

Summary
Dietary nutrients interact with gene networks to orchestrate adaptive responses during metabolic stress. Here we identify Baf60a as a diet-sensitive subunit of the SWI/SNF chromatin-remodeling complexes in the mouse liver that links the consumption of fat- and cholesterol-rich diet to elevated plasma cholesterol levels. Baf60a expression was elevated in the liver following feeding with a western diet. Hepatocyte-specific inactivation of Baf60a reduced bile acid production and cholesterol absorption, and attenuated diet-induced hypercholesterolemia and atherosclerosis in mice. Baf60a stimulates expression of genes involved in bile acid synthesis, modification, and transport through a CAR/Baf60a feedforward regulatory loop. Baf60a is required for the recruitment of the SWI/SNF chromatin-remodeling complexes to facilitate an activating epigenetic switch on target genes. These studies elucidate a regulatory pathway that mediates the hyperlipidemic and atherogenic effects of western diet consumption.
Graphical abstract
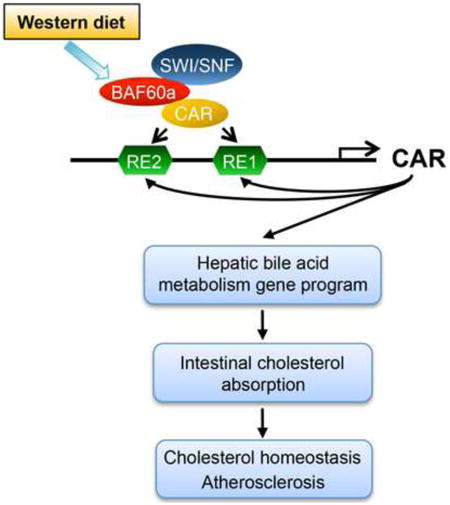
Introduction
Elevated plasma low-density lipoprotein (LDL) cholesterol is a major risk factor for atherosclerosis and its associated cardiovascular mortality and morbidity (Glass and Witztum, 2001; Ross, 1993; Steinberg, 2002). The cholesterol pool in the body is tightly regulated by feedback mechanisms that impinge on endogenous cholesterol biosynthesis, catabolism and excretion as bile acid (Chiang, 2009; Goldstein and Brown, 2015). Accumulation of intracellular sterol prevents the proteolytic activation of the sterol-response element binding protein (Srebp), transcriptional regulators of cholesterol biosynthesis, while stimulating the expression of genes involved in bile acid formation and excretion (Brown and Goldstein, 2009; Chiang, 2009). Pharmacological targeting of these pathways has proven effective in lowering LDL-cholesterol and reducing the risk of atherosclerosis (2001; Grundy et al., 2004; Waters et al., 2009).
Nuclear hormone receptors have been implicated in sensing diverse metabolites in the cell, including lipids, oxysterols, bile acids, and xenobiotic compounds (Evans and Mangelsdorf, 2014). Hepatocytes sense the enterohepatic flux of cholesterol and bile acids in part through engaging liver X receptor (LXR) and farnesoid X receptor (FXR) (Calkin and Tontonoz, 2012; Matsubara et al., 2013). Oxysterols are oxygenated derivatives of cholesterol that serve as LXR ligands. A major target gene of LXR is Cyp7a1, which catalyzes the first step of the classic bile acid synthesis pathway (Lehmann et al., 1997; Peet et al., 1998). An alternative pathway initiated by sterol-27 hydroxylase (Cyp27a1) also contributes to cholesterol catabolism to bile acids (Schwarz et al., 2001). Bile acids are efficiently recycled through the enterohepatic circulation to facilitate intestinal absorption of dietary fats (Thomas et al., 2008). Accumulation of bile acids in hepatocytes results in FXR activation and induction of its target gene small heterodimer partner (Shp), which mediates the feedback inhibition of bile acid synthesis (Goodwin et al., 2000; Lu et al., 2000). In addition, constitutive androstane receptor (CAR) and pregnane X receptor (PXR), best known as xenobiotic sensors, regulate bile acid detoxification by stimulating the expression of hepatic genes responsible for the modification, conjugation, and transport of bile acids (Li and Chiang, 2013; Pascussi et al., 2008). Dietary intake of cholesterol is known to stimulate bile acid synthesis, and increase bile acid pool and fecal excretion in rodents and humans; however, the nature of dietary regulation of bile acid homeostasis and intestinal lipid absorption remains elusive (Duane, 1994; Tiemann et al., 2004; Xu et al., 1999).
Nuclear receptors activate or repress gene transcription through recruiting various chromatin-remodeling complexes to alter the epigenetic landscape of target genomic loci (Chen and Roeder, 2011; Dasgupta et al., 2014; Mottis et al., 2013). Despite this, the significance of the nucleosome-remodeling complexes, such as the SWI/SNF complexes, in nuclear receptor signaling and metabolic physiology remains poorly understood. The SWI/SNF complexes are composed of one of two catalytic ATPase subunits (Brg1 or Brm) and additional subunits known as Brg/Brm-associated factors (Bafs) (Phelan et al., 1999; Sudarsanam and Winston, 2000; Wang et al., 1996; Wu et al., 2009). While Baf47, Baf170, and Baf155 form part of a core complex with Brg1/Brm, incorporation of other Baf subunits confers diversity and specificity of SWI/SNF complexes in transcriptional control. Recent studies have demonstrated that the Baf60 family members Baf60a and Baf60c recruit SWI/SNF complexes to regulate metabolic gene programs in the liver and skeletal muscle (Li et al., 2008; Meng et al., 2013; Meng et al., 2014). In this study, we identify Baf60a as a diet-sensitive factor in the liver that controls a hepatic gene program responsible for bile acid synthesis and intestinal cholesterol absorption through a Baf60a/CAR feedforward regulatory loop. Disruption of this pathway by liver-specific inactivation of Baf60a protects mice from diet-induced hypercholesterolemia and atherosclerosis.
Results
Hepatic Baf60a is a diet-sensitive regulator of cholesterol homeostasis
Chromatin-remodeling factors link nutrient signaling to metabolic gene programs through altering the epigenetic state of chromatin. The SWI/SNF complexes play an important role in differentiation, development, and tumorigenesis (Puri and Mercola, 2012; Wilson and Roberts, 2011); however, their role in diet-induced hyperlipidemia has not been explored. We analyzed the expression of core SWI/SNF subunits in the liver from mice fed standard chow or Western diet (WD); the latter has been commonly used to induce hypercholesterolemia. Immunoblotting analysis of liver nuclear extracts indicated that protein levels of core SWI/SNF subunits, including Brg1, Baf170, Baf155, and Baf53a, were similar between two groups (Figure 1A and 1B). In contrast, mRNA and protein levels of Baf60a were significantly elevated following WD- feeding. The expression levels of Baf60b and Baf60c, two Baf60 family members closely related to Baf60a, remained largely insensitive to diets. In addition, hepatic expression of Baf60a was similar between control and genetic obese mice (Fig. S1A). Previous studies have demonstrated that Baf60a and Baf60c mediate the recruitments of the SWI/SNF complexes to specific target genes through their interaction with transcription factors and cofactors (Debril et al., 2004; Li et al., 2008; Meng et al., 2013). As such, Baf60a may serve as a diet-sensitive component of the SWI/SNF complexes in the liver that links dietary fat intake to lipid metabolism.
Figure 1. BaLKO mice are resistant to diet-induced hypercholesterolemia.
