 2 | 475 ± 3 | 470 ± 6 |
| 1AbV171C/F371A (Figure 3E) | 492 ± 1 | 472 ± 0 | 480 ± 3 |
| 1AbF371C (Figure 3F) | 492 ± 0 | 466 ± 5 | 474 ± 1 |
However, for the double mutant (Figure 3B), where the protein was labeled with acrylodan in domain I (position 171), the protein was able to insert into the SUVs and was also protected from proteinase K in these artificial vesicles as seen by protection of the label in the protease treated vesicles. Upon proteinase K treatment of the toxin bound to BBMV, there was a loss of fluorescence intensity.
Voltage Clamping Analysis
To confirm the lack of pore formation of the non-inserting mutants, we carried out voltage clamping of M. sexta guts and measured the percentage of remaining short circuit current in the gut upon addition of Cry1Ab and its mutant proteins used in this study. The results as shown in Figure 4 demonstrate that while the domain I mutant V171C can form ion channels better than Cry1Ab wild type, the domain II mutant and the double mutant proteins have completely lost their ability to form pores in the midgut membrane. These data, while they do not indicate toxicity directly, correlate the loss in formation of ion channel to the loss in toxicity seen in the bioassay results in Table 1.
Figure 4.
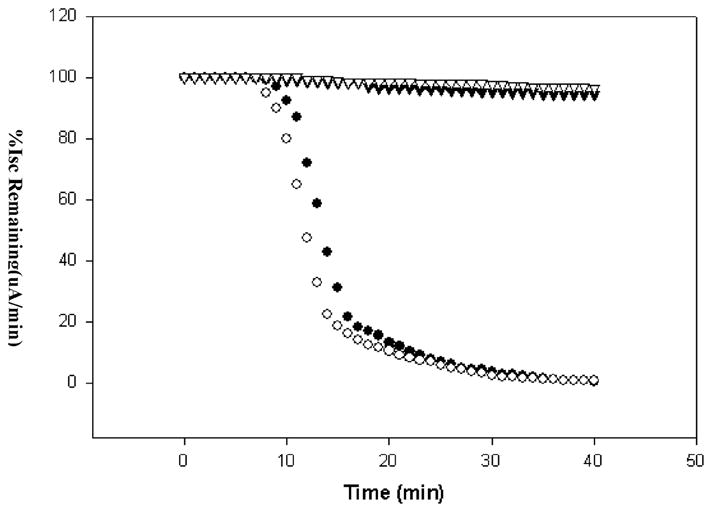
Voltage clamp response of Cry1Abwt (●) compared to that of BBMV inserting mutant 1AbV171C (○) and non-BBMV inserting mutants 1AbF371C (▼) and 1AbF371A/V171C (▽).
DISCUSSION
The process of insertion of Cry toxins has been studied actively since 1994. Most studies have limited their focus to the fate of domain I of the toxin in the inserted state in membranes, despite work that has shown protection of the whole toxin inside the membrane (7, 12, 14, 15). Our study present mutants in domain II of the Cry1Ab toxin, at position F371, that allow receptor binding but prevent insertion of the toxin into the membrane. Proteinase K protection combined with steady state fluorescence measurements of labeled toxin molecules effectively demonstrates that the residue F371 plays a role in the mechanism of insertion, at least in Cry1Ab and for M. sexta.
