

Abstract
Aberrant activation of the three-layered protein kinase cascade, Raf/MEK/ERK, is often detected in human cancer, which is mainly attributed to the oncogenic alterations of RAF, or its upstream activators RAS or cell surface receptor tyrosine kinases. Deregulated activity of the Raf/MEK/ERK pathway drives uncontrolled tumor cell proliferation and survival, thus providing a rational therapeutic target for the treatment of many cancers. While Raf, MEK1/2, and ERK1/2 are equally important targets for the design of therapeutic small molecular weight inhibitors, the effort to develop MEK1/2-specific inhibitors has been greatly successful. Particularly, MEK1/2 have been relatively advantageous for the design of highly selective ATP-noncompetitive inhibitors. Indeed, a plethora of highly selective and potent MEK1/2 inhibitors are now available and many of those inhibitors have been evaluated for their therapeutic potential. Herein, we review different MEK1/2 inhibitors that have been studied for their inhibitory mechanisms and therapeutic potential in cancer. Some of the key structural features of MEK1/2 that are important for the efficacy of these inhibitors are also discussed. In addition, we discuss current challenges and future prospective in using these advanced MEK1/2 inhibitors for cancer therapy.
Introduction
Although the first mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) was discovered in mammalian cells only a few decades ago 1–3, the significance of MAPK/ERK-mediated signal transduction has been rapidly established in a number of biological contexts spanning from early development to various diseases with tremendous implication in cancer. MAPK/ERK serves as the key effector of a three-layered kinase cascade called the Raf/MEK/ERK pathway, which relays various signals transmitted from cell surface receptors to cytosolic and nuclear targets. The ubiquitously expressed Ser/Thr kinases ERK1 and ERK2 (collectively referred to as ERK1/2) are specific effectors of the Raf/MEK/ERK pathway that also consists of the Ser/Thr kinase Raf (i.e., A-Raf, B-Raf, or C-Raf/Raf-1) and the dual-specificity kinases MEK1 and its homologue MEK2 4. Upon activation, Raf phosphorylates MEK1/2, which in turn sequentially phosphorylate Tyr and Thr on the activation loop of their only substrates, ERK1/2. ERK1/2 then activate/inactivate many proteins to mediate diverse cellular processes 5, 6 (Fig. 1A). The Raf/MEK/ERK pathway is controlled by a complex network of regulators, including the small GTPase Ras and Rap, phosphatases, scaffolds, and other kinases, which affect the magnitude, duration, and compartmentalization of the pathway activity 4, 7–9. The Raf/MEK/ERK pathway plays pivotal roles in regulating cell survival, cell cycle progression and differentiation, and its deregulated activity is a central signature of many epithelial cancers [reviewed in 10–13].
Figure 1.

The Raf/MEK/ERK pathway and MEK1/2 inhibition. (A) Extracellular stimuli such as growth factors regulate diverse physiological processes by activating the cell surface receptors, e.g., receptor tyrosine kinases (RTK), which relay the signals to the three-layered kinase cascade, Raf/MEK/ERK, typically via the adapter protein, Growth factor receptor-bound protein 2 (Grb2), the guanine nucleotide exchange factor, Son of sevenless (Sos), and the small GTPase, Ras. Upon activation, ERK1/2 not only activate/inactivate various cytosolic and nuclear substrates but also feedback-inhibit Raf activity to modulate the pathway activity in cells. (B) MEK1/2 inhibition relieves ERK1/2-mediated feedback inhibition of C-Raf by inactivating ERK1/2. Certain MEK1/2 inhibitors (I) increase the interaction between MEK1/2 and C-Raf, and, this can promote MEK1/2 phosphorylation by C-Raf, resulting in the rebound of MEK/ERK activity in RAS mutant tumors. However, newer MEK1/2 inhibitors (II) that avoid this feedback rebound of MEK/ERK activity in RAS mutant tumors are becoming available (see text for details).
Aberrant activation of the Raf/MEK/ERK pathway is mainly driven by mutations in BRAF or its upstream activator, RAS (i.e., KRAS, HRAS, or NRAS), although other genetic and epigenetic alterations also drive the pathway. For example, mutations in BRAF, mainly affecting the Val600 codon (i.e., Val600 to Glu, Lys, or Arg), are detected at high frequencies in different cancers, including melanoma (50–70%), papillary thyroid cancers (40%), hairy cell leukemia (100%), Langerhans cell histiocytosis (57%), low-grade ovarian carcinomas (>30%), colorectal cancers (8%), non-small cell lung cancers (2–3%), and multiple myeloma (4%) 14–21. RAS mutations, mainly affecting Gly12 or Glu61, are among the most often detected genetic alterations in human cancers, including the malignancies of pancreas (63%), colon (36%), biliary tract (33%), skin (27%), small intestine (20%), lung (19%), ovary (18%), salivary gland (18%), urinary tract (18%), cervix (17%), endometrium (16%), upper aero-digestive tract (16%), prostate (15%), hematopoietic cells/lymphoid (15%), and thyroid (14%) 22. Mutations in BRAF and RAS are mutually exclusive in cancer, which suggests that activation of the MEK/ERK cascade is a critical process in mediating Ras- or Raf-driven carcinogenesis 12, 21, 23–25.