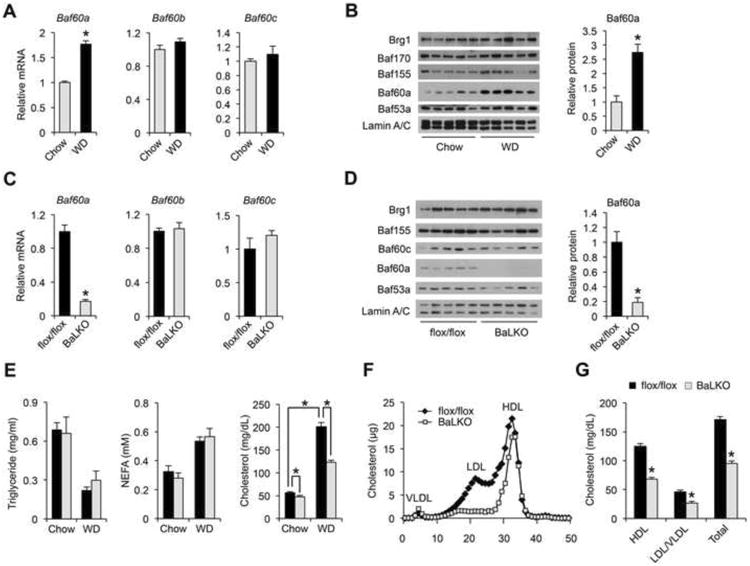
(A) qPCR analysis of liver gene expression in mice fed standard chow orWestern diet (WD) for 8 weeks (n = 5).
(B) Left, immunoblots of liver nuclear extracts; Right, quantitation of Baf60a protein levels after normalization to Lamin A/C.
(C) qPCR analysis of gene expression in livers from flox/flox and BaLKO mice (n = 5).
(D) Left, immunoblots of liver nuclear extracts; Right, quantitation of Baf60a protein levels after normalization to Lamin A/C.
(E-G) Plasma lipid levels (E), lipoprotein profile analysis (F), and concentrations of HDL-, LDL/VLDL-, and total-cholesterol (G) in flox/flox and BaLKO mice following 12 weeks of WD feeding (n = 5). Values are mean ± s.e.m.; *P < 0.05 by two-tailed Student's t-test.
See also Figure S1.
To dissect the role of Baf60a in mediating diet-gene interaction, we generated mice with conditional Baf60a deficiency in the liver. Mice containing floxed Baf60a alleles were crossed with Albumin-CRE (Alb-CRE) transgenic mice to generate control (flox/flox) and liver-specific Baf60a knockout mice (BaLKO, flox/flox:Alb-CRE) (Figure S1B). As expected, Baf60a mRNA and protein expression was markedly reduced in BaLKO mouse livers (Figure 1C and 1D). The expression levels of Baf60b, Baf60c, and other SWI/SNF subunits remained largely unaffected by Baf60a deficiency. Further, Baf60a expression in other metabolic tissues, including skeletal muscle, adipose tissues, and small intestine, was unaffected in BaLKO mice (Figure S1C and S1D). BaLKO mice were born at expected Mendelian ratio and appeared indistinguishable from control littermates. Plasma lipid concentrations were similar between two groups when fed standard chow, and as expected, control mice exhibited elevated plasma cholesterol levels following 12 weeks of WD-feeding (Figure 1E). In contrast, BaLKO mice were partially resistant to diet-induced hypercholesterolemia despite similar weight gain (Figure 1E and Figure S1E); total plasma cholesterol concentration was approximately 40% lower in BaLKO mice than control. Plasma triglycerides (TAG), non-esterified fatty acids (NEFA), and fasting blood glucose levels were similar between two groups following WD-feeding (Figure 1E and Figure S1F). Lipoprotein profile analysis and quantitative measurements of cholesterol in different lipoprotein fractions revealed that both LDL/VLDL and HDL cholesterol were lower in mice lacking Baf60a in the liver (Figure 1F and 1G). Together, these findings support a uniquely important role of Baf60a in mediating diet-induced elevation of plasma cholesterol levels.
Baf60a deficiency in hepatocytes impairs bile acid synthesis and cholesterol absorption
We next performed transcriptional profiling on the livers from WD-fed control and BaLKO mice to globally define hepatic target genes of Baf60a (GEO accession number GSE73709). Pathway analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) revealed that a cluster of genes involved in primary bile acid biosynthesis was the most significantly down-regulated gene set in response to Baf60a inactivation in hepatocytes (Figure 2A and 2B). Several nuclear hormone receptors, including FXR, LXR, CAR, and PXR, have been reported as important regulators of cholesterol and bile acid metabolism in the liver (Beaven and Tontonoz, 2006; Chiang, 2009; Evans and Mangelsdorf, 2014; Gao and Xie, 2012). While mRNA levels for PXR, LXR, and FXR remained largely unaltered by Baf60a deficiency, the expression of CAR was markedly lower in BaLKO livers than control (Figure 2C). This decrease in CAR expression correlated with lower expression of genes involved in bile acid synthesis, including Cyp8b1, Cyp27a1, Cyp7b1, BAAT, and Hsd3b5, and those involved in xenobiotic responses and bile acid modification and transport, such as Cyp2b10, Cyp3a11, Sult5a1, Ugt1a1, Oatp2, Mrp3, and Mdr2 (Figure 2D). In contrast, the expression of Shp, a canonical target gene of FXR, and Cyp7a1, Abca1, Abcg5, and Abcg8, target genes of LXR, remained similar between two groups (Figure 2E). The expression of hepatic genes involved in cholesterol biosynthesis (Srebp1a, Srebp1c, Srebp2, Hmgcr, and Hmgcs1) appeared largely unaffected by Baf60a deficiency. Further, mRNA and protein levels of LDLR, which is responsible for the uptake of plasma LDL (Brown and Goldstein, 1986), and Pcsk9, an important modulator of LDLR protein levels (Horton et al., 2009), were comparable between control and BaLKO mice (Figure 2E-2G). Consistently, lipoprotein secretion analysis indicated that the rise of plasma cholesterol concentrations following tail vein injection of tyloxapol (500 mg/kg) was unaffected by Baf60a deficiency (Figure S2A). As such, hepatic inactivation of Baf60a appears to exert modest effects on cholesterol synthesis, LDL uptake, and lipoprotein secretion. To rule out the possibility that Baf60a deficiency may impair liver function and cause liver injury, we measured plasma alanine transaminase (ALT) and aspartate aminotransferase (AST) levels, and the expression of marker genes involved in liver fibrosis and inflammation. Plasma concentrations of ALT and AST were significantly lower in BaLKO mice than control, whereas the mRNA expression of genes involved in liver fibrosis and inflammation remained similar between two groups (Figure S2B and S2C).
Figure 2. Baf60a regulates CAR and its target genes involved in bile acid metabolism.
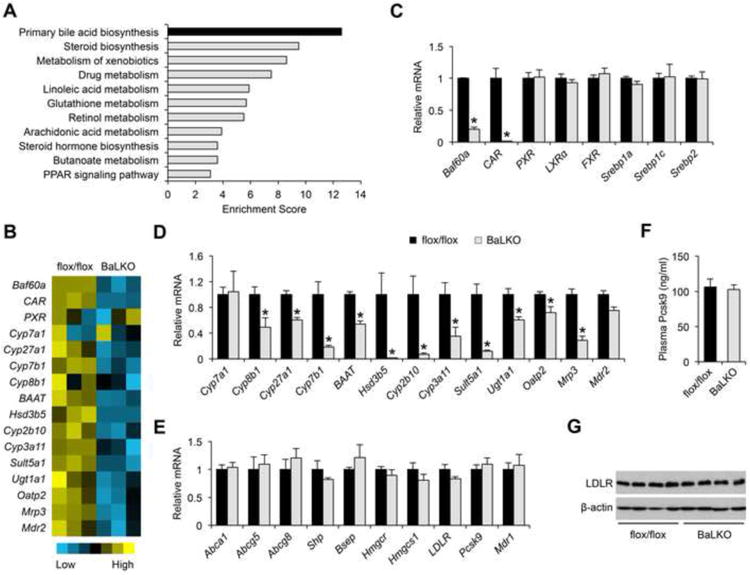
(A) DAVID pathway analysis of down-regulated genes in BaLKO mouse livers identified by microarray.