Rajamohan, et. al (17) reported that mutating residue F371 to a number of amino acids did not affect its competition binding to M. sexta BBMV, but affected toxicity and irreversible binding in a manner inversely related to the hydrophobicity of the replacement amino acid. At the time of that study, it was unclear whether irreversible binding was due to tightness of binding to the receptor or proficiency of insertion into the membrane. Our binding studies of the mutant protein 1Ab-F371C and the double mutant protein 1Ab-F371A/V171C to cadherin receptor (Dorsch TBR sequence of repeats 11 and 12 (29)) using surface plasmon resonance support the view that these mutants do not suffer significant loss (2.6-fold for F371C and 3.6 fold for F371C/V171C) in binding to M. sexta cadherin when compared to the wild type protein. This loss of binding (Table 2) is unlikely to account for the >100-fold loss in toxicity (Table 1) and complete loss in ion channel activity (Figure 4). However, the data from toxicity bioassays and the voltage clamping studies reflect the inability of the mutant forms to retain their toxicity. This indicates a role for the residue F371 in associations of the toxin with the BBMV post-receptor-binding membrane insertion. Steady state fluorescence measurements of the bound and protected toxin show a blue shift in the maximal wavelength of the domain I mutant protein, suggesting the displacement of the label to a hydrophobic (membrane) environment (27). The hydrophobicity could hypothetically be due to binding of toxin to receptor (assuming the receptor is sufficiently hydrophobic); however, since binding is not greatly affected, this seems unlikely. In addition, one would expect proteinase K treatment would be able to access and digest the region of the receptor exposed outside the membrane and thereby also digest the toxin, given the incubation conditions of 30 min. Furthermore, when V171C, a domain I residue that is not believed to make contact with the receptor is labeled with acrylodan, we observe that the label migrates to a more hydrophobic environment in BBMV (Figure 3A). Our data from the proteinase K for the domain I mutant protein 1AbV171C and other mutants (manuscript in progress) dispersed across the toxin confirm that the label is in an environment not accessible by the protease suggesting that it is embedded into the bilayer. However, when the label is attached to the cysteine on position 371, the toxin was unable to enter the vesicles, leaving the protein exposed to proteinase K even after binding to the receptors on BBMV. This is also the case for the double mutant where the fluorescent label is on the domain I residue but also incorporates a mutation in position 371 in domain II. Non-receptor-mediated residual partitioning of the regions of domain I (around position 171) into the vesicles occurs in BBMV explaining the residual fluorescence associated with the proteinase K treated BBMV, in the case of double mutant 1Ab-F371A/V171C. SDS–PAGE gels show a 60 kDa protected form of toxin for the wild type and 1AbV171C, which is also seen in case of several other mutations in 1Ab toxin (Figure 5). But an absence of that form for the F371C or F371A double mutant corroborates the pattern of fluorescence data.
Figure 5.
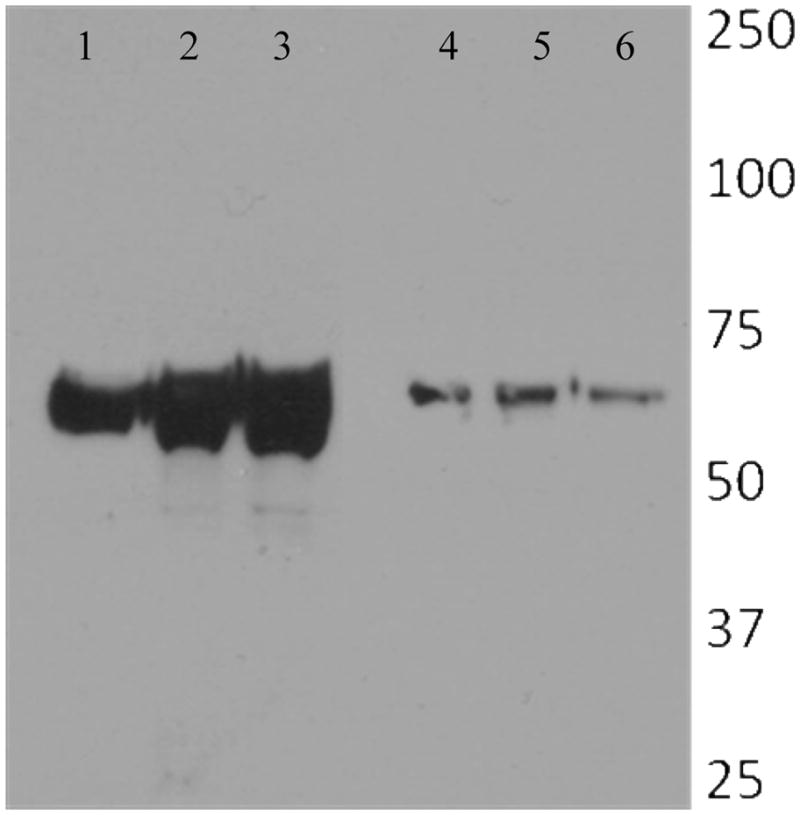
Proteinase K protection assay of mutants in 3 domains of Cry toxin. Mutations 1Ab S176C (domain I), 1Ab S443C (domain II), and 1Ab F461C (domain III) were expressed and purified (lanes 1, 2, and 3). Proteinase K protection assay was performed, and SDS–PAGE showing a 60 kDa protected form is observed for 1Ab S176C (lane 4), 1Ab S443C (lane 5), and 1Ab F461C (lane 6).