MEK1/2 is a key therapeutic target in cancer
Although MEK1 and MEK2 are rarely mutated in cancer, expression of constitutively active forms of their mutants (i.e., MEK1-ΔN3/S218E/S222D and MEK2-ΔN4/S222D/S226D) was sufficient to induce oncogenic transformation of normal cells 26, 27. This demonstrates the pivotal roles of MEK1/2 in malignant transformation, rationalizing therapeutic targeting of upregulated MEK1/2 activity in cancer. In addition, there are unique characteristics of MEK1 and MEK2 that might support the advantage of therapeutic development of MEK1/2 inhibition. First, MEK1/2 have very narrow substrate specificity, thus MEK1/2 inhibition specifically shuts off ERK1/2 signaling without directly affecting other signaling pathways. Second, MEK1/2 have a unique structural advantage for the design of highly selective ATP-noncompetitive inhibitors, which induce conformational changes that lock MEK1/2 into a catalytically inactive state 28, 29. ATP-noncompetitive inhibitors are advantageous over ATP-competitive inhibitors in that they have relatively low chances to induce the undesired adverse effects associated with inadvertent inhibition of the highly conserved ATP-binding pockets in protein kinases and they avoid the challenge of competing with ATP that are abundantly present (usually at mM ranges) in cells 28. Development of the ATP-noncompetitive inhibitors has been particularly successful with MEK1/2, and some of the MEK1/2 inhibitors have shown therapeutic efficacy in cancers when tested in a clinical setting. Some of the key features of MEK1/2 structure that are important for their inhibition are highlighted below.
Structural basis for MEK1/2 activation and catalysis
MEK1 and MEK2 were first identified in 199130, 31. MEK1 (45 kDa) and MEK2 (46 kDa) are encoded by MAP2K1 and MAP2K2 respectively, among the seven MAP2K genes in humans, exhibiting 85% peptide sequence homology with 86% identity being detected in their catalytic domains. The typical MEK1/2 secondary structures feature the N-terminal ~70 amino acid residues, the protein kinase domain (~290 amino acids), and the C-terminal ~30 amino acid residues (Fig. 2). The N-terminal sequence consists of the “D-domain” that interacts with the “common docking site” of ERK1/2, the nuclear export sequence (NES), and the inhibitory segment, whereas the role of C-terminal residues are less known although it has been previously demonstrated to affect cytoplasmic localization of MEK1 27, 32–37 (Fig. 2). Tertiary structures of MEK1/2 revealed by X-ray crystallography are also highly homologous although the N- and C-termini of MEK2 are more disordered than those of MEK1 28. These tertiary structures consist of a small N-terminal lobe and a large C-terminal lobe, which contain several α-helices and β-strands that are conserved in most protein kinases 38 (Fig. 3A).
Figure 2.
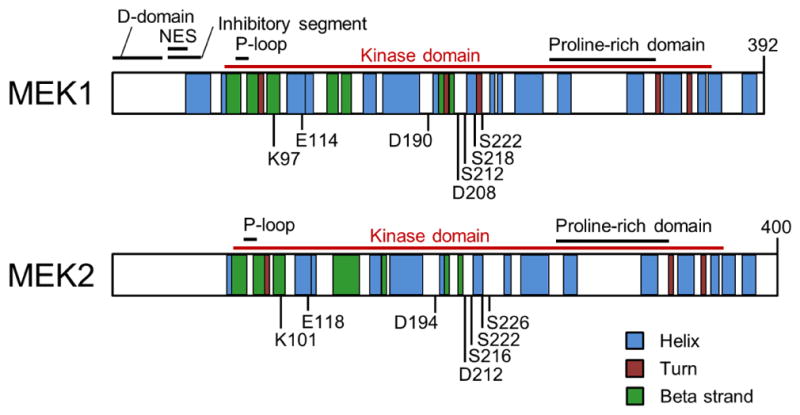
Secondary structures of MEK1/2. Illustrated are key domains and motifs, including D-domain, the nuclear export sequence (NES), the glycine-rich loop (P-loop), kinase domains, the proline–rich domains, and several key residues for MEK1/2 activation and catalysis (see text for details). Structural information obtained from the UniProtKB database (accession number: MEK1:Q02750; MEK2:P36507).
Figure 3.

Tertiary structure of MEK1. (A) Crystal structure of ATP-bound unphosphorylated MEK1 (PDB:3EQD). (B) Unphosphorylated MEK1 bound by PD318088 (PDB:1S9J). αC-helix, red; hinge region, green; P-loop, yellow; activation loop, orange; catalytic loop, purple; ATP and PD318088, blue. Images produced by the UCSF Chimera software.
The key structural features in the small lobe include a conserved five-stranded antiparallel β-sheet (β1-β5), the αC-helix, and the glycine-rich (Gly-Xaa-Gly-Xaa-Xaa-Gly) loop (also referred to as “P-loop”), which are important for binding and positioning ATP for catalysis (Fig. 3A). The key structural features in the large lobe include six conserved α-helixes (αD-αI) and four short conserved β-strands (β6-β9) that contain most of the catalytic residues required for catalysis and for interaction with the substrate, ERK1/2 39. MEK1/2 also have a unique prolin-rich insert (Fig. 2), which is not present in other MEK family members. This structurally flexible region facilitates efficient activation of ERK1/2, although it does not promote Raf-MEK interaction 40.
The catalytic site of MEK1/2 is formed in the deep cleft located between the small and large lobes (Fig. 3A). The β1 and β2 strands in the small lobe harbor the adenine ring of ATP and the proximal glycine-rich loop positions γ-phosphate of ATP for kinase reaction. Three conserved amino acids, known as the K/D/D (Lys/Asp/Asp) motif, are the key to subsequent phosphoryl transfer reaction 41. Briefly, a lysine (Lys97/101 of MEK1/2) in the β3 strand couples α- and β-phosphates of ATP, which facilitates the formation of a salt-bridge between the lysine and a conserved glutamate (Glu114/118 in MEK1/2) near the center of αC-helix in the small lobe. The presence of this bond indicates a kinase in a catalytically active state, and vice versa 41. A base in the catalytic loop (Asp190/194 of MEK1/2), which is present between the β6- and β7-strands, abstracts the proton from the hydroxyl group of tyrosine or threonine in the activation loop of EKR1/2, facilitating the nucleophilic attack of oxygen on the γ-phosphate of ATP. For coordination of these α-, β- and γ-phosphates of ATP, two Mg2+ ions are bound by an aspartate (Asp208/212 of MEK1/2) in the activation loop, which begins and ends with the DFG (Asp-Phe-Gly) and APE (Ala-Pro-Glu, Ser-Pro-Glu in MEK1/2) sequences that are conserved in kinases 41.