(B) Heat map representation of genes involved in bile acid metabolism.
(C-E) qPCR analysis of the mRNA expression of transcriptional factors (C), CAR target genes (D), and FXR, LXR and PXR target genes (E) involved in bile acid metabolism in livers from flox/flox and BaLKO mice fed with WD for 12 weeks (n = 4-5).
(F) Plasma concentrations of Pcsk9 (n = 4-5).
(G) Immunoblots of total liver lysates. Data in C-F are mean ± s.e.m.; *P < 0.05 by two-tailed Student's t-test.
See also Figure S2.
As bile acids play an important role in the digestion and absorption of dietary fats, impaired intestinal cholesterol absorption may contribute to resistance to diet-induced hypercholesterolemia in BaLKO mice. In support of this, total bile volume and biliary bile acid content were approximately 37% lower in BaLKO mice than control, while plasma and liver bile acid concentrations remained similar between two groups (Figure 3A and 3B). Measurements of bile acid pool indicated that liver/gallbladder and intestine bile acid pool sizes were both significantly lower in BaLKO mice, resulting in lower whole body bile acid content (Figure 3C). However, the composition of different bile acid species in bile from gallbladder remained similar between two groups (Figure 3D). In parallel, liver cholesterol content is elevated in BaLKO mice compared to control (Figure 3E). Consistent with dietary cholesterol being an important source of the lipid, cholesterol-lowering effect of hepatic Baf60a deficiency was abolished following overnight starvation (Figure 3F). While food intake remained similar between two groups, cholesterol excretion in feces was significantly higher in BaLKO mice (Figure 3G), suggesting that hepatic Baf60a inactivation impaired intestinal cholesterol absorption. Direct measurements of cholesterol absorption by oral gavage of 3H-labeled cholesterol indicated that the amounts of radiolabeled cholesterol in plasma and the liver were significantly lower in BaLKO mice than control (Figure 3H). Consequently, cholesterol-lowering effects of ezetimibe, an inhibitor of cholesterol absorption, failed to further reduce plasma cholesterol levels in BaLKO mice (Figure 3I). These results illustrate that impaired cholesterol absorption secondary to reduced bile acid synthesis likely mediates resistance to diet-induced hypercholesterolemia in BaLKO mice.
Figure 3. Hepatic Baf60a regulates bile acid metabolism and cholesterol absorption.
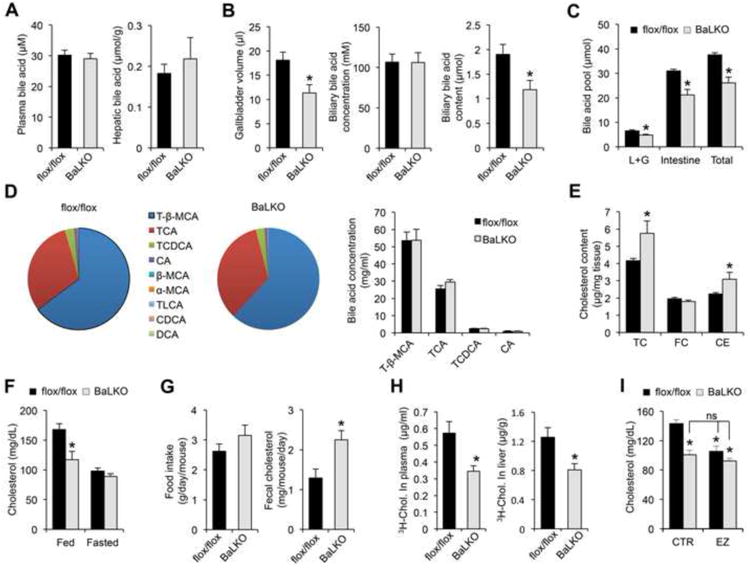
flox/flox and BaLKO mice were fed with standard chow or WD for 12 weeks.
(A-B) Plasma and hepatic bile acid levels (A), and gallbladder volume, biliary bile acid concentrations, and biliary bile acid content (B) (n = 6-8).
(C) Bile acid pool size in liver plus gallbladder (L+G), intestine and whole body (Total) (n = 5).
(D) Bile acid composition in the bile from gallbladder. Left, percentile compositionof different bile acids in the bile from flox/flox and BaLKO mice; Right,concentrations of four major bile acids in the bile. T-β-MCA, tauro-β-muricholicacid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid; CA, cholicacid; β-MCA, β-muricholic acid; α-MCA, α-muricholic acid; TLCA, taurolithocholicacid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid.
(E) Concentrations of total cholesterol (TC), free cholesterol (FC), andcholesterol ester (CE) in livers from control (dark bars) and BaLKO (gray bars)mice (n = 6-9).
(F) Plasma cholesterol levels in mice under fed and fasted conditions (n = 6).
(G) Food intake and fecal cholesterol content in WD-fed control and BaLKO mice (n = 4-5).
(H) Cholesterol uptake into plasma and liver following oral gavage of 3H-cholesterol (3H-chol.) for 2 h.
(I) Plasma cholesterol concentrations in WD-fed control and BaLKO mice treated with vehicle alone (CTR) or ezetimibe (EZ) for 4 days (n = 6); *P < 0.05 vs. flox/flox mice treated with vehicle alone; ns, not significant. In all panels, values are mean ± s.e.m.; *P < 0.05 by two-tailed Student's t-test.
Baf60a cell-autonomously regulates the bile acid metabolism gene program
The marked reduction of CAR expression in the BaLKO mouse livers suggests that its deficiency may be responsible for lower expression of genes involved in bile acid metabolism. We next examined whether Baf60a regulates CAR gene expression in a cell-autonomous manner. Gene expression analysis of primary hepatocytes isolated from flox/flox and BaLKO mice demonstrated that mRNA expression of CAR was approximately 74% lower in Baf60a-deficient hepatocytes (Figure 4A and 4B). In parallel, mRNA expression of several genes involved in the alternative pathway of bile acid synthesis, including Cyp8b1, Cyp27a1, and Cyp7b1, and bile acid modification and transport, including BAAT, Hsd3b5, Cyp2b10, Cyp3a11, Sult5a1, Ugt1a1, Oatp2, Mrp3, and Mdr2, was also significantly lower in Baf60a-deficient hepatocytes. Treatments of hepatocytes with TCPOBOP, a synthetic CAR agonist, significantly induced mRNA expression of Cyp2b10, a canonical CAR target gene, and CAR itself. However, the induction of these two genes by TCPOBOP was nearly abolished in hepatocytes lacking Baf60a (Figure 4C), indicating that Baf60a is required for maintaining the expression and transcriptional activity of CAR in hepatocytes.
Figure 4. Baf60a regulates the bile acid metabolism gene program in a cell-autonomous manner.
