

Abstract
Large-scale genetic analysis of lethal phenotypes has elucidated the molecular underpinnings of many biological processes. Using the bacterial clustered regularly interspaced short palindromic repeats (CRISPR) system, we constructed a genome-wide single-guide RNA (sgRNA) library to screen for genes required for proliferation and survival in a human cancer cell line. Our screen revealed the set of cell-essential genes, which was validated by an orthogonal gene-trap-based screen and comparison with yeast gene knockouts. This set is enriched for genes that encode components of fundamental pathways, are expressed at high levels, and contain few inactivating polymorphisms in the human population. We also uncovered a large group of uncharacterized genes involved in RNA processing, a number of whose products localize to the nucleolus. Lastly, screens in additional cell lines showed a high degree of overlap in gene essentiality, but also revealed differences specific to each cell line and cancer type that reflect the developmental origin, oncogenic drivers, paralogous gene expression pattern, and chromosomal structure of each line. These results demonstrate the power of CRISPR-based screens and suggest a general strategy for identifying liabilities in cancer cells.
The systematic identification of essential genes in microorganisms has provided critical insights into the molecular basis of many biological processes (1). Similar studies in human cells have been hindered by the lack of suitable tools. Moreover, little is known about how the set of cell essential genes differs across cell types and genotypes. Differentially essential genes are likely to encode tissue-specific modulators of key cellular processes and important targets for cancer therapies. Here, we used two independent approaches for inactivating genes at the DNA level to define the cell-essential genes of the human genome.
The first approach employs the CRISPR/Cas9-based gene editing system, which has emerged as a powerful tool to engineer the genomes of cultured cells and whole organisms (2, 3). We and others have shown that lentiviral (single-guide RNA) sgRNA libraries can enable pooled loss-of-function screens and have used the technology to uncover mediators of drug resistance and pathogen toxicity (4-6). To systematically identify cell-essential genes, we constructed a novel library, which was optimized for high cleavage activity, and performed a proliferation-based screen in the near-haploid human KBM7 chronic myelogenous leukemia (CML) cell line (Fig. 1; Note S1).
Fig. 1. Two approaches for genetic screening in human cells.

CRISPR/Cas9 (top): Cells are transduced with a genome-wide sgRNA lentiviral library. Gene inactivation via Cas9-mediated genomic cleavage is directed by the 20-bp sequence at the 5′ end of the sgRNA. Cells bearing sgRNAs targeting essential genes are depleted in the final population. Gene-trap (bottom): KBM7 cells are transduced with a gene-trap retrovirus which integrates in an inactivating or “harmless” orientation at random genomic loci. Essential genes contain fewer insertions in the inactivating orientation. Sample data for two neighboring genes RPL14, encoding an essential ribosomal protein, and ZNF619, encoding a dispensable zinc finger protein, are displayed. For CRISPR/Cas9, sgRNAs are plotted according to their target position along each gene with the height of each bar indicating the level of depletion. Boxes indicate individual exons. For gene-trap, the intronic insertion sites in each gene are plotted according to their orientation and genomic position. The height of each point is randomized.
The unusual karyotype of these cells also allows for an independent method of genetic screening. In this approach, null mutants are generated at random through retroviral gene-trap mutagenesis, selected for a phenotype, and monitored by sequencing the viral integration sites to pinpoint the causal genes (7). Positive selection-based screens using this method have identified genes underlying processes such as epigenetic silencing and viral infection (7-9). We extended this technique by developing a strategy for negative selection and conducted a screen for cell-essential genes (Fig. 1; Note S2).
For both methods, we computed a score for each gene that reflects the fitness cost imposed by inactivation of the gene. We defined the CRISPR score (CS) as the average log2 fold-change in the abundance of all sgRNAs targeting a given gene, with replicate experiments showing a high degree of reproducibility (r=0.90) (Fig. 2A, S1A; Table S2). Of the 18,166 genes targeted by the library, 1,878 scored as essential for optimal proliferation in our screen, although this precise number depends on the cutoff chosen (Fig. 2A; Table S2-3). Overall, this fraction represents ∼10% of genes within our dataset or roughly 9.2% of the entire genome (many of the genes not targeted by our library encode olfactory receptors that are unlikely to be cell-essential). Gene products that act in a non-cell-autonomous manner are not expected to score as essential in this pooled setting (Fig. S1B).