Other domain II residues that have the characteristics of F371 in affecting toxicity and some aspect of irreversible binding but not affecting competition binding have been described. Rajamohan (30) describes several Cry1Ab mutations in the region of residues 371–375 that affect toxicity to various degrees and concomitantly reduce irreversible binding, but not competition binding. These include F371, N372, G374, I375, and a deletion mutant (residues 370–375 deleted). The mutant residues that have the greatest effect on irreversible binding are F371A, the deletion mutant and G374A. A follow-up paper (17) focuses on F371. Substitutions at this position that modified the hydrophobic side chains at this position showed a correlation between hydrophobicity, toxicity, and the irreversible binding to BBMV. Wu, et al. (31), studying mutations in domain II, loop 1 of Cry3Aa, identify two residues, Y350 and Y351 that when mutated to phenylalanine increase toxicity. Neither of these mutations affect competition binding significantly, but do increase the irreversible binding of the mutant toxins to BBMV. Thus, a number of published mutations show the effect of domain II residues on irreversible binding. We show in the present article that, in the case of Cry1Ab F371 mutants, irreversible binding is correlated to membrane insertion. If this correlation is confirmed with other mutations and Cry toxins, it would support a direct role for domain II loops in membrane insertion.
An interesting observation in these studies is that Cry1Ab membrane partitioning into BBMVs is different from its partitioning into SUVs. Labeling the toxin on α-helix 5 of domain I (residue 171) with acrylodan showed a blue shift in both BBMV and SUV when domain II residue 371 is wild-type (Phe). When residue 371 is mutated to alanine, the acrylodan-labeled domain I undergoes a blue shift in SUV and is protected from proteinase K, but not in BBMV. This indicates that domain I, at least residue 171 or α-helix 5, is able to enter the artificial SUV membrane but not the BBMV membrane, even if residue 371 is alanine or cysteine. The choice of lipids in our study were similar to the phospholipids (and ratios) used in earlier studies on Cry toxin or synthetic peptide insertion into lipid bilayers or vesicles (10, 11, 32, 33), in support of the umbrella model. As early as 1988, Yunovitz and Yawitz (34) showed that phosphatidyl choline, phosphatidyl ethanolamine, and cholesterol allowed Cry proteins to partition into liposomes. Other combinations of lipids did not. Our results indicate that domain I alone of Cry proteins can partition into artificial SUVs, but not into BBMVs when F371C and F371A mutants block partitioning of domain II. This demonstrates that domain I can spontaneously partition in to artificial lipids but not into native membranes. This observation calls into question the conclusions of previous studies using artificial vesicles as support for the umbrella and penknife models.
A Formal Critique of the Penknife and Umbrella Models
Hodgeman and Ellar (35) proposed the penknife model, where α-helices 5 and 6 partition into the apical membrane to form the pore. This model was based on the highly conserved and hydrophobic sequences of α-helix 5 being predicted to be the membrane-spanning region. Li, et al. (2), in their landmark paper on the crystal structure of Cry3Aa, proposed the umbrella model, where α-helices 4 and 5, or 6 and 7 would insert at the helical hairpin and form the pore. These helices were chosen because the domain II end of domain I was expected to oriented toward the apical membrane. These models were nicely reviewed by Knowles (9), with drawings of the inserted forms of the models. Both of the models assumed that “domain II and its apical region are most likely to bind the membrane receptors” (2, 9), and it was assumed that it remained bound to the receptors. Supporting data for these models have focused exclusively on the α-helices of domain II (36–39). However, several studies have appeared that do not agree with models proposing that only domain I α-helices partitioned into the membrane. There was an early indication of a role for domain III in ion channel activity (40–42). Proteinase protection studies have indicated that virtually the whole toxin (missing α-helix 1, in the case of Cry1Aa, or α-helices 1–3, in the case of Cry3Aa) is protected from digestion with broad spectrum proteinases that completely digest the toxin in solution (7, 12, 14, 15). The present data suggests that domain II plays a direct role in membrane partitioning. There is sufficient current evidence to question the validity of the penknife and umbrella models as they were originally stated.