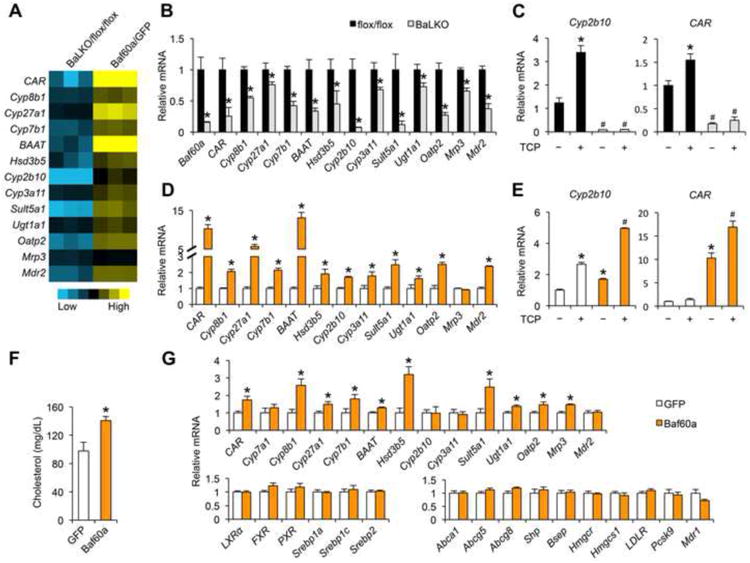
(A) Heat map representation of gene regulation in response to Baf60a deficiency (BaLKO/flox/flox) and Baf60a overexpression (Baf60a/GFP) in primary hepatocytes.
(B-C) qPCR analysis of gene expression in primary hepatocytes isolated from flox/flox and BaLKO mice in the presence or absence of TCPOBOP treatment (TCP, 1 μM) for 24 h. In c, *P < 0.05 vs. vehicle treatment alone (the first bar); #P < 0.05 vs. TCP treatment alone (the second bar).
(D-E) qPCR analysis of gene expression in primary hepatocytes transduced with GFP or Baf60a adenoviruses and treated with or without TCP 1 μM for 24 h. *P < 0.05 vs. GFP expression alone; #P < 0.05 vs. TCP treatment or Baf60a expression alone.
(F) Plasma cholesterol levels in WD-fed WT mice transduced with adenovirusesexpressing GFP or Baf60a.
(G) qPCR analysis of hepatic gene expression in WT mice transduced withadenoviruses expressing GFP or Baf60a. Data in B-E represent mean ± s.d. andare representative of three independent experiments; Data in F and G representmean ± s.e.m. (n = 6); #P < 0.05, and *P < 0.05 by two-tailed Student's t-test.
To determine whether Baf60a serves as an upstream regulator of CAR, we transduced primary hepatocytes with control GFP or Baf60a adenoviruses. Gene expression analysis indicated that adenoviral-mediated overexpression of Baf60a resulted in over 10-fold induction of endogenous CAR (Figure 4A and 4D). Importantly, Baf60a stimulated the expression of Cyp8b1, Cyp27a1, Cyp7b1, and a panel of genes involved in bile acid modification and transport. Baf60a-mediated induction of CAR and Cyp2b10 was further augmented by TCPOBOP treatments, suggesting that Baf60a acts in concert with CAR to regulate the expression of CAR target genes (Figure 4E). To assess the effects of hepatic Baf60a activation on cholesterol absorption in mice, we transduced WD-fed wild type mice with recombinant adenoviruses expressing GFP or Baf60a via tail vein injection. Compared to control, mice transduced with Baf60a adenovirus had significantly elevated total plasma cholesterol levels (Figure 4F). Gene expression analysis indicated that tail vein transduction of Baf60a adenovirus significantly increased mRNA levels of CAR and its target genes in the liver (Figure 4G). The expression of LXR, FXR, PXR, Srebp1a, Srebp1c, Srebp2, and their target genes remained similar between two groups.
Baf60a and CAR form a feedforward regulatory loop through a SWI/SNF-mediated epigenetic mechanism
The SWI/SNF chromatin-remodeling complexes are recruited to specific chromatin loci through physical interaction with transcription factors. The BAF60 subunit has been demonstrated to serve as a molecular link between the core complexes and DNA-binding factors (Li et al., 2008; Meng et al., 2013; Meng et al., 2014). To test whether Baf60a physically interacts with CAR, we performed protein-protein interaction assay using coimmunoprecipitation (co-IP). Baf60a and CAR formed a physical complex when they were co-expressed in HEK293 cells following transient transfection (Figure 5A). The physical association between CAR and Baf60a was further confirmed in HepG2 hepatoma cells. We transduced HepG2 cells with adenoviruses expressing GFP or Flag/HA-tagged CAR (FH-CAR), and performed immunoprecipitation using anti-HA agarose beads. Immunoblotting analyses indicated that, in addition to endogenous Baf60a, other subunits of the SWI/SNF complexes, including Brg1, Baf170, Baf155, and Baf53a, were readily detectable in the CAR immunocomplexes (Figure 5B). These findings suggest that Baf60a mediates the recruitment of the SWI/SNF chromatin-remodeling complexes to the genomic targets occupied by CAR.
Figure 5. Baf60a and CAR form a feedforward regulatory loop.
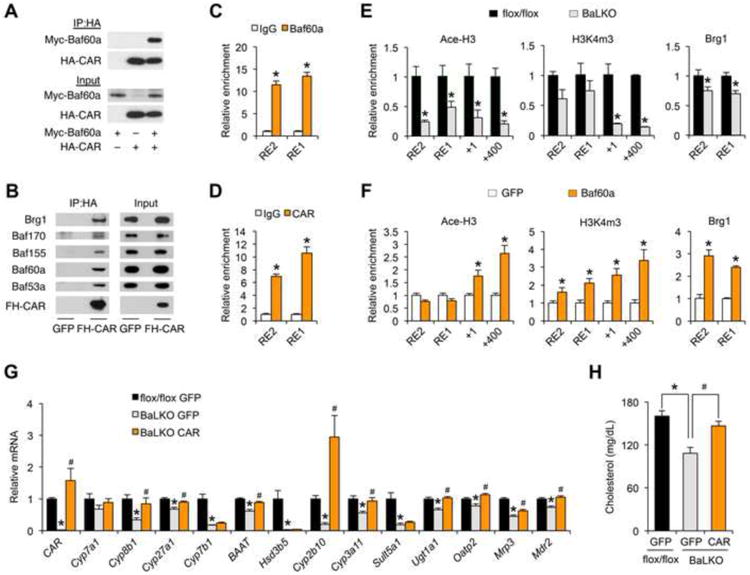
(A) Physical interaction between CAR and Baf60a in transiently transfected 293T cells. IP, immunoprecipitation; Myc-Baf60a, Myc-tagged Baf60a; HA-CAR, HA-tagged CAR.
(B) Immunoblots of immunoprecipitated proteins or total protein lysates from HepG2 cells transduced with adenoviruses expressing Flag/HA-tagged CAR (FH-CAR). Note that FH-CAR interacts with endogenous SWI/SNF subunits.
(C-D) Relative enrichment of Baf60a (C) and CAR (D) at the putative CAR binding site on the proximal CAR promoter. RE1, CAR response element 1; RE2, CAR response element 2.
(E) ChIP assay with chromatin lysates prepared from flox/flox and BaLKO mouselivers using indicated antibodies.
(F) ChIP assay with chromatin lysates prepared from primary hepatocytestransduced with GFP or Baf60a adenoviruses.
(G-H) WD-fed flox/flox and BaLKO mice were transduced with adenoviruses expressing GFP or CAR for 1 week. Shown are qPCR analysis of hepatic gene expression (G) and measurement of plasma cholesterol levels (H); *P < 0.05, BaLKO GFP vs. flox/flox GFP; #P < 0.05, BaLKO CAR vs. BaLKO GFP. Data in C-F represent mean ± s.d. and are representative of three independent experiments; Data in G and H represent mean ± s.e.m. (n = 4-5); #P < 0.05, and *P < 0.05 by two-tailed Student's t-test.
See also Figure S3.