Fig. 2. Identification and characterization of human cell-essential genes.

(A) CRISPR scores (CS) of all genes in the KBM7 cells. Similar proportions of cell-essential genes were identified on all autosomes. (B) KBM7 gene-trap score (GTS) distributions. No low GTS genes were detected on the diploid chromosome 8. (C) CS and GTS of overlapping genes. (D) Yeast homolog essentiality prediction analysis. (E) Broader retention of essential genes across species. (F) Higher sequence conservation of essential genes. (G) Genes with deleterious stop-gain variants are less likely to be essential. (H) Greater connectivity of proteins encoded by essential genes. (I) Higher mRNA transcript levels of essential genes. (J) Genes with paralogs are less likely to be essential. *** denotes p<0.001 from Kolmogorov-Smirnov test.
We defined the gene-trap score (GTS) as the fraction of insertions in a given gene occurring in the inactivating orientation. Because the accuracy of this score depends on the depth of insertional coverage, we set a requirement on the minimum number (n=65) of anti-sense inserts in a gene needed for inclusion in our analysis by measuring the concordance between replicate experiments (Fig. 2B, S1C-D; Table S4; Note S2). For the 7,370 genes on the haploid chromosomes that exceeded this threshold, the GTS was well-correlated with the CS and with results from a co-published study employing a similar gene-trap approach (r=0.68) (Fig. 2C, S1E-F; Note S3). The strong correspondence between the overlapping sets of cell-essential genes defined by the two methods provides support for the accuracy of the CRISPR scores for the full set of 18,166 genes.
The two methods differed with respect to the diploid chromosome 8. Whereas the gene-trap screen failed to detect any cell-essential genes on this chromosome, the CRISPR screen uncovered a similar proportion of cell-essential genes on all the autosomes (Fig. 2A-C). These observations indicate that (i) the vast majority of cell-essential genes are haplosufficient and (ii) biallelic inactivation occurs at high frequency in our CRISPR screen (4).
To assess the accuracy of our scores with other measures of gene essentiality, we relied on functional profiling experiments conducted in yeast S. cerevisiae as a benchmark (1, 10). Specifically, we ranked genes common to all datasets by their scores in each dataset –CRISPR, gene trap, scores from similar loss-of-function RNA interference (RNAi) screens (11), and, as a naïve proxy for gene essentiality, gene expression levels determined by RNA-seq– and compared these rankings with the essentiality of yeast homologs. The CRISPR and gene-trap methods had significantly stronger correlations with the yeast results than did the RNAi screens or gene expression, which performed similarly to each other (both methods: p<10-4, permutation test) (Fig. 2D). Based on additional comparisons with yeast gene essentiality, we also found that (i) our new optimized sgRNA library gave better results than those from screens using older unoptimized libraries (4, 5) and (ii) the coverage of this library (∼10 constructs per gene) approaches saturation, as evidenced by down-sampling (decreasing the coverage by randomly eliminating subsets of data) (Fig. S2A-B). Together, our results suggest that scores from the CRISPR and gene-trap screens both provide accurate measures of the cell-essentiality of human genes.
Essential genes should be under strong purifying selection and should thus show greater evolutionary constraint than non-essential genes (12). Consistent with this expectation, the essential genes found in our screens were more broadly retained across species, showed higher levels of conservation between closely related species, and contain fewer inactivating polymorphisms within the human species, as compared to their dispensable counterparts (Fig. 2E-G). Essential genes also tend to have higher expression and encode proteins that engage in more protein-protein interactions (13-15). These patterns were also observed in our CRISPR dataset (Fig. 2H-I).
In S. cerevisiae, genes with paralogous copies in the genome show a lower degree of essentiality –due presumably to at least partial functional overlap (16). Surprisingly, meta-analysis of knockout mouse collections has suggested that there is no such correlation in mammals (17, 18). However, others have challenged this interpretation because the genes analyzed were far from a random sample (19). Using the results from our genome-wide screens, we revisited this question and observed that genes with paralogs are indeed less likely to be essential, consistent with the idea that paralogs can provide functional redundancy at the cellular level (Fig. 2J).