In summary, the data presented in this article indicate a role for the domain II residue, phenylalanine 371, in a post-receptor-binding step in the mechanism of action of the toxin. On the basis of the available data from studies of the crystal toxin, after receptor binding, the toxin undergoes insertion into the vesicles to form ion channels. We postulate phenylalanine 371 to participate directly in the membrane insertion step, or in some postbinding step that leads to insertion.
Our observation that the mutant 1AbF371C is able to bind to the cadherin receptor, Bt-R1, but has lost all measurable toxicity to M. sexta, has implications on other current models for the mechanism of action of Cry toxins. It is not in agreement with the conclusions of Zhang, et al. (43), who propose that cytotoxicity is correlated to binding of Cry toxin to Bt-R1. In their model, cell death is mediated only by a Mg2+-dependent cellular response. The difference between our studies and those of Zhang, et al. (43) possibly lie in the cellular system utilized. Our results were gathered on M. sexta BBMV for insertion studies. The insects used in the bioassay were M. sexta, and the electrophysiological studies were also done on M. sexta midguts, while Zhang, et al. (43) have used a High Five (Invitrogen, Inc.) cell line from Trichoplusia ni expressing a Bt-R1 fragment.
The larger goal of this study is to determine the mechanism of insertion of the toxin. The ability of domain II mutant to partition into the membranes of these vesicles is in agreement with our hypothesis proposed in our recent article (44) that while α-helices from domain I are able to insert, the mechanism of insertion is not based solely on individual helices entering the membrane. Other studies (13) suggest a buried unchanged structure model for the toxin where almost all of the toxin is buried into the membrane. Our data support the view that the mode of entry of the toxin into the membrane is in the form of an intact 60 kDa monomer or oligomeric toxin that lacks α-helix 1 at its N-terminus.
Acknowledgments
We thank Dr. Oscar Alzate for the mutant Cry1AbV171C and Dr. David Stetson for the use of his automated voltage clamp apparatus. We also thank Drs. Dan Ziegler, Oscar Alzate, and David Stetson for reading the manuscript.
Footnotes
This work was supported by a National Institutes of Health grant (R01A129092 to D.H.D.) and Michael J. Adang.
Abbreviations: BBMV, brush border membrane vesicles; SUV, small unilamelar vesicles; CD, circular dichroism; Isc, inhibition of short circuit current; NHS, N-hydroxysuccinimide; EDC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; CM5, carboxymethylated dextran matrix.
References
- 1.Schnepf E, Crickmore N, VanRie J, Lereclus D, Baum J, Feitelson J, Zeigler DR, Dean DH. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev. 1998;62:775–806. doi: 10.1128/mmbr.62.3.775-806.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J, Carroll J, Ellar DJ. Crystal structure of insecticidal δ-endotoxin from Bacillus thuringiensis at 2.5 Å resolution. Nature. 1991;353:815–821. doi: 10.1038/353815a0. [DOI] [PubMed] [Google Scholar]
- 3.Grochulski P, Borisova S, Pusztai-Carey M, Masson L, Cygler M. 3D Crystal Structure of Lepidopteran-Specific Delta-Endotoxin Cry1A(a). Proc. VIth Internatl. Coll. Invert. Path. Micro. Control; Montpellier, France. 1994. [Google Scholar]
- 4.Knight PJK, Crickmore N, Ellar DJ. The receptor for Bacillus thuringiensis CryIA(c) delta-endotoxin in the brush border membrane of the lepidopteran Manduca sexta is aminopeptidase N. Mol Microbiol. 1994;11:429–436. doi: 10.1111/j.1365-2958.1994.tb00324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Francis BR, Bulla LA., Jr Further characterization of BT-R1, the cadherin-like receptor for Cry1Ab toxin in tobacco hornworm (Manduca sexta) midguts. Insect Biochem Mol Biol. 1997;27:541–550. doi: 10.1016/s0965-1748(97)00029-5. [DOI] [PubMed] [Google Scholar]