The active site in the cleft opens and closes during the catalytic cycle. Whereas the open form allows the access of ATP and the release of ADP, the closed form aligns the key residues in catalytically active positions. These dynamic movements are driven by a few conserved segments that undergo significant conformational changes. Whereas the αC-helix is the key regulatory segment in the small lobe, the activation loop in the large lobe dramatically changes its inactive versus active orientations in response to Raf-mediated phosphorylation of the two serine residues (i.e., Ser218/222 for MEK1 and Ser222/226 for MEK2) in this segment 42. In the inactive conformation, the aspartate side chain (Asp208/212 of MEK1/2) of the conserved DFG sequence in the activation segment faces away from the active site, which is called the “DFG-out” conformation 43, 44. In contrast, this aspartate side chain faces into the active site in the active conformation, which is called the “DFG-in” conformation. In the inactive (DFG-out) conformation, the activation loop blocks the ERK1/2 binding sites on MEK1/2 28. Noteworthy is that movement of the activation loop to the “DFG-out” conformation exposes an additional hydrophobic site (also referred to as the allosteric site) directly adjacent to the ATP binding pocket, which is separated by the side chains of the conserved Lys97 on the β3-strand and Met143 in the hinge region 28. Because the amino acids surrounding this region are less conserved than those in the ATP binding pocket, this region provides an advantage for the design of selective inhibitors that can lock kinases in an inactive state 28.
Types of kinase inhibitors
Protein kinases share conserved subdomains that fold into a typical bi-lobed structure that contains the catalytic site in the deep cleft created between these two lobes 45–47. Many kinase inhibitors take effect by targeting the catalytic site or a surrounding region that is important for enzyme activation. Most kinase inhibitors that form non-covalent reversible bonds are mainly classified as follows.
Type 1 inhibitors typically consist of a heterocyclic ring system that mimic the adenine ring structure of ATP, constituting the majority of ATP-competitive inhibitors. Type 1 inhibitors function by interrupting the molecular interactions in the specific compartments of the catalytic site, i.e., the adenine region, the ribose region and the phosphate-binding region, and the adjacent hydrophobic regions I and II 46. In these regions, type 1 inhibitors act as a scaffold for the side chains of amino acids that occupy the hydrophobic regions I and II. The resulting hydrogen bonds mimic those normally formed by the exocyclic amino group of adenine. Therefore, type 1 kinase inhibitors compete with ATP for the catalytic site of kinase and are most potent on active kinase conformation 43, 46. While all type 1 inhibitors invariably occupy the adenine region, these inhibitors can specifically affect those other compartments of the catalytic site, which provides the basis for their selectivity among different kinases 48. Type 1 inhibitors are generally used to inhibit many kinases, whereas ATP-noncompetitive inhibitors such as type 2 or allosteric inhibitors have been successful for MEK1/2 inhibition.
Type 2 inhibitors occupy the hydrophobic site created directly adjacent to the ATP binding pocket when the activation loop of a kinase undergoes “DFG-out” conformational changes. This is contrasted with the binding mechanism of type 1 inhibitors, which does not require the “DFG-out” conformation of the activation loop. Type 2 kinase inhibitors typically form hydrogen bonds with the amino acids in the region, i.e., one with the conserved glutamate in the αC-helix (Glu114/118 of MEK1/2) and another with the aspartate in the DFG motif 43. In addition to these hydrogen bonds, type 2 inhibitors also form van der Waals interactions with other residues in this region. This results in locking the kinase in an inactive state. Although occupancy of this site characterizes all the type 2 inhibitors, many of these inhibitors also extend into the adenine region in the ATP binding pocket and exert similar effects as type 1 inhibitors 43. Because type 2 inhibitors recognize and bind to inactive enzymes, they usually exhibit more potent cellular activity although certain type 1 kinase inhibitors are among the most selective inhibitors 49, 50.
Allosteric inhibitors bind at an allosteric site outside the ATP-binding pocket and modulate kinase activity in an allosteric manner. Allosteric inhibitors exhibit the highest degree of kinase selectivity because they inhibit the regulatory mechanism specific to a kinase 43. Allosteric MEK1/2 inhibitors usually constrain the movement of the activation loop and thus decrease the rate of Raf-mediated MEK phosphorylation, and/or lock the kinase in a catalytically inactive state.
MEK1/2 inhibitors developed to date
A number of highly selective MEK1/2 inhibitors have been developed, and many of them have been tested in a clinical setting. Notable examples of these inhibitors (Fig. 4, Table 1) are described below for known inhibitory mechanisms. Clinical efficacy of these inhibitors has been extensively reviewed elsewhere 51–53. Table 1 lists only recent and ongoing clinical trials.
Figure 4.

Chemical structures of MEK1/2 inhibitors. WX-554 not shown.
Table 1.