To examine the functions of the cell-essential genes we used gene set enrichment analysis (GSEA) and found strong enrichment for many fundamental biological processes, such as DNA replication, RNA transcription and mRNA translation (Fig. S3A) (20). Whereas most of the genes could be assigned to such well-defined pathways, no function has been ascribed to about 330 of the cell-essential genes (18%) (Fig. 3A). For this set of uncharacterized genes, an analysis of the domains within their encoded gene products and comparisons to proteomic datasets from organellar purifications revealed substantial enrichment in proteins found in the nucleolus and those containing domains associated with RNA processing (Fig. S3B-C) (21).
Fig. 3. Functional characterization of novel cell-essential genes.
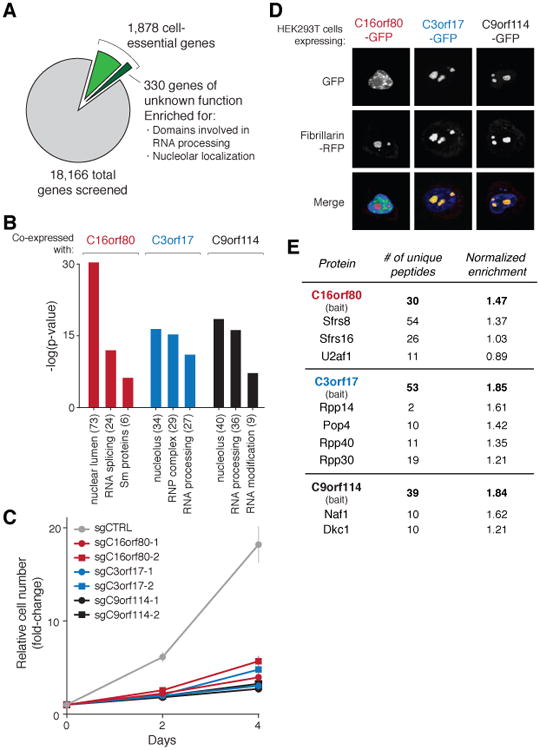
(A) Of the 1,878 cell-essential genes identified in the KBM7 cell line, 330 genes had no known function. (B) Genes co-expressed with C16orf80, C3orf17, and C9orf114 across CCLE cell lines were associated with RNA processing. Parentheses denote the number of genes in the set. (C) Proliferation of KBM7 cells transduced with sgRNAs targeting C16orf80, C3orf17, and C9orf114, or a non-targeting control. Error bars denote SD (n=4). (D) C16orf80 localized to the nucleus and C3orf17 and C9orf114 to the nucleolus. Fibrillarin-RFP was used as a nucleolar marker. (E) Multiple subunits of the spliceosome, RNAse P/MRP and H/ACA ribonucleoprotein complexes interact with C16orf80, C3orf17, and C9orf114, respectively.
We characterized three such genes, C16orf80, C3orf17, and C9orf114, whose mRNA expression patterns across the cancer cell line encyclopedia (CCLE) were correlated with that of genes involved in RNA processing (Fig. 3B). We validated the essentiality of these genes in short-term proliferation assays and detected localization of their products to the nucleus (C16orf80) or nucleolus (C3orf17 and C9orf114) (Fig. 3C-D) (22). Additionally, mass spectrometric analyses of anti-FLAG-immunoprecipitates prepared from KBM7 cells expressing FLAG-tagged C16orf80, C3orf17, and C9orf114 revealed interactions with multiple subunits of the spliceosome, ribonuclease (RNAse) P/MRP, and H/ACA small nucleolar ribonucleoprotein (snoRNP) complexes, respectively (Fig. 3E). These results implicate C16orf80 in splicing, consistent with its association with mRNAs; C3orf17 in rRNA/tRNA processing; and C9orf114 in RNA modification (23). More broadly, our results indicate that the molecular components of many critical cellular processes, especially RNA processing, have yet to be fully defined in mammalian cells.