- 6.Sangadala S, Walters FS, English LH, Adang MJ. A mixture of Manduca sexta aminopeptidase and phosphatase enhances Bacillus thuringiensis insecticidal CryIA(c) toxin binding and 86Rb+-K+ efflux in vitro. J Biol Chem. 1994;269:10088–10092. [PubMed] [Google Scholar]
- 7.Aronson AI, Geng C, Wu L. Aggregation of Bacillus thuringiensis Cry1A toxins upon binding to target insect larval midgut vesicles. Appl Environ Microbiol. 1999;65:2503–2507. doi: 10.1128/aem.65.6.2503-2507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bravo A, Gomez I, Conde J, Munoz-Garay C, Sanchez J, Miranda R, Zhuang M, Gill SS, Suboron M. Oligomerization triggers binding of a Bacillus thuringiensis Cry1Ab pore-forming toxin to aminopeptidase N receptor leading to insertion into membrane microdomains. Biochem Biophys Acta. 2004;1667:38–46. doi: 10.1016/j.bbamem.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Knowles BH. Mechanism of action of Bacillus thuringiensis insecticidal δ-endotoxins. Adv Insect Physiol. 1994;24:275–308. [Google Scholar]
- 10.Gazit E, Shai Y. The assembly and organization of the α5 and α7 helices from the pore-forming domain of Bacillus thuringiensis δ-endotoxin. J Biol Chem. 1995;270:2571–2578. doi: 10.1074/jbc.270.6.2571. [DOI] [PubMed] [Google Scholar]
- 11.Masson L, Tabashnik BE, Liu YB, Brousseau R, Schwartz JL. Helix 4 of the Bacillus thuringiensis Cry1Aa toxin lines the lumen of the ion channel. J Biol Chem. 1999;274:31996–32000. doi: 10.1074/jbc.274.45.31996. [DOI] [PubMed] [Google Scholar]
- 12.Aronson A. Incorporation of protease K into larval insect membrane vesicles does not result in disruption of function of the pore-forming Bacillus thuringiensis δ-endotoxins. Appl Environ Microbiol. 2000;66:4568–4570. doi: 10.1128/aem.66.10.4568-4570.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loseva OI, Tiktopulo EI, Vasiliev VD, Nikulin AD, Dobritsa AP, Potekhin SA. Structure of Cry3A-endotoxin within phospholipid membranes. Biochemistry. 2001;40:14143–14151. doi: 10.1021/bi010171w. [DOI] [PubMed] [Google Scholar]
- 14.Tomimoto K, Hayakawa T, Hori H. Pronase digestion of brush border membrane-bound Cry1Aa shows that almost the whole activated Cry1Aa molecule penetrates into the membrane. Comp Biochem Physiol, B. 2006;144:413–422. doi: 10.1016/j.cbpb.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Arnold S, Curtiss A, Dean DH, Alzate O. The role of a proline-induced broken-helix motif in α-helix 2 of Bacillus thuringiensis δ-endotoxins. FEBS Lett. 2001;490:70–74. doi: 10.1016/s0014-5793(01)02139-1. [DOI] [PubMed] [Google Scholar]
- 16.Lee MK, Rajamohan F, Gould F, Dean DH. Resistance to Bacillus thuringiensis CryIA δ-endotoxins in a laboratory-selected Heliothis virescens strain is related to receptor alteration. Appl Environ Microbiol. 1995;61:3836–3842. doi: 10.1128/aem.61.11.3836-3842.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajamohan F, Cotrill JA, Gould F, Dean DH. Role of domain II, loop 2 residues of Bacillus thuringiensis CryIAb 104-endotoxin in reversible and irreversible binding to Manduca sexta and Heliothis virescens. J Biol Chem. 1996;271:2390–2397. doi: 10.1074/jbc.271.5.2390. [DOI] [PubMed] [Google Scholar]
- 18.Lee MK, Milne RE, Ge AZ, Dean DH. Location of a Bombyx mori receptor binding region on a Bacillus thuringiensis δ-endotoxin. J Biol Chem. 1992;267:3115–3121. [PubMed] [Google Scholar]