Small molecule inhibitors of MEK1/2
| Name | Other name | Company a | Year b | Mechanism | Binding site | IC50 c | Clinical trials d |
|---|---|---|---|---|---|---|---|
| PD98059 | Warner-Lambert | 1995 | ATP-noncompetitive | Not overlap with ATP or ERK binding sites | 2 μM for MEK1 (Ref. 56) | None | |
| U0126 | DuPont | 1998 | ATP-noncompetitive and allosteric | An allosteric site that overlaps with PD98059 binding site | 72 nM for MEK1 58 nM for MEK2 (Ref. 55) |
None | |
| Ro 09-2210 | Roche | 1998 | N.D. e | N.D. | 59 nM for MEK1 (Ref. 61) | None | |
| CI-1040 | PD184352 | Warner-Lambert/Pfizer | 1999 | ATP-noncompetitive and allosteric | An allosteric site near the kinase subdomains III and IV, and adjacent to the ATP binding site. Interacts with Val127 in the hinge region and Ser212 in the activation loop. (PDB:1S9J and 1S9I) | 17 nM for MEK1 (Ref. 62) |
NCT00033384 (Phase II, completed) NCT00034827 (Phase II, completed) |
| PD0325901 | Pfizer | 2004 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with Ser212 in the activation loop. | 1 nM for MEK1/2 (Ref. 65) |
NCT00174369 (Phase II, terminated) NCT00147550 (Phase I/II, terminated) |
|
| Selumetinib | AZD6244 ARRY-142886 |
Array Biopharma/AstraZeneca | 2005 | ATP-noncompetitive and allosteric | An allosteric site | 14 nM for MEK1 (Ref. 70) |
NCT01843062 (Phase III, recruiting) NCT01933932 (Phase III, recruiting) NCT01974752 (Phase III, active) |
| Cobimetinib | GDC-0973 XL518 |
Exelixis/Genentech | 2007 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with γ-phosphate of ATP, Ser212 in the activation loop and Asp190 in the catalytic loop. (PDB:4LMN) | 1 nM for MEK1 (Ref. 78) | NCT01689519 (Phase III, active) |
| AZD8330 | ARRY-424704 | Array BioPharma/AstraZeneca | 2009 | ATP-noncompetitive | N.D. | 7 nM for MEK1/2 (Ref. 103) | NCT00454090 (Phase I, completed) |
| E6201 | Eisai | 2009 | ATP-competitive | ATP binding site | 5.2 nM for MEK1 (Ref. 100) | NCT00794781 (Phase I, active) | |
| Pimasertib | AS703026 MSC1936369B |
EMD Serono | 2009 | ATP-noncompetitive and allosteric | An allosteric site | N.D. |
NCT00957580 (Phase II, terminated) NCT01085331 (Phase II, completed) NCT01693068 (Phase II, active) NCT01936363 (Phase II, active) |
| Refametinib | RDEA119 BAY 869766 |
Ardea Biosciences/Bayer | 2009 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with α-and γ-phosphates of ATP, Lys97 in the β3-strand, Val127 in the hinge region, DFG motif, Ser212 and Met219 in the activation loop. (PDB:3E8N) | 19 nM for MEK1 47 nM for MEK2 (Ref. 80) |
NCT01915589 (Phase II, not yet recruiting) NCT01915602 (Phase II, recruiting) |
| Trametinib | JTP-74057 GSK1120212 |
GlaxoSmithKline | 2009 | ATP-noncompetitive and allosteric | An allosteric site | 14.9 nM for MEK1 (Ref. 81) | FDA approved in 2013 |
| RO4987655 | CH4987655 | Hoffmann La Roche |
2009 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with ATP, Lys97 in the β3-strand, Val127 in the hinge region, Val211, Ser212 and Asn221 in the activation loop. (PDB:3ORN) | 5.2 nM for MEK1 (Ref. 89) | NCT00817518 (Phase I, completed) |
| WX-554 | Wilex | 2009 | ATP-noncompetitive | N.D. | N.D. |
NCT01581060 (Phase I/II, terminated) NCT01859351 (Phase I, terminated) |
|
| G-573 | Genentech | 2010 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with Ser212 in the activation loop. | N.D. | None | |
| Binimetinib | ARRY-438162 MEK162 |
Array BioPharma/Novartis | 2011 | ATP-noncompetitive | N.D. | N.D. |
NCT01763164 (Phase III, recruiting) NCT01849874 (Phase III, recruiting) NCT01909453 (Phase III, recruiting) |
| TAK733 | Millenium Pharmaceuticals | 2011 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with Lys97 in the β3-strand and Ser212 in the activation loop. (PDB:3PP1) | 3.2 nM for MEK1 (Ref. 96) |
NCT00948467 (Phase I, completed) NCT01613261 (Phase Ib, withdrawn) |
|
| GDC-0623 | Genentech | 2013 | ATP-noncompetitive and allosteric | An allosteric site. Interacts with Ser212 in the activation loop. | N.D. | NCT01106599 (Phase I, completed) | |
| RO5126766 | CH5126766 | Hoffmann La Roche |
2013 | Allosteric | An allosteric site. Interacts with Ser212, Ser222, and Asn221 in the activation loop. (PDB:3WIG) | 160 nM for MEK1 (Ref. 90) | NCT00773526 (Phase I, completed) |
Invented or licensed by the company listed.
Year in which MEK1/2 inhibitory activity of the listed compound was discovered.
IC50 obtained from in vitro kinase assays.
Information obtained from ClinicalTrials.gov (as of March 11, 2015)
Not determined.
PD98059 and U0126 are the first generation small molecule inhibitors of MEK1/2. These inhibitors were discovered from the small molecule compound library screening to identify selective inhibitors of the constitutively active MEK1 mutant or of PMA (phorbol 12-myristate 13-acetate)-induced AP-1 transcription 54, 55. PD98059 and U0126 are potent ATP- and ERK1/2-noncompetitive inhibitors 54–56, and occupy similar regions in MEK1/2 while U0126 exhibited ~100-fold higher affinity to the constitutively active MEK1 mutant than PD98059 55. Although potently inhibiting ERK1/2 phosphorylation, both compounds did not affect serum-induced MEK1/2 phosphorylation in NIH3T3 and WM35 cells, which suggests that theses inhibitors do not interfere with Raf access to MEK1/2 57. PD98059 and U0126 effectively suppressed in vitro Ras transformation 54, 55. When examined against a panel of protein kinases, U0126 and PD98059 showed very high selectivity to MEK1/2 with very low off-target effects 58. Given their high specificity, these two inhibitors have been invaluable reagents for academic research. However, both PD98059 and U0126 can also inhibit MEK5, albeit at a higher IC50 59. A recent study identified additional off-target effects of these inhibitors in cells 60. Therefore, data obtained using these inhibitors should be carefully interpreted.
Ro 09-2210 is another earlier MEK1/2 inhibitor, which was identified from the screening of microbial extracts for the activity to inhibit anti-CD3-induced T cell activation 61. Ro 09-2210 is a highly selective MEK1 inhibitor, exhibiting an IC50 below 60 nM in vitro. The mode of its inhibitory mechanism is not clear.