To determine how the set of essential genes differs among cell lines, we screened another CML cell line (K562) and two Burkitt's lymphoma cell lines (Raji and Jiyoye) using the CRISPR system (Table S2-3). Overall, the sets of essential genes in the four cell lines showed a high degree of overlap (Fig. 4A). Importantly, out of these four cell lines, the KBM7 CRISPR results showed the highest correlation with the KBM7 gene-trap dataset, suggesting that the few differences observed are likely to be biologically meaningful (Fig. S4A).
Fig. 4. Comparisons of gene essentiality across four cell lines.
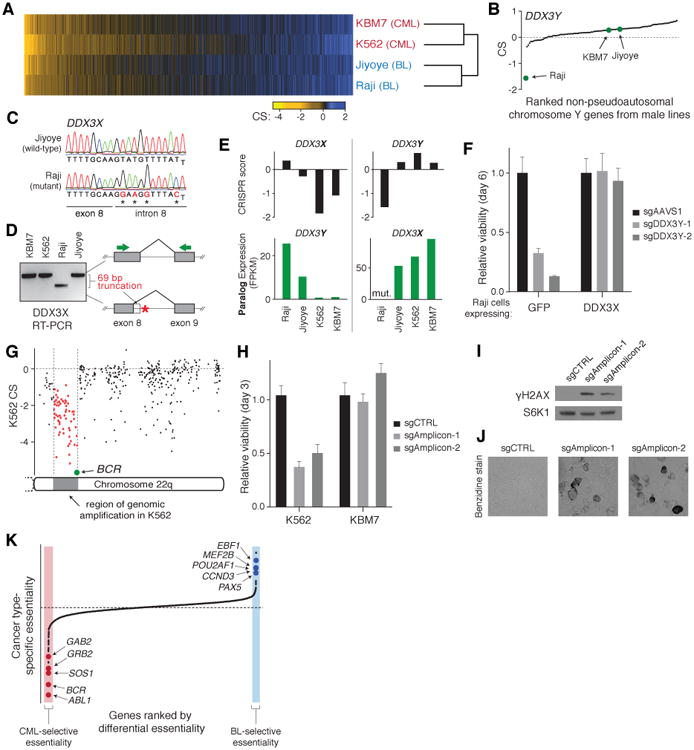
(A) Heatmap of CS across four cell lines sorted by average CS. (B) CS of genes residing in the non-pseudoautosomal region of chromosome Y for male cell lines (Raji, Jiyoye and KBM7). (C) Sanger sequencing of DDX3X in Raji cells reveals mutations in the 5′ splice site of intron 8. (D) Splice-site mutations in DDX3X result in a 69-bp truncation of the mRNA. PCR primers spanning exons 8 and 9 of DDX3X (denoted by green arrows) were used to amplify cDNA from each line. (E) Essentiality of DDX3X and DDX3Y is determined by the expression and mutation status of their paralogs. (F) Proliferation of GFP- and DDX3X-expressing Raji cells transduced with sgRNAs targeting DDX3Y or an AAVS1-targeting control. Error bars denote SD (n=4). (G) Analysis of genes on chromosome 22q reveals apparent ‘essentiality’ of 61 contiguous genes in K562 residing in a region of high-copy tandem amplification. (H) Proliferation of K562 and KBM7 cells transduced with sgRNAs targeting a non-genic region within the BCR-ABL amplicon in K562 cells or a non-targeting control. Error bars denote SD (n=4). (I) γH2AX (phospho-S139 H2AX) immunoblot analysis of K562 transduced with sgRNAs as in (H). S6K1 was used as a loading control. (J) Cleavage within amplified region induced erythroid differentiation in K562 cells as assessed by 3,3′-dimethoxybenzidine hemoglobin staining. (K) Comparison of gene essentiality between the two cancer types reveals oncogenic drivers and lineage specifiers. Genes were ranked by the difference between the average CS of each cancer type.