- 19.Zhuang M, Oltean DI, Gómez I, Pullikuth AK, Soberon M, Bravo A, Gill SS. Heliothis virescens and Manduca sexta lipid rafts are involved in Cry1A toxin binding to the midgut epithelium and subsequent pore formation. J Biol Chem. 2002;277:13863–13872. doi: 10.1074/jbc.M110057200. [DOI] [PubMed] [Google Scholar]
- 20.Grochulski P, Masson L, Borisova S, Pusztai-Carey M, Schwartz JL, Brousseau R, Cygler M. Bacillus thuringiensis CryIA(a) insecticidal toxin: crystal structure and channel formation. J Mol Biol. 1995;254:447–464. doi: 10.1006/jmbi.1995.0630. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz JL, Lu YJ, Sohnlein P, Brousseau R, Laprade R, Masson L, Adang MJ. Ion channels formed in planar lipid bilayers by Bacillus thuringiensis toxins in the presence of Manduca sexta midgut receptors. FEBS Lett. 1997;412:270–276. doi: 10.1016/s0014-5793(97)00801-6. [DOI] [PubMed] [Google Scholar]
- 22.Pitcher WH, III, Huestis WH. Preparation and analysis of small unilamellar phospholipid vesicles of a uniform size. Biochem Biophys Res Commun. 2002;296:1352–1355. doi: 10.1016/s0006-291x(02)02092-2. [DOI] [PubMed] [Google Scholar]
- 23.Liebig B, Stetson DL, Dean DH. Quantification of the effect of Bacillus thuringiensis toxins on short-circuit current in the midgut of Bombyx mori. J Insect Physiol. 1995;41:17–22. [Google Scholar]
- 24.Wolfersberger M, Lüthy P, Maurer A, Parenti P, Sacchi FV, Giordana B, Hanozet GM. Preparation and partial characterization of amino acid transporting brush border membrane vesicles from the larval midgut of the cabbage butterfly (Pieris brassicae) Comp Biochem Physiol, A. 1987;86:301–308. [Google Scholar]
- 25.Hua G, Jurat-Fuentes JL, Adang MJ. Bt-R1a extracellular cadherin repeat 12 mediates Bacillus thuringiensis Cry1Ab binding and cytotoxicity. J Biol Chem. 2004;279:28051–28056. doi: 10.1074/jbc.M400237200. [DOI] [PubMed] [Google Scholar]
- 26.Chamberlin ME. Developmental changes in midgut ion transport and metabolism in the tobacco hornworm (Manduca sexta) Physiol Zool. 1994;67:82–94. [Google Scholar]
- 27.Valeva A, Walev I, Gerber A, Klein J, Palmer M, Bhakdi S. Staphylococcal alpha-toxin: repair of a calcium-impermeable pore in the target cell membrane. Mol Microbiol. 2000;36:467–476. doi: 10.1046/j.1365-2958.2000.01865.x. [DOI] [PubMed] [Google Scholar]
- 28.Prendergast FG, Meyer M, Carlson GL, Iida S, Potter JD. Synthesis, spectral properties, and use of 6-acryloyl-2-dimethylaminonaphthalene (Acrylodan). A thiol-selective, polarity-sensitive fluorescent probe. J Biol Chem. 1983;258:7541–7544. [PubMed] [Google Scholar]
- 29.Dorsch JA, Candas M, Griko NB, Maaty WSA, Midboe EG, Vadlamudi RK, Bulla LA. Cry1A toxins of Bacillus thuringiensis bind specifically to a region adjacent to the membrane-proximal extracellular domain of BT-R1 in Manduca sexta: Involvement of a cadherin in the entomopathogenicity of Bacillus thuringiensis. Insect Biochem Mol Biol. 2002;32:1025–1036. doi: 10.1016/s0965-1748(02)00040-1. [DOI] [PubMed] [Google Scholar]
- 30.Rajamohan F, Alcantara E, Lee MK, Chen XJ, Curtiss A, Dean DH. Single amino acid changes in domain II of Bacillus thuringiensis CryIAb δ-endotoxin affect irreversible binding to Manduca sexta midgut membrane vesicles. J Bacteriol. 1995;177:2276–2282. doi: 10.1128/jb.177.9.2276-2282.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu SJ, Koller CN, Miller DL, Bauer LS, Dean DH. Enhanced toxicity of Bacillus thuringiensis Cry3A δ-endotoxin in coleopterans by mutagenesis in a receptor binding loop. FEBS Lett. 2000;473:227–232. doi: 10.1016/s0014-5793(00)01505-2. [DOI] [PubMed] [Google Scholar]
- 32.Vie V, Van Mau N, Pomarede P, Dance C, Schwartz JL, Laprade R, Frutos R, Rang C, Masson L, Heitz F, LeGrimellec C. Lipid-induced pore formation of the Bacillus thuringiensis Cry1Aa insecticidal toxin. J Membr Biol. 2001;180:195–203. doi: 10.1007/s002320010070. [DOI] [PubMed] [Google Scholar]