CI-1040 (PD184352) was identified in 1999 as a highly selective, potent allosteric inhibitor that is ATP- and ERK1/2-noncompetitive 62. A genetic screen identified five key amino acid residues that bind to CI-1040. These residues were mapped to the Hank’s conserved kinase subdomains III and IV, which form a potential hydrophobic binding pocket adjacent to the ATP binding site 63. The remarkable selectivity of CI-1040 and its analogs is attributed to the low sequence homology of their binding sites in MEK1/2 relative to other kinases, excluding MEK5 28. The occupancy of this unique pocket by an inhibitor locks MEK1/2 in an inactive conformation and disrupts the molecular interactions required for catalysis and proper access of the active site of MEK1/2 to the activation loop of ERK1/2, although binding of ATP and ERK1/2 still occurs. Crystal structures of human MEK1 and MEK2 bound to the CI-1040 analogs, PD318088 and PD334581, revealed that concurrent binding of these inhibitors and ATP to MEK1/2 causes several conformational changes, which account for their unique non-competitive mechanisms 28 (Fig. 3B). CI-1040 is the first MEK1/2 inhibitor that was tested in a clinical trial 64.
PD0325901 is a structural analog of CI-1040 with an optimized diphenylamine core and a modified hydroxamate side chain (Fig. 4). PD0325901 is a highly selective allosteric inhibitor that does not compete with ATP or ERK1/2, and its interaction with MEK1/2 does not perturb the ATP binding site 65. PD325901 presents a synergistic stabilizing effect in the formation of the ternary complex of the inhibitor-MEK1-nucleotide 66. PD0325901 exhibits significantly increased potency, i.e., IC50 of 1 nM for activated MEK1/2 65, and considerably improved oral bioavailability and metabolic stability compared with CI-1040 67. Consistent with this, PD0325901 inhibited proliferation of tumor cells at ~100 fold increased efficacy compared with CI-1040 65, 68. PD0325901 potently suppressed B-RafV600E tumor xenografts in mice, but it showed only partial effects on RAS-mutant tumor xenografts 68. Indeed, PD0325901 increased the interaction between MEK1/2 and C-Raf in KRAS mutant tumor cells, although not in BRAF mutant tumor cells 69. Because MEK1/2 inhibition relieved the ERK1/2-mediated feedback-inhibition of C-Raf, which resulted in increased C-Raf activity in RAS mutant tumor cells, the increased Raf-MEK interaction led to rebound of MEK/ERK activity in those tumor cells 69 (Fig. 1B). These unexpected effects partly account for the relatively low efficacy of PD0325901 in RAS mutant tumors.
Selumetinib (AZD6244/ARRY-142886) is an ATP-noncompetitive allosteric inhibitor. Selumetinib inhibits enzymatic activity of MEK1/2 but not MEK1/2 phosphorylation by Raf 70. Molecular modeling suggests that selumetinib binds to a unique allosteric site in MEK1/2, which confers a high selectivity. Indeed, selumetinib, even used at 10 μM, did not show any significant off-target effects in a test using more than 40 protein kinases including MEK5. Selumetinib robustly inhibited ERK1/2 phosphorylation (IC50 < 40 nM) in RAS or BRAF mutant tumor cells, and its dose-dependent antitumor activity was observed in a panel of colorectal, pancreatic, liver, skin, and lung cancer xenografts 70–73. In a single-arm phase II study, selumetinib has demonstrated significant efficacy for recurrent low-grade serous carcinoma of the ovary or peritoneum and biliary cancers 74, 75, which are the tumor types that present a high basal MEK/ERK activity. Similarly as PD0325901, selumetinib also increased MEK1/2 interaction with C-Raf and subsequently MEK1/2 activation in KRAS mutant tumor cells, although not in BRAF mutant tumor cells 69. This partly accounts for its lower potency in KRAS mutant tumor cells than in B-RafV600E tumor cells 68, 76.
Cobimetinib (GDC-0973/XL518/RG7421) is an ATP-noncompetitive allosteric inhibitor. It is a structural analog of CI-1040 optimized for metabolic stability by replacing the hydroxamate ester in CI-1040 with a carboxamide 77 (Fig. 4). The cyclic aminoethanol terminus of cobimetinib is predicted to form a highly complex network of interactions with both the catalytic loop and the γ-phosphate of ATP 78. Although similarly effective in BRAF mutant tumor cells, cobimetinib exhibited much less potency in a panel of KRAS mutant tumor cells than two newer allosteric inhibitors, GDC-0623 and G-57379. Comparison of their binding modes by structural analysis led to the prediction that the affinity of the inhibitor to Ser212 (MEK1) in the activation loop determines the degree of inhibition of MEK1/2 activation by Raf 79. In addition to its effect on MEK1/2, cobimetinib induced translocation of C-Raf to plasma membrane in KRAS mutant tumor cells 79. This partly accounts for a mechanism by which C-Raf is activated and subsequently MEK/ERK activity is rebound in cobimetinib-treated KRAS mutant tumor cells.
Refametinib (RDEA119/BAY869766) is an ATP-noncompetitive allosteric inhibitor 80. Structural analysis has revealed that refametinib binds to an allosteric site adjacent to the ATP binding pocket and interacts with ATP, the activation loop, and surrounding amino acids in the region through hydrogen bonding and hydrophobic interactions in a similar manner to PD318088 28, 80 (Fig. 3B). In addition, the unique cyclopropyl group of refametinib makes direct hydrophobic contacts with the side chain of Met219 in the activation loop, further stabilizing the inhibitor in the closed, DFG-in “active” conformation of MEK1/2 80. This imparts strong potency in inhibiting ERK1/2 phosphorylation and suppressing cell proliferation in a panel of human cancer cell lines in vitro and in mouse xenografts 80.