Genomatix motif analysis predicted two putative CAR Response Elements (CARRE) located approximately 2.0 kb (RE2) and 0.9 kb (RE1) upstream of the transcriptional start site of CAR gene. Chromatin immunoprecipitation (ChIP) assays demonstrated that epitope-tagged Baf60a and CAR were recruited to these two sites on the native CAR promoter (Figure 5C and 5D). As such, Baf60a and CAR appear to form a feedforward regulatory loop to drive the expression of CAR and its target genes. A prediction of this model is that Baf60a levels may dictate the recruitment of the SWI/SNF complexes, which in turn impacts on the local epigenetic landscape. To test this, we performed ChIP assay in control and Baf60a-deficient primary hepatocytes. Baf60a deficiency significantly reduced the recruitment of SWI/SNF core subunit Brg1 to the chromatin near the CAR binding sites, leading to reduced levels of acetyl-histone H3 (Ace-H3) and trimethylation of H3 lysine 4 (H3K4m3), two epigenetic markers associated with transcriptional activation (Figure 5E). Conversely, adenoviral-mediated expression Baf60a augmented the recruitment of Brg1 and increased Ace-H3 and H3K4m3 levels on the proximal CAR promoter (Figure 5F). There results demonstrate that Baf60a modulates the transcriptional activity of CAR through its recruitment of SWI/SNF-mediated chromatin remodeling.
The marked reduction of CAR expression in BaLKO mouse livers suggest that Baf60a deficiency may disrupt the feedforward regulatory loop and impair the transcription of CAR target genes. We next performed rescue studies to determine the significance of CAR in mediating the effects of Baf60a deficiency on plasma cholesterol. Consistent with previous transgenic studies (Dong et al., 2009; Saini et al., 2004), tail vein injection of CAR adenovirus significantly increased the expression of its target genes involved in bile acid metabolism in wild type mice (Figure S3A-3C). Adenoviral-mediated rescue of CAR expression in BaLKO mice to slightly above its endogenous levels restored the expression of several CAR target genes, including Cyp8b1, Cyp27a1, BAAT, Cyp2b10, Cyp3a11, Ugt1a1, Oatp2, Mrp3, and Mdr2 (Figure 5G), and more importantly, reversed plasma cholesterol levels in BaLKO mice (Figure 5H). Although the expression PXR was not significantly altered in livers from BaLKO mice, our co-IP experiments indicated that Baf60a could also physically interact with PXR (Figure S3D). As such, PXR may mediate the transcriptional effects of Baf60a on a subset of target genes involved in bile acid metabolism and xenobiotic detoxification. In support of this possibility, adenoviral expression of CAR failed to restore the expression of Cyp7b1, Hsd3b5, and Sult5a1 in BaLKO mouse livers. We conclude from these studies that Baf60a activates a CAR-dependent program of gene expression in the liver to facilitate bile acid production and intestinal lipid absorption.
Liver-specific inactivation of Baf60a protects mice from diet-induced atherosclerosis
Elevated LDL cholesterol is a major risk factor for atherosclerosis. ApoE plays an important role in the regulation of plasma cholesterol through its interaction with LDLR to facilitate the uptake of cholesterol-rich lipoproteins (Mahley, 1988). ApoE null (ApoE-/-) mice have markedly elevated plasma cholesterol levels and are a commonly used model of atherosclerosis (Ishibashi et al., 1994; Piedrahita et al., 1992). To determine the significance of Baf60a in cholesterol homeostasis and atherosclerosis, we generated cohorts of flox/flox:ApoE-/- and BaLKO:ApoE-/- mice and subjected them to WD feeding. Plasma lipid measurements indicated that hepatocyte-specific inactivation of Baf60a lowered total plasma cholesterol levels by approximately 30% in ApoE-/- background (Figure 6A). The cholesterol lowering effect was only observed in BaLKO:ApoE-/- mice under fed conditions. To elucidate the significance of hepatic Baf60a in atherosclerosis, we performed Oil Red O staining on the dissected arterial trees from flox/flox:ApoE-/- and BaLKO:ApoE-/- mice following 12 weeks of WD-feeding. Remarkably, hepatocyte-specific inactivation of Baf60a significantly ameliorated atherosclerotic lesion formation (Figure 6B). Compared to control, lesion area was approximately 53% smaller in BaLKO mice (Figure 6C). Gene expression analysis indicated that the expression of CAR and its target genes involved in bile acid metabolism was significantly lower in BaLKO:ApoE-/- mouse livers (Figure 6D). Together, these results illustrate a diet-sensitive epigenetic pathway in the liver that plays a critical role in lipid absorption, cholesterol homeostasis, and atherosclerosis.
Figure 6. Hepatic inactivation of Baf60a protects mice from diet-induced atherosclerosis.
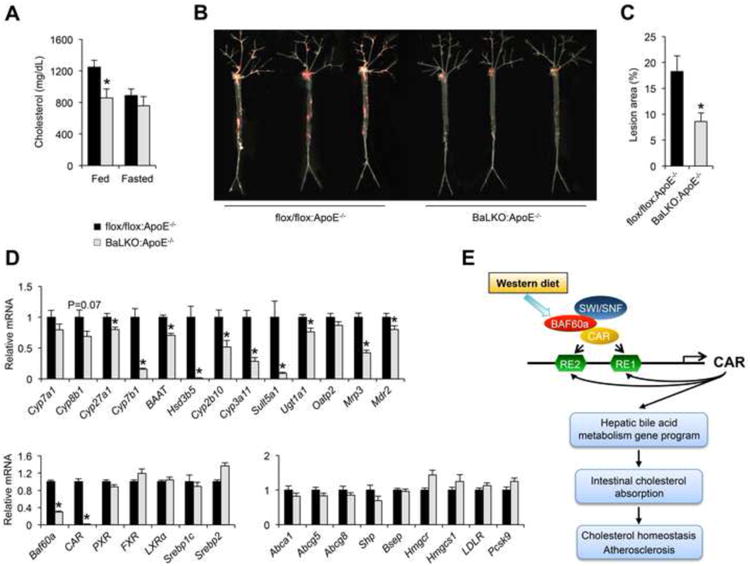
(A) Fed and fasting plasma cholesterol levels in flox/flox:ApoE-/- and BaLKO:ApoE-/- mice fed with WD for 12 weeks (n = 6).
(B) Representative images of en face Oil Red O stained aortic arches.
(C) Quantification of lesion area (n = 10-15).
(D) qPCR analysis of hepatic gene expression in flox/flox:ApoE-/- and BaLKO:ApoE-/- mouse livers (n = 6-9).
(E) Schematic model of the Baf60a/CAR feedforward regulatory loop inmediating diet-induced hypercholesterolemia and atherosclerosis. Data shown inA, C, and D represent mean ± s.e.m.; *P < 0.05 by two-tailed Student's t-test.
Discussion
The interaction between dietary nutrients and the gene networks is pivotal for metabolic homeostasis and contributes to the pathogenesis of complex diseases. Chronic consumption of fat- and cholesterol-rich diet is known to elevate plasma cholesterol levels and increase the risk for cardiovascular diseases (Glass and Witztum, 2001; Ross, 1993; Steinberg, 2002). In this study, we identified Baf60a as a diet-sensitive subunit of the SWI/SNF chromatin-remodeling complexes in the liver that serves a critical role in elevating plasma cholesterol levels following WD feeding. Hepatocyte-specific inactivation of Baf60a partially protects mice from WD-induced elevation of plasma cholesterol levels and atherosclerosis. At the mechanistic level, hepatic Baf60a is required for maintaining bile acid homeostasis and intestinal cholesterol absorption through a CAR/Baf60a feedforward regulatory loop in the liver (Figure 6E).