- 33.Peyronnet O, Vachon V, Brousseau R, Baines D, Schwartz JL, Laprade R. Effect of Bacillus thuringiensis toxins on the membrane potential of lepidopteran insect midgut cells. Appl Environ Microbiol. 1997;63:1679–1684. doi: 10.1128/aem.63.5.1679-1684.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yunovitz H, Sneh B, Schuster S, Oron U, Broza M, Yawetz A. A new sensitive method for determining the toxicity of a highly purified fraction from delta-endotoxin produced by Bacillus thuringiensis var. entomocidus on isolated larval midgut of Spodoptera littoralis (Lepidoptera, Noctuidae) J Invert Pathol. 1986;48:223–231. [Google Scholar]
- 35.Hodgman TC, Ellar DJ. Models for the structure and function of the Bacillus thuringiensis δ-endotoxins determined by compilational analysis. DNA Seq. 1990;1:97–106. doi: 10.3109/10425179009016037. [DOI] [PubMed] [Google Scholar]
- 36.Gazit E, Bach D, Kerr ID, Sansom MSP, Chejanovsky N, Shai Y. The alpha-5 segment of Bacillus thuringiensis delta-endotoxin: in vitro activity, ion channel formation and molecular modeling. Biochem J. 1994;304:895–902. doi: 10.1042/bj3040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gazit E, LaRocca P, Sansom MSP, Shai Y. The structure and organization within the membrane of the helices composing the pore-forming domain of Bacillus thuringiensis d-endotoxin are consistent with an “umbrella-like” structure of the pore. Proc Natl Acad Sci USA. 1998;95:12289–12294. doi: 10.1073/pnas.95.21.12289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shai Y. Molecular recognition between membrane-spanning polypeptides. TIBS. 1995;20:100–104. doi: 10.1016/s0968-0004(00)89101-x. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz JL, Juteau M, Grochulski P, Cygler M, Préfontaine G, Brousseau R, Masson L. Restriction of intramolecular movements within the CrylAa toxin molecule of Bacillus thuringiensis through disulfide bond engineering. FEBS Lett. 1997;410:397–402. doi: 10.1016/s0014-5793(97)00626-1. [DOI] [PubMed] [Google Scholar]
- 40.Chen XJ, Lee MK, Dean DH. Site-directed mutations in a highly conserved region of Bacillus thuringiensis δ-endotoxin affect inhibition of short circuit current across Bombyx mori midguts. Proc Natl Acad Sci USA. 1993;90:9041–9045. doi: 10.1073/pnas.90.19.9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolfersberger MG, Chen XJ, Dean DH. Site-directed mutations in the third domain of Bacillus thuringiensis δ-endotoxin CryIAa affects its ability to increase the permeability of Bombyx mori midgut brush border membrane vesicles. Appl Environ Microbiol. 1996;62:279–282. doi: 10.1128/aem.62.1.279-282.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz JL, Potvin L, Chen XJ, Brousseau R, Laprade R, Dean DH. Single site mutations in the conserved alternating arginine region affect ionic channels formed by CryIAa, a Bacillus thuringiensis toxin. Appl Environ Microbiol. 1997;63:3978–3984. doi: 10.1128/aem.63.10.3978-3984.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, Candas M, Griko NB, Rose-Young L, Bulla JLA. Cytotoxicity of Bacillus thuringiensis Cry1Ab toxin depends on specific binding of the toxin to the cadherin receptor BT-R1 expressed in insect cells. Cell Death Differ. 2005;12:1407–1416. doi: 10.1038/sj.cdd.4401675. [DOI] [PubMed] [Google Scholar]
- 44.Alzate O, You T, Claybon M, Osorio C, Curtiss A, Dean DH. Effects of disulfide bridges in domain I of Bacillus thuringiensis Cry1Aa delta-endotoxin on ion-channel formation in biological membranes. Biochemistry. 2006;45:13597–13605. doi: 10.1021/bi061474z. [DOI] [PubMed] [Google Scholar]