Trametinib (GSK1120212/JTP-74057) is an ATP-noncompetitive inhibitor. Trametinib binds to MEK1/2 in a manner similar to PD0325901, inducing allosteric inhibition of MEK1/2 catalysis 81. Importantly, unlike PD0325901, trametinib does not increase the interaction between MEK1/2 and C-Raf in KRAS mutant tumor cells 69. On the contrary, trametinib promotes dissociation of MEK1/2 from C-Raf, which reduces the rebound of ERK1/2 phosphorylation observed in KRAS mutant tumor cells 69. Therefore, trametinib inhibits not only MEK1/2 activity but also MEK1/2 activation by Raf. Indeed, trametinib demonstrated substantial antitumor effects against not only BRAF but also KRAS mutant tumor cells in mouse xenografts 81.
Trametinib is the only MEK1/2 inhibitor currently approved by the US Food and Drug Administration for clinical applications as a monotherapy and as a combination therapy with the B-Raf inhibitor dabrafenib. Trametinib exhibited the highest efficacy in patients with BRAF mutant melanoma 82, although it also showed efficacy in patients with NRAS mutant melanoma, uveal melanoma, pancreatic cancer, and non-small cell lung carcinoma 82, 83. Of note, when used in combination with dabrafenib, trametinib significantly improved survival of the patients with B-RafV600E,K melanomas compared with the dabrafenib monotherapy, indicating that simultaneous inhibition of B-RafV600E,K and MEK1/2 has a therapeutic advantage 84. This improvement is partly attributed to the ability of trametinib to suppress the rebound of MEK/ERK activity in BRAF mutant tumors, which is a major mechanism of acquired resistance to B-Raf inhibitors triggered via alternate upstream activators such as N-Ras, C-Raf, and Cot/Tpl-2 85. Trametinib has also been evaluated in combination with other anticancer agents, including gemcitabine 86, everolimus 87, and the AKT inhibitor afuresertib 88. These combination therapies are reviewed elsewhere 53.
RO4987655 (CH4987655) is an ATP-noncompetitive inhibitor. This compound potently and selectively inhibits MEK1 at a very low IC50 (5 nM), while not inhibiting 400 other kinases even at 10 μM 89. Co-crystal structure revealed that RO4987655 binds to an allosteric inhibitor binding site in ATP analog-bound MEK1 via interactions with Lys97, Val127, Val211, Ser212, and the nucleotide 89. RO4987655 exhibited anti-proliferative activity in a panel of KRAS and BRAF mutant tumor cell lines in vitro and in vivo 89.
RO5126766 (CH5126766) is a novel allosteric inhibitor. Co-crystal structure revealed that RO512677 binds to an allosteric inhibitor binding site in ATP analog-bound MEK1 69, which overlaps with the PD0325901 binding site 90. Specifically, RO512677 interacts with Ser212, Ser222 and Asn221, and induces a conformational change in the activation loop, which prevents MEK1 phosphorylation by C-Raf 69. RO512677 increased the affinity between inactive MEK and Raf, and the resulting Raf-MEK complex was so stable that it became a dominant-negative inhibitor of Raf 69. Of note, RO512677 inhibited not only MEK1 but also C-Raf, B-Raf, and B-RafV600E in an in vitro kinase assay, although it did not inhibit 254 other kinases 90. Therefore, RO5126766 is a first-in-class dual Raf and MEK inhibitor. Because of these properties, RO5126766 prevented C-Raf-mediated MEK/ERK reactivation 69, and exhibited improved tumor suppressive effects in KRAS mutant tumor cells, including PD0325901-resistant KRAS mutant tumor cells 90.
Binimetinib (MEK162/ARRY-438162) is an ATP-noncompetitive inhibitor 91. Binimetinib suppressed NRAS mutant melanoma cells 92. Consistent with this, 10% of patients with NRAS mutant melanoma and 5% of patients with BRAF mutant melanoma responded to binimetinib 93. Binimetinib also suppressed pancreatic cancer cells expressing K-RasD12, R12, or C12, although it was less effective in pancreatic cancer cells expressing K-RasV12 or increased copy numbers of wild type KRAS 91.
Pimasertib (AS703026/MSC1936369B) is an ATP-noncompetitive allosteric inhibitor which did not inhibit 217 other kinases 94. Pimasertib significantly suppressed human multiple myeloma and other solid tumor xenografts in mice 94. Pimasertib also suppressed KRAS-mutated DLD-1 colorectal cancer cells and their cetuximab-resistant variants in mouse xenografts 95.
TAK733 is an ATP-noncompetitive allosteric inhibitor 96. Co-crystal structure revealed that TAK733 binds to an allosteric inhibitor binding site in ATP-bound MEK1, wherein it interacts with Lys97 and Ser212 96. TAK733 exhibited very low IC50 (3.2 nM to MEK1), while not inhibiting other kinases, receptors, or ion channels 96. In a preclinical study, TAK733 showed antitumor activity for human breast carcinoma, colorectal cancer, melanoma, non-small cell lung cancer, and pancreatic cancer 96. Of note, despite its efficacy in BRAF mutant melanoma cells, TAK733 upregulated MEK1/2 phosphorylation in melanoma cells harboring NRAS, GNAQ, or GNA11 mutations 97, indicating its ability to trigger the rebound of MEK/ERK activity.
GDC-0623 and G-573 are two recently developed ATP-noncompetitive allosteric inhibitors 79, 98. Molecular modeling suggests that GDC-0623 and G-573 bind to MEK1 in a similar manner as cobimetinib 79. Noteworthy is that GDC-0623 and G-573 form stronger interaction with Ser212 in the activation loop than cobimetinib, which account for their superiority in blocking MEK1/2 phosphorylation by Raf 79. Unlike cobimetinib, these inhibitors did not increase B-Raf and C-Raf hetero-dimerization 79. Moreover, G-573 blocked Raf translocation to the plasma membrane in KRAS mutant tumor cells, suggesting that these inhibitors have an additional role other than MEK inhibition 79. Consistent with this, GDC-0623 and G-573 did not induce the rebound of MEK/ERK activity in KRAS mutant tumor cells while exhibiting much higher potency than cobimetinib 79.