Several lines of evidence support a crucial role of Baf60a in bile acid production and intestinal cholesterol absorption. Transcriptional profiling and pathway analysis indicated that the genes involved in primary bile acid biosynthesis ranked at the top among those down-regulated by Baf60a deficiency. Interestingly, the expression of Cyp7a1, a target gene of LXR in the classic bile acid synthesis pathway, remained largely unchanged. In contrast, mRNA levels for the genes responsible for the alternative pathway of bile acid biosynthesis were significantly reduced in BaLKO mouse livers. Hepatic expression of CAR and a panel of genes involved in bile acid modification and transport were also lower in BaLKO mice. Consistently, adenoviral-mediated Baf60a overexpression was sufficient to induce this set of genes in cultured primary hepatocytes and in vivo in the liver. The significance of the alternative pathway was illustrated by significantly reduced bile acid synthesis and pool size in Cyp27a1 null mice (Repa et al., 2000; Rosen et al., 1998). Interestingly, inactivation of Cyp7a1 abolished the primary bile acid biosynthesis pathway and led to induction of the alternative pathway (Ishibashi et al., 1996; Schwarz et al., 1996). Whether Baf60a is required for this compensatory response when the primary bile acid biosynthesis pathway is defective remains unknown. Gain- and loss-of-function studies indicate that Baf60a regulates the expression of the bile acid metabolism gene program in a cell-autonomous manner. At the physiological level, hepatic Baf60a deficiency impaired bile acid production and intestinal cholesterol absorption, resulting in elevated fecal excretion of cholesterol.
Previous studies demonstrated that Baf60a forms a transcriptional complex with PPARα and PGC-1α to regulate fatty acid β-oxidation in hepatocytes (Li et al., 2008). Consistently, mRNA expression of several genes involved in hepatic fat oxidation was reduced in BaLKO mouse livers (data not shown). A somewhat unexpected finding in the current work is that hepatocyte-specific deficiency of Baf60a markedly reduced the expression of CAR. Our gain- and loss-of-function studies demonstrated that Baf60a cell-autonomously induces the expression of CAR through a feedforward regulatory loop. Accordingly, Baf60a deficiency impaired the recruitment of the SWI/SNF complexes, leading to reduced levels of chromatin marks characteristic of transcriptional activation in hepatocytes. The SWI/SNF ATPase subunit Brm has been shown to interact with Shp and modulate feedback regulation of Cyp7a1 (Kemper et al., 2004). Surprisingly, Baf60a deficiency does not appear to alter Cyp7a1 gene expression. Previous studies demonstrated that TCPOBOP treatments lowered plasma cholesterol levels in mice, in part through increased cholesterol catabolism and inhibition of lipogenesis (Masson et al., 2008; Sberna et al., 2011a; Sberna et al., 2011b). It is possible that the discrepancy between these studies and ours is due to whole body CAR activation vs. liver-specific reduction of CAR signaling. Alternatively, it is possible that Baf60a may target other additional transcription factors in the liver to modulate cholesterol metabolism. A future use of the CAR null mice will help determine the significance of CAR in the regulation of bile acid and cholesterol metabolism by hepatic Baf60a in vivo. Nevertheless, our results strongly suggest that hepatic Baf60a/CAR pathway plays a unique role in bile acid and cholesterol homeostasis. Therapeutic intervention of this pathway in the liver may alter the course of diet-induced hyperlipidemia and reduce the risk of atherosclerosis.
Experimental Procedures
Animal studies
For the generation of Baf60a liver specific knockout (BaLKO) mice, a targeting vector that carries two loxP sites flanking exons 3 to 5 of mouse Baf60a gene was constructed using BAC recombineering (Figure S1a). C57BL/6J ES cell line (Bruce 4) from Transgenic Animal Model Core at the University of Michigan was chosen for homologous recombination to facilitate breeding into C57BL/6J genetic background for metabolic studies. Transgenic mice expressing the CRE recombinase driven by liver specific albumin prompter (Alb-CRE) were used to cross with Baf60a flox/flox mice to achieve hepatic specific deletion of Baf60a. Mice were maintained in 12/12 h light/dark cycles and fed with normal rodent chow or Western diet (D12079B, Research Diets) as indicated. All animal studies were performed according to procedures approved by the University Committee on Use and Care of Animals. For in vivo adenovirus transduction, recombinant adenoviruses expressing GFP alone, FH-Baf60a, or FH-CAR were generated using AdEasy adenoviral vector system (Stratagene), and transduced into mice through tail vein injection. Prior to the final experiments, all adenoviruses were titrated in mice and monitored for the expression of GFP and adenoviral gene AdE4 to ensure similar doses were administered. For ezetimide treatment, ezetimide was suspended in vehicle (0.5% Hydroxyethylcellulose + 0.2% Tween-80), and administrated to mice through oral gavage for 4 days. Vehicle alone was used for the control treatment.
Primary hepatocyte isolation and adenovirus transduction
Primary hepatocytes were isolated using collagenase (Type II, Worthington CLS2), as previously described (Li et al., 2008). Cells were maintained in DMEM medium containing 10% bovine growth serum, and transduced with recombinant adenoviruses at similar moiety of infection (MOI) for 24 h, followed by RNA isolation and gene expression analysis, or ChIP assays.
Coimmunoprecipitation and ChIP
HEK293T cells were transiently transfected with HA-CAR and Myc-Baf60a for 24 h. Total lysates or immunoprecipitated proteins were analyzed by western blot using antibodies against c-Myc (Sigma, C3956; 1:2,000) or HA (Santa Cruz, sc-66181; 1:2,000). ChIP assay was performed according to the protocol developed by Upstate Biotechnology as described (Meng et al., 2014). Briefly, liver nuclei or primary hepatocytes were fixed with 1% formaldehyde and sonicated to produce chromatin lysates. The lysates were pre-cleared with Protein-G agarose beads and immunoprecipitated overnight with antibodies against Ace-H3 (Millipore, 06-599), H3K4m3 (Millipore, 07-473), Brg1 (Santa Cruz, sc-17796×), or control IgG in the presence of BSA and salmon sperm DNA. The next day, Protein-G agarose beads were added to each immunoprecipitation reaction for 1 h, followed by extensively wash and reverse crosslink. DNA was eluted from the beads and purified using a PCR Purification Kit (Invitrogen) and subsequently analyzed by qPCR using primers located on the proximal CAR promoter. Primers are available upon request.
Gene expression and Western blot analyses
TRIzol reagent (Life Technologies) was used to extract total RNA from tissues and cultured primary hepatocytes. Gene expression analyses were performed using SYBR Green reagent (Life Technologies). Relative abundance of mRNA was normalized to ribosomal protein 36B4. For total protein extracts, frozen liver samples were homogenized in a lysis buffer containing 50 mM Tris (pH = 7.5), 150 mM NaCl, 5 mM NaF, 25 mM β-glycerol phosphate, 1 mM sodium orthovanadate, 10% glycerol, 1% Triton X-100, 1 mM dithiothreitol (DTT), and protease inhibitor cocktail (Roche). For liver nuclear protein extracts, frozen livers were homogenized using a Dounce homogenizer in ice-cold homogenization buffer containing 0.6% NP40, 150 mM NaCl, 10 mM HEPES (pH = 7.9), 1 mM EDTA and protease inhibitor cocktail (Roche). The homogenates were centrifuged at 700 rpm for 1 min at 4 °C to remove tissue debris. The suspension was transferred to a new 15 ml tube and centrifuged at 3,000 rpm for 5 min at 4 °C. The nuclei pellet was washed with homogenization buffer and resuspended in a low-salt buffer containing 20 mM Tris (pH = 7.5), 25% glycerol, 1.5 mM MgCl2, 200 μM EDTA, 20 mM KCl, and protease inhibitor cocktail, followed by the addition of a high-salt buffer (1/2 volume) containing 20 mM Tris (pH = 7.5), 1.5 mM MgCl2, 200 μM EDTA, 1.2 M KCl and protease inhibitor cocktail. The homogenates were rotated for 2 h at 4 °C, and centrifuged at 13,000 rpm for 20 min at 4 °C to remove the cell debris. For immunoblotting assays, total protein lysates or liver nuclear extracts were separated by SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane (Millipore), followed by immunoblotting with the following primary antibodies. Rabbit polyclonal antibody to Baf60c was generated with the recombinant GST fusion mouse Baf60c protein and affinity purified. Antibody against Baf60a (1:1,000, 611728) was from BD Transduction Laboratories. Antibody against Baf53a (1:1,000, 10341-1-AP) was from Proteintech. Antibody against Lamin A/C (1:2,000, 2032) was from Cell Signaling. Antibodies against c-Myc (1:2000, C3956) and β-actin (1:2000, A4700) were from Sigma. Antibodies against HA (1:2,000, sc-66181), Brg1 (1:1,000, sc-17796×), Baf155 (1:2,000, sc-10756×), and Baf170 (1:1,000, sc-10757×) were from Santa Cruz. Antibody against LDLR (1:1,000, ab52818) was from Abcam.