We focused first on genes found to be essential in only one of the four cell lines. The Raji, Jiyoye, and KBM7 cell lines had 6, 7, and 19 such genes, respectively (Fig. S4B; Table S5). One example was DDX3Y, which resides in the non-pseudoautosomal region of the Y chromosome and was required only in Raji cells (Fig. 4B). Its X-linked paralog, DDX3X, was essential in KBM7 and K562 cells (Fig 4E). Both genes encode DEAD-box helicases that likely have similar cellular functions (24). Thus, the dependence on one paralog might reflect functional absence of the other paralog. Indeed, DNA sequencing of DDX3X in Raji cells revealed hemizygous mutations in the 5′-splice site of intron 8 that resulted in the production of a truncated mRNA transcript (Fig. 4C-D). Conversely, DDX3Y was not expressed in KBM7 cells and was not present in K562 cells, which are of female origin (Fig. 4E). Introduction of wild-type DDX3X cDNA into Raji cells fully rescued the proliferation defect resulting from DDX3Y loss, indicating that the paralogous genes are essential and functionally overlapping (Fig. 4F). Essential paralogous gene pairs, involved in glucose metabolism (HK1/2 and SLC2A1/3) and cell-cycle regulation (CDK4/6), were also observed in the Jiyoye line (Fig S4C; Note S4). Vulnerabilities due to the loss of a paralogous partner may serve as targets for highly personalized anti-tumor therapies (25).
In some cases, cell line-specific essentiality of paralogous genes did not reflect differential expression. For example, the transcription factors GATA1 and GATA2 are expressed in both K562 and KBM7 cells, but the first is specifically essential in K562 cells and the second in KBM7 cells (Fig. S4D). These master regulators are known to promote proliferation and survival during distinct developmental stages in the hematopoietic lineage – GATA1 is required for the survival of erythroid progenitors and GATA2 for the maintenance and proliferation of immature hematopoietic progenitors (26). These two cell types likely correspond to the cells-of-origin of the two CML lines (27, 28). We also identified similar instances of genes required for cell line-specific functions such as nf-κb pathway regulation and homology-directed DNA repair in Raji and KBM7 cells, respectively (Fig. S4E-F; Note S5).
Whereas the other three cell lines showed only a few cell line-specific essential genes, the K562 cell line was an outlier, with 63 such genes (Fig. S4B). Oddly, these genes showed no discernible common functions and many were not even expressed in the K562 cell line. (Additionally, a few encoded secreted factors whose loss would not be expected to be lethal in a pooled screen.) This mystery was resolved when we examined the chromosomal location of the genes: strikingly, the majority (39 of the 63 genes) reside near 9q34 and 22q11. These two regions are translocated to produce the BCR-ABL oncogene and are present in a high-copy tandem amplification in K562 cells (Fig. S5A-B) (29). Notably, all 61 contiguous genes within the amplicon on 22q11 scored as essential, suggesting that sgRNA-mediated cleavage in this repeated region induces cytotoxicity in a manner unrelated to the function of the target genes themselves (Fig. 4G, S5F). Indeed, two sgRNAs targeting non-genic sites within the amplicon were toxic to K562, but not KBM7 cells. They increased the abundance of phosphorylated histone H2AX (γH2AX), a marker of DNA damage, and induced erythroid differentiation, which occurs upon DNA damage in this cell line (Fig. 4H-J, S5C) (30). We obtained similar results in another cell line, HEL, which contains a highly amplified region surrounding the JAK2 tyrosine kinase (Fig. S5D-E). Together, these findings indicate that lethality upon Cas9-mediated cutting may also reflect chromosome structure and therefore should be evaluated in light of copy-number information.
Finally, we looked for consistent differences in essential genes between the two CML and two Burkitt's lymphoma lines. Such genes might represent attractive targets for anti-neoplastic therapies as their inhibition is less likely to be broadly cytotoxic. Overall, we identified 33 genes that were specifically essential in the CML lines and 15 genes in the Burkitt's lymphoma lines (Fig. 4K; Table S6). As a control, permuted comparisons – that is, a set containing of one CML and one Burkitt's line vs. the complementary sets – showed roughly half as many “set-specific” essential genes (Fig. S6A-C).