E6201 is unique in that it is an ATP-competitive inhibitor. Molecular docking indicates its positioning in the ATP-binding pocket 99. E6201 exhibited very low IC50 (5.2 nM) to MEK1 and effectively suppressed B-RafV600E melanoma cells 99–101. Of note, E6201 effectively blocked MEK/ERK activity and cell growth in melanoma cells expressing MEK1-C121S 99. Cys121 to Ser switch in MEK1 is a mechanism that imparts vemurafenib resistance to BRAF mutant melanomas 102. MEK1-C121S is also resistant to selumetinib, suggesting that the binding site for selumetinib is altered in this MEK1 mutant 102. This effect of E6201 indicates a potential benefit of targeting ATP-binding pocket using a type 1 inhibitor when a drug-resistant mutation occurs in the binding sites of ATP-noncompetitive inhibitors.
Other recently developed MEK inhibitors include AZD8330/ARRY-424704 103 and WX-554 104. These inhibitors are currently evaluated in phase I clinical trials (Table 1). The inhibitory mechanisms for these inhibitors are not yet clear.
Current challenges and future perspectives
A plethora of MEK1/2 inhibitors are now available and some of them are expected to improve the treatment of Raf/MEK/ERK-driven cancers. Nevertheless, there are obstacles that limit the use of these specific inhibitors. First, various genetic alterations have been detected in tumor cells that have acquired resistance to MEK1/2 inhibitors. Most of these alterations converge into reactivation of the Raf/MEK/ERK pathway, e.g., amplification of BRAF 105 and MEK1 mutations 106, 107, although mutations in other pathways are also detected, e.g., STAT3 upregulation 108. Different MEK1 mutations detected in the tumor specimen from selumetinib-treated patients 107 and in the tumor cells resistant to PD0325901 and G-573 109 indicate the ability of tumor cells to bypass the effect of advanced MEK1/2 inhibitors by genetically altering the allosteric inhibitor binding sites in MEK1/2 or enhancing intrinsic kinase activity of MEK1/2. Noteworthy is that most of these tumor cells remain addicted to MEK/ERK activity. Therefore, the MEK/ERK pathway continuously provides the target to treat these tumors. Indeed, it has been demonstrated that use of ERK1/2 inhibitors overcomes acquired resistance to MEK1/2 inhibitors 109, although it is predicted that different ERK1/2 mutations that confer drug resistance would eventually arise in response to ERK1/2 inhibitors. In support of this notion, it has been demonstrated that different ERK2 mutations bypass trametinib effects in BRAF mutant melanoma cells 110. Alternatively, type 1 inhibitors may be used to treat the tumors that developed resistance to the ATP-noncompetitive inhibitors, as exemplified by E6201 in melanoma cells expressing MEK1-C121S 99. ATP-competitive inhibitors have lower chances to develop resistance mutations because mutations in the ATP-binding pocket are likely to disable the catalytic activity of a kinase, thereby not providing selection advantage.
Second, intrinsic resistance of tumor cells limits the therapeutic window of advanced MEK1/2 inhibitors. Similarly to the cases of B-Raf inhibitors, the intrinsic resistance to MEK1/2 inhibitors is mainly attributed to rapid rebound of MEK/ERK activity after inhibitor treatment, for which an ERK1/2-mediated feedback loop is critical. ERK1/2 feedback inhibits C-Raf via phosphorylation in RAS mutant tumor cells 111 (Fig. 1B). Inhibition of MEK1/2 relieves this feedback loop by inactivating ERK1/2, subsequently leading to C-Raf activation when Ras activity is available 111. Certain ME1/2 inhibitors promote this phenomenon by facilitating C-Raf-MEK interactions. Development of newer inhibitors such as trametinib and RO5126766 has shown promise to prevent this reactivation mechanism 69, 112.
MEK1/2 inhibitors have potential to confer significant therapeutic benefits when combined with other treatments, e.g., combination with dabrafenib 84. Similarly a number of combination therapies using MEK1/2 inhibitors are currently evaluated and are likely to expand their clinical applications. Of note, combination of MEK1/2 inhibitors with certain drugs induces synthetic lethality in cancer, e.g., combination with Bcl-XL inhibitors in KRAS mutant tumor cells 113. Further identification of such strategies will require better understanding of the functional relationships between the Raf/MEK/ERK pathway and other key pathways for signal transduction and metabolic regulation in cancer.
Acknowledgments
This work was supported in part by the National Cancer Institute (R01CA138441) and American Cancer Society (RSGM-10-189-01-TBE) to J.I.P. The authors wish to apologize to those whose work is not cited owing to space limitations.