Plasma analysis
For the lipoprotein profiling, pooled plasma samples from three flox/flox or three BaLKO mice were fractionated by fast protein liquid chromatography. The concentrations of cholesterol in each fraction were measured. HDL- and VLDL/LDL-cholesterol in individual plasma samples were separated and measured using HDL and LDL/VLDL Quantification Colorimetric Kit (BioVision). Plasma concentrations of triglycerides and NEFA were measured using commercial assay kits from Sigma and Wako Diagnostics, respectively. Plasma concentrations of total cholesterol, ALT, and AST were measured using assay kits from Stanbio Laboratory. Plasma Pcsk9 level was measured using the mPcsk9 ELISA kit (Circulex, Woburn, MA).
3H-Cholesterol absorption assay
Acute cholesterol absorption assay was performed as described (Li et al., 2014). Mice were oral gavage with [1,2-3H(N)]-cholesterol (5 mCi and 0.1 mg unlabeled cholesterol in 0.2 ml corn oil). Two hour later, the mice were euthanized, and plasma and liver samples were harvested. Livers were weighed (100 mg/mouse) and homogenized in 1 ml chloroform/methanol (2:1, v/v), followed by rotation at 37°C for 4 h, and spin at 2,000 g for 10 min. Radioactivity in triplicate aliquots of plasma and liver extracts was measured by liquid scintillation counting.
Bile acid analysis
Control and BaLKO mice fed with WD for 12 weeks were anesthetized. Plasma samples were collected via heart puncture. The gallbladder was removed after common bile duct ligation and transferred to a pre-weighed eppendorf tube. The bile content in the gallbladder was weighed and converted into volume assuming a density of 1 mg/μl. Liver was dissected, weighed, and homogenized in 75% ethanol. The homogenate was incubated at 50°C for 2 h to extract bile acids and centrifuged at 6,000 g for 10 min. Bile acid content in the supernatant was measured. Total bile acid concentrations in plasma, bile, and liver extracts were determined using a Total Bile Acid Assay Kit (BQ 092A-EALD, BQKits).
The concentration of different bile acids in the gallbladder was measured using liquid chromatography–tandem mass spectrometry (LC–MS/MS) as described previously (Perwaiz et al., 2001). Briefly, bile samples were diluted 1:200 and 1:10000 in methanol/water (1:1, v/v). Standard curve was generated with ACS grade analytical standard bile acid compounds in methanol/water (1:1, v/v) via serial dilution using propyl hydroxyl benzoate as internal standard. Ten μL of each sample was loaded to a Thermo Scientific Betabasic-18 100 × 2.1 mm, pore size 5 μm column, eluted with 0.1% formic acid in water as mobile phase A and methanol as mobile phase B in 20 min at 0.2 ml/min, and analyzed with a ABSciex 3200 QTRAP mass spectrometer using negative ESI MRM mode.
The bile acid pool size was measured as previously described (Li et al., 2011), Briefly, after the mice were weighed, anesthetized, and exsanguinated, the fresh organs and their contents were collected, minced, and extracted in 20 ml of 95% ethanol at 50 °C overnight, followed by spin at 6,000 g at 4 °C for 10 min and collection of the supernatant as extract. The extraction was repeated with another 20 ml of 85% ethanol and 20 ml of 75% ethanol at 50 °C for 2 h, sequentially. All the three extracts were combined and subjected for bile acid measurement using the Bile Acid Assay Kit (BQ 092A-EALD, BQKits).
Evaluation of Atherosclerosis
Atherosclerosis studies were conducted as described(Chang et al., 2012). Briefly, mice were anesthetized and subjected to whole body perfusion to fix the tissues. The entire aorta was micro-dissected from each mouse. Atherosclerosis was identified by en face Oil Red O staining, followed by imaging and lesion area quantification using Image J software.
Statistics
Data were analysed using the unpaired two-tailed Student's t-test for independent groups. A P value of less than 0.05 was considered statistically significant.
Acknowledgments
We thank the staff at the University of Michigan Transgenic Animal Core for the generation of Baf60a flox/flox mice and A. D. Jones (Michigan State University) for help with bile acid analysis. This work was supported by NIH grants (DK095151 and DK077086 to J.D.L; HL068878 and HL105114 to Y.E.C.; HL122664 to L.C.). Z.X.M is supported by the Scientist Development Grant from American Heart Association (15SDG22970032).
Footnotes
Accession Numbers. The accession number for the microarray datasets reported in this paper is GEO: GSE73709.