In the CML lines, the top two genes were BCR and ABL1, consistent with the known essentiality of the BCR-ABL translocation product and the therapeutic effect of BCR-ABL inhibitors such as imatinib (31). Additional members of the BCR-ABL signaling pathway, son of sevenless homolog 1 (SOS1), growth factor receptor-bound protein (GRB2), and GRB2-associated binding protein 2 (GAB2) scored strongly as well (ranked 3, 4, and 7, respectively). Network analysis of the other top hits also uncovered several genes encoding assembly factors for the electron transport chain, as well as enzymes involved in folate-mediated one-carbon metabolism. These results suggest additional potential targets for CML therapy (Table S6).
In the B cell-derived Burkitt's lymphoma cell lines, the top genes included three B cell lineage transcription factors early B-cell factor 1 (EBF1), POU class 2 associating factor 1 (POU2AF1), and paired box 5 (PAX5) (ranked 3, 6, and 8, respectively). Each of these genes is the target of recurrent translocations in lymphoma (32-34). Enhancers of the corresponding three gene loci all show a high level of bromodomain containing 4 (BRD4) occupancy in Ly1 cells, a related diffuse large B cell lymphoma cell line, suggesting bromodomain inhibitors such as JQ1 as potential treatments (35). Other selectively essential genes included MEF2B, a transcriptional activator of BCL6, B-cell lymphoma 6, and CCND3, cyclin D3, both of which are frequently mutated and implicated in the pathogenesis of various lymphomas (36). Intriguingly, the top two hits, CHM and RPP25L, do not appear to have specific roles in B cells; rather, their differential essentiality is likely explained by the lack of expression of their paralogs, CHML and RPP25, in both of the Burkitt's lymphoma cell lines studied (Fig. S6D).
In conclusion, we used two complementary and concordant approaches, CRISPR and gene-trap, to define the cell-essential genes in the human genome. Although the gene-trap method is suitable only for loss-of-function screening in rare haploid cell lines, the CRISPR method is broadly applicable. Extending our analysis across different cell lines and tumor types, we developed a framework to assess differential gene essentiality and identify potential drivers of the malignant state. The method can be readily applied to more cell lines per cancer type to eliminate idiosyncrasies particular to a given cell line and to more cancer types to systematically uncover tumor-specific liabilities that might be exploited for targeted therapies.
Supplementary Material
Acknowledgments
We thank T. Mikkelsen for assistance with oligonucleotide synthesis; Z. Tsun for assistance with figures; C. Hartigan, G. Guzman, M. Schenone, and S. Carr for mass spectrometric analysis; J. Down and J. Chen for reagents for hemoglobin staining. This work was supported by the National Institutes of Health (CA103866) (D.M.S.), the National Human Genome Research Institute (2U54HG003067-10) (E.S.L.), an award from the National Science Foundation (T.W.), an award from the MIT Whitaker Health Sciences Fund (T.W.). D.M.S. is an investigator of the Howard Hughes Medical Institute. T.W., D.M.S., and E.S.L. are inventors on a patent application for functional genomics using the CRISPR-Cas system and T.W. and D.M.S. are in the process of forming a company using this technology. The sgRNA plasmid library and other plasmids described here have been deposited in Addgene.