Author Contributions: J.D.L. and Z.X.M. conceived the project and designed research. Z.X.M., L.W. L.C, J.S., Y.L., J.B., and Y.E.C. performed the studies. Z.X.M. and J.D.L. analyzed the data and wrote the manuscript.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) Jama. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annual review of medicine. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. Journal of lipid research. 2009;50(Suppl):S15–27. doi: 10.1194/jlr.R800054-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nature reviews Molecular cell biology. 2012;13:213–224. doi: 10.1038/nrm3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C, Zhang J, Wu J, Zeng R, Chen YE. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation. 2012;126:1067–1078. doi: 10.1161/CIRCULATIONAHA.112.104489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Roeder RG. Mediator-dependent nuclear receptor function. Seminars in cell & developmental biology. 2011;22:749–758. doi: 10.1016/j.semcdb.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JY. Bile acids: regulation of synthesis. Journal of lipid research. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Lonard DM, O'Malley BW. Nuclear receptor coactivators: master regulators of human health and disease. Annual review of medicine. 2014;65:279–292. doi: 10.1146/annurev-med-051812-145316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debril MB, Gelman L, Fayard E, Annicotte JS, Rocchi S, Auwerx J. Transcription factors and nuclear receptors interact with the SWI/SNF complex through the BAF60c subunit. The Journal of biological chemistry. 2004;279:16677–16686. doi: 10.1074/jbc.M312288200. [DOI] [PubMed] [Google Scholar]
- Dong B, Qatanani M, Moore DD. Constitutive androstane receptor mediates the induction of drug metabolism in mouse models of type 1 diabetes. Hepatology. 2009;50:622–629. doi: 10.1002/hep.23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duane WC. Effects of lovastatin and dietary cholesterol on bile acid kinetics and bile lipid composition in healthy male subjects. Journal of lipid research. 1994;35:501–509. [PubMed] [Google Scholar]
- Evans RM, Mangelsdorf DJ. Nuclear Receptors, RXR, and the Big Bang. Cell. 2014;157:255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Xie W. Targeting xenobiotic receptors PXR and CAR for metabolic diseases. Trends in pharmacological sciences. 2012;33:552–558. doi: 10.1016/j.tips.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell. 2015;161:161–172. doi: 10.1016/j.cell.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Molecular cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Merz CN, Brewer HB, Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC, Jr, Stone NJ. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. Journal of lipid research. 2009;50(Suppl):S172–177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Schwarz M, Frykman PK, Herz J, Russell DW. Disruption of cholesterol 7alpha-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. The Journal of biological chemistry. 1996;271:18017–18023. doi: 10.1074/jbc.271.30.18017. [DOI] [PubMed] [Google Scholar]
- Kemper JK, Kim H, Miao J, Bhalla S, Bae Y. Role of an mSin3A-Swi/Snf chromatin remodeling complex in the feedback repression of bile acid biosynthesis by SHP. Molecular and cellular biology. 2004;24:7707–7719. doi: 10.1128/MCB.24.17.7707-7719.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. The Journal of biological chemistry. 1997;272:3137–3140. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- Li PS, Fu ZY, Zhang YY, Zhang JH, Xu CQ, Ma YT, Li BL, Song BL. The clathrin adaptor Numb regulates intestinal cholesterol absorption through dynamic interaction with NPC1L1. Nature medicine. 2014;20:80–86. doi: 10.1038/nm.3417. [DOI] [PubMed] [Google Scholar]
- Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. Genome-wide coactivation analysis of PGC-1alpha identifies BAF60a as a regulator of hepatic lipid metabolism. Cell metabolism. 2008;8:105–117. doi: 10.1016/j.cmet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiang JY. Nuclear receptors in bile acid metabolism. Drug metabolism reviews. 2013;45:145–155. doi: 10.3109/03602532.2012.740048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Matozel M, Boehme S, Kong B, Nilsson LM, Guo G, Ellis E, Chiang JY. Overexpression of cholesterol 7alpha-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology. 2011;53:996–1006. doi: 10.1002/hep.24107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Molecular cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Masson D, Qatanani M, Sberna AL, Xiao R, Pais de Barros JP, Grober J, Deckert V, Athias A, Gambert P, Lagrost L, et al. Activation of the constitutive androstane receptor decreases HDL in wild-type and human apoA-I transgenic mice. J Lipid Res. 2008;49:1682–1691. doi: 10.1194/jlr.M700374-JLR200. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Molecular and cellular endocrinology. 2013;368:17–29. doi: 10.1016/j.mce.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ZX, Li S, Wang L, Ko HJ, Lee Y, Jung DY, Okutsu M, Yan Z, Kim JK, Lin JD. Baf60c drives glycolytic metabolism in the muscle and improves systemic glucose homeostasis through Deptor-mediated Akt activation. Nature medicine. 2013;19:640–645. doi: 10.1038/nm.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng ZX, Wang L, Xiao Y, Lin JD. The Baf60c/Deptor pathway links skeletal muscle inflammation to glucose homeostasis in obesity. Diabetes. 2014;63:1533–1545. doi: 10.2337/db13-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes & development. 2013;27:819–835. doi: 10.1101/gad.214023.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annual review of pharmacology and toxicology. 2008;48:1–32. doi: 10.1146/annurev.pharmtox.47.120505.105349. [DOI] [PubMed] [Google Scholar]
- Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- Perwaiz S, Tuchweber B, Mignault D, Gilat T, Yousef IM. Determination of bile acids in biological fluids by liquid chromatography-electrospray tandem mass spectrometry. Journal of lipid research. 2001;42:114–119. [PubMed] [Google Scholar]
- Phelan ML, Sif S, Narlikar GJ, Kingston RE. Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Molecular cell. 1999;3:247–253. doi: 10.1016/s1097-2765(00)80315-9. [DOI] [PubMed] [Google Scholar]
- Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri PL, Mercola M. BAF60 A, B, and Cs of muscle determination and renewal. Genes & development. 2012;26:2673–2683. doi: 10.1101/gad.207415.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Lund EG, Horton JD, Leitersdorf E, Russell DW, Dietschy JM, Turley SD. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. The Journal of biological chemistry. 2000;275:39685–39692. doi: 10.1074/jbc.M007653200. [DOI] [PubMed] [Google Scholar]
- Rosen H, Reshef A, Maeda N, Lippoldt A, Shpizen S, Triger L, Eggertsen G, Bjorkhem I, Leitersdorf E. Markedly reduced bile acid synthesis but maintained levels of cholesterol and vitamin D metabolites in mice with disrupted sterol 27-hydroxylase gene. The Journal of biological chemistry. 1998;273:14805–14812. doi: 10.1074/jbc.273.24.14805. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- Saini SP, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W. A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Molecular pharmacology. 2004;65:292–300. doi: 10.1124/mol.65.2.292. [DOI] [PubMed] [Google Scholar]
- Sberna AL, Assem M, Gautier T, Grober J, Guiu B, Jeannin A, Pais de Barros JP, Athias A, Lagrost L, Masson D. Constitutive androstane receptor activation stimulates faecal bile acid excretion and reverse cholesterol transport in mice. Journal of hepatology. 2011a;55:154–161. doi: 10.1016/j.jhep.2010.10.029. [DOI] [PubMed] [Google Scholar]
- Sberna AL, Assem M, Xiao R, Ayers S, Gautier T, Guiu B, Deckert V, Chevriaux A, Grober J, Le Guern N, et al. Constitutive androstane receptor activation decreases plasma apolipoprotein B-containing lipoproteins and atherosclerosis in low-density lipoprotein receptor-deficient mice. Arteriosclerosis, thrombosis, and vascular biology. 2011b;31:2232–2239. doi: 10.1161/ATVBAHA.110.222497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Lund EG, Setchell KD, Kayden HJ, Zerwekh JE, Bjorkhem I, Herz J, Russell DW. Disruption of cholesterol 7alpha-hydroxylase gene in mice. II. Bile acid deficiency is overcome by induction of oxysterol 7alpha-hydroxylase. The Journal of biological chemistry. 1996;271:18024–18031. doi: 10.1074/jbc.271.30.18024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Russell DW, Dietschy JM, Turley SD. Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. Journal of lipid research. 2001;42:1594–1603. [PubMed] [Google Scholar]
- Steinberg D. Atherogenesis in perspective: hypercholesterolemia and inflammation as partners in crime. Nat Med. 2002;8:1211–1217. doi: 10.1038/nm1102-1211. [DOI] [PubMed] [Google Scholar]
- Sudarsanam P, Winston F. The Swi/Snf family nucleosome-remodeling complexes and transcriptional control. Trends Genet. 2000;16:345–351. doi: 10.1016/s0168-9525(00)02060-6. [DOI] [PubMed] [Google Scholar]
- Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nature reviews Drug discovery. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- Tiemann M, Han Z, Soccio R, Bollineni J, Shefer S, Sehayek E, Breslow JL. Cholesterol feeding of mice expressing cholesterol 7alpha-hydroxylase increases bile acid pool size despite decreased enzyme activity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1846–1851. doi: 10.1073/pnas.0308426100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes & development. 1996;10:2117–2130. doi: 10.1101/gad.10.17.2117. [DOI] [PubMed] [Google Scholar]
- Waters DD, Brotons C, Chiang CW, Ferrieres J, Foody J, Jukema JW, Santos RD, Verdejo J, Messig M, McPherson R, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation. 2009;120:28–34. doi: 10.1161/CIRCULATIONAHA.108.838466. [DOI] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nature reviews Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136:200–206. doi: 10.1016/j.cell.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Salen G, Shefer S, Tint GS, Nguyen LB, Chen TS, Greenblatt D. Increasing dietary cholesterol induces different regulation of classic and alternative bile acid synthesis. The Journal of clinical investigation. 1999;103:89–95. doi: 10.1172/JCI4414. [DOI] [PMC free article] [PubMed] [Google Scholar]