References and Notes
- 1.Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 2.Cong L, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H, et al. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shalem O, et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera MDC, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotech. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 7.Carette JE, et al. Haploid Genetic Screens in Human Cells Identify Host Factors Used by Pathogens. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]
- 8.Carette JE, et al. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tchasovnikarova IA, et al. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science. 2015;348:1481–1485. doi: 10.1126/science.aaa7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kastenmayer JP, et al. Functional genomics of genes with small open reading frames (sORFs) in S. cerevisiae. Genome Research. 2006;16:365–373. doi: 10.1101/gr.4355406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley GS, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Scientific Data. 2014;1 doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson AC, Carlson SS, White TJ. Biochemical Evolution. Annual Review of Biochemistry. 1977;46:573–639. doi: 10.1146/annurev.bi.46.070177.003041. [DOI] [PubMed] [Google Scholar]
- 13.Ishihama Y, et al. Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics. 2008;9:102. doi: 10.1186/1471-2164-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 15.Hart T, Brown KR, Sircoulomb F, Rottapel R, Moffat J. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Molecular Systems Biology. 2014;10:n/a–n/a. doi: 10.15252/msb.20145216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu Z, et al. Role of duplicate genes in genetic robustness against null mutations. Nature. 2003;421:63–66. doi: 10.1038/nature01198. [DOI] [PubMed] [Google Scholar]
- 17.Liang H, Li WH. Gene essentiality, gene duplicability and protein connectivity in human and mouse. Trends in Genetics. 2007;23:375–378. doi: 10.1016/j.tig.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Liao BY, Zhang J. Mouse duplicate genes are as essential as singletons. Trends in Genetics. 2007;23:378–381. doi: 10.1016/j.tig.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Makino T, Hokamp K, McLysaght A. The complex relationship of gene duplication and essentiality. Trends in Genetics. 2009;25:152–155. doi: 10.1016/j.tig.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian A, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmad Y, Boisvert FM, Lundberg E, Uhlen M, Lamond AI. Systematic Analysis of Protein Pools, Isoforms, and Modifications Affecting Turnover and Subcellular Localization. Molecular & Cellular Proteomics. 2012;11 doi: 10.1074/mcp.M111.013680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–307. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexander G, Baltz, et al. The mRNA-Bound Proteome and Its Global Occupancy Profile on Protein-Coding Transcripts. Molecular Cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 24.Sekiguchi T, Iida H, Fukumura J, Nishimoto T. Human DDX3Y, the Y-encoded isoform of RNA helicase DDX3, rescues a hamster temperature-sensitive ET24 mutant cell line with a DDX3X mutation. Experimental Cell Research. 2004;300:213–222. doi: 10.1016/j.yexcr.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 25.Muller FL, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–342. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohneda K, Yamamoto M. Roles of Hematopoietic Transcription Factors GATA-1 and GATA-2 in the Development of Red Blood Cell Lineage. Acta Haematologica. 2002;108:237–245. doi: 10.1159/000065660. [DOI] [PubMed] [Google Scholar]
- 27.Andersson LC, Nilsson K, Gahmberg CG. K562—A human erythroleukemic cell line. International Journal of Cancer. 1979;23:143–147. doi: 10.1002/ijc.2910230202. [DOI] [PubMed] [Google Scholar]
- 28.Andersson BS, et al. KBM-7, a human myeloid leukemia cell line with double Philadelphia chromosomes lacking normal c-ABL and BCR transcripts. Leukemia. 1995;9:2100–2108. [PubMed] [Google Scholar]
- 29.Wu SQ, et al. Extensive amplification of bcr/abl fusion genes clustered on three marker chromosomes in human leukemic cell line K-562. Leukemia. 1995;9:858–862. [PubMed] [Google Scholar]
- 30.Constantinou A, Kiguchi K, Huberman E. Induction of Differentiation and DNA Strand Breakage in Human HL-60 and K-562 Leukemia Cells by Genistein. Cancer Research. 1990;50:2618–2624. [PubMed] [Google Scholar]
- 31.Scappini B, et al. Changes associated with the development of resistance to imatinib (STI571) in two leukemia cell lines expressing p210 Bcr/Abl protein. Cancer. 2004;100:1459–1471. doi: 10.1002/cncr.20131. [DOI] [PubMed] [Google Scholar]
- 32.Iida S, et al. The t(9;14)(p13;q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX-5 gene. Blood. 1996;88:4110–4117. [PubMed] [Google Scholar]
- 33.Bouamar H, et al. A capture-sequencing strategy identifies IRF8, EBF1, and APRIL as novel IGH fusion partners in B-cell lymphoma. Blood. 2013;122:726–733. doi: 10.1182/blood-2013-04-495804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galiegue-Zouitina S, et al. Fusion of the LAZ3/BCL6 and BOB1/OBF1 genes by t(3; 11) (q27; q23) chromosomal translocation. C R Acad Sci III. 1995;318:1125–1131. [PubMed] [Google Scholar]
- 35.Chapuy B, et al. Discovery and Characterization of Super-Enhancer-Associated Dependencies in Diffuse Large B Cell Lymphoma. Cancer Cell. 2013;24:777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.