Footnotes
Conflict of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Pui-Kei Wu, Postdoctoral Research Fellow, Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin, 53226, USA
Jong-In Park, Associate Professor, Department of Biochemistry, Medical College of Wisconsin, Milwaukee, Wisconsin, 53226, USA
References
- 1.Ray LB, Sturgill TW. Rapid stimulation by insulin of a serine/threonine kinase in 3T3-L1 adipocytes that phosphorylates microtubule-associated protein 2 in vitro. Proc Natl Acad Sci U S A. 1987;84:1502–6. doi: 10.1073/pnas.84.6.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ray LB, Sturgill TW. Characterization of insulin-stimulated microtubule-associated protein kinase. Rapid isolation and stabilization of a novel serine/threonine kinase from 3T3-L1 cells. J Biol Chem. 1988;263:12721–7. [PubMed] [Google Scholar]
- 3.Boulton TG, Yancopoulos GD, Gregory JS, Slaughter C, Moomaw C, Hsu J, et al. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science. 1990;249:64–7. doi: 10.1126/science.2164259. [DOI] [PubMed] [Google Scholar]
- 4.Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773:1213–26. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 6.von Kriegsheim A, Baiocchi D, Birtwistle M, Sumpton D, Bienvenut W, Morrice N, et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol. 2009;11:1458–64. doi: 10.1038/ncb1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 8.Wortzel I, Seger R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–43. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 10.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3:954–87. doi: 10.18632/oncotarget.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 12.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence MC, Jivan A, Shao C, Duan L, Goad D, Zaganjor E, et al. The roles of MAPKs in disease. Cell Res. 2008;18:436–42. doi: 10.1038/cr.2008.37. [DOI] [PubMed] [Google Scholar]
- 14.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 15.Puxeddu E, Moretti S, Elisei R, Romei C, Pascucci R, Martinelli M, et al. BRAF(V599E) mutation is the leading genetic event in adult sporadic papillary thyroid carcinomas. J Clin Endocrinol Metab. 2004;89:2414–20. doi: 10.1210/jc.2003-031425. [DOI] [PubMed] [Google Scholar]
- 16.Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–15. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 18.Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–23. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. N Engl J Med. 2009;361:98–9. doi: 10.1056/NEJMc0904160. [DOI] [PubMed] [Google Scholar]
- 20.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singer G, Oldt R, 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–6. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 22.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 24.Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 25.Sieben NL, Macropoulos P, Roemen GM, Kolkman-Uljee SM, Jan Fleuren G, Houmadi R, et al. In ovarian neoplasms, BRAF, but not KRAS, mutations are restricted to low-grade serous tumours. J Pathol. 2004;202:336–40. doi: 10.1002/path.1521. [DOI] [PubMed] [Google Scholar]
- 26.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–52. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 27.Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–70. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 28.Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–7. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 29.Fischmann TO, Smith CK, Mayhood TW, Myers JE, Reichert P, Mannarino A, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48:2661–74. doi: 10.1021/bi801898e. [DOI] [PubMed] [Google Scholar]
- 30.Ahn NG, Seger R, Bratlien RL, Diltz CD, Tonks NK, Krebs EG. Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J Biol Chem. 1991;266:4220–7. [PubMed] [Google Scholar]
- 31.Gomez N, Cohen P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature. 1991;353:170–3. doi: 10.1038/353170a0. [DOI] [PubMed] [Google Scholar]
- 32.Mansour SJ, Candia JM, Matsuura JE, Manning MC, Ahn NG. Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry. 1996;35:15529–36. doi: 10.1021/bi961854s. [DOI] [PubMed] [Google Scholar]
- 33.Xu B, Wilsbacher JL, Collisson T, Cobb MH. The N-terminal ERK-binding site of MEK1 is required for efficient feedback phosphorylation by ERK2 in vitro and ERK activation in vivo. J Biol Chem. 1999;274:34029–35. doi: 10.1074/jbc.274.48.34029. [DOI] [PubMed] [Google Scholar]
- 34.Fukuda M, Gotoh I, Gotoh Y, Nishida E. Cytoplasmic localization of mitogen-activated protein kinase kinase directed by its NH2-terminal, leucine-rich short amino acid sequence, which acts as a nuclear export signal. J Biol Chem. 1996;271:20024–8. doi: 10.1074/jbc.271.33.20024. [DOI] [PubMed] [Google Scholar]
- 35.Fukuda M, Gotoh I, Adachi M, Gotoh Y, Nishida E. A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade. Role of nuclear export signal of MAP kinase kinase. J Biol Chem. 1997;272:32642–8. doi: 10.1074/jbc.272.51.32642. [DOI] [PubMed] [Google Scholar]
- 36.Jaaro H, Rubinfeld H, Hanoch T, Seger R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc Natl Acad Sci U S A. 1997;94:3742–7. doi: 10.1073/pnas.94.8.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cha H, Lee EK, Shapiro P. Identification of a C-terminal region that regulates mitogen-activated protein kinase kinase-1 cytoplasmic localization and ERK activation. J Biol Chem. 2001;276:48494–501. doi: 10.1074/jbc.M107601200. [DOI] [PubMed] [Google Scholar]
- 38.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roskoski R., Jr MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem Biophys Res Commun. 2012;417:5–10. doi: 10.1016/j.bbrc.2011.11.145. [DOI] [PubMed] [Google Scholar]
- 40.Dang A, Frost JA, Cobb MH. The MEK1 proline-rich insert is required for efficient activation of the mitogen-activated protein kinases ERK1 and ERK2 in mammalian cells. J Biol Chem. 1998;273:19909–13. doi: 10.1074/jbc.273.31.19909. [DOI] [PubMed] [Google Scholar]
- 41.Kornev AP, Haste NM, Taylor SS, Eyck LF. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci U S A. 2006;103:17783–8. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13:1610–9. doi: 10.1002/j.1460-2075.1994.tb06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–64. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 44.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9:268–72. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 45.Johnson LN, Lowe ED, Noble ME, Owen DJ The Eleventh Datta Lecture. The structural basis for substrate recognition and control by protein kinases. FEBS Lett. 1998;430:1–11. doi: 10.1016/s0014-5793(98)00606-1. [DOI] [PubMed] [Google Scholar]
- 46.Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol Ther. 1999;82:195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- 47.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 48.Gray NS, Wodicka L, Thunnissen AM, Norman TC, Kwon S, Espinoza FH, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281:533–8. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]
- 49.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–45. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 51.Grimaldi AM, Simeone E, Ascierto PA. The role of MEK inhibitors in the treatment of metastatic melanoma. Curr Opin Oncol. 2014;26:196–203. doi: 10.1097/CCO.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 52.Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol. 2014;15:e371–81. doi: 10.1016/S1470-2045(14)70072-5. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 54.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–9. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 56.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 57.Ahn NG, Nahreini TS, Tolwinski NS, Resing KA. Pharmacologic inhibitors of MKK1 and MKK2. Methods Enzymol. 2001;332:417–31. doi: 10.1016/s0076-6879(01)32219-x. [DOI] [PubMed] [Google Scholar]
- 58.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem. 1999;274:26563–71. doi: 10.1074/jbc.274.37.26563. [DOI] [PubMed] [Google Scholar]
- 60.Wauson EM, Guerra ML, Barylko B, Albanesi JP, Cobb MH. Off-target effects of MEK inhibitors. Biochemistry. 2013;52:5164–6. doi: 10.1021/bi4007644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams DH, Wilkinson SE, Purton T, Lamont A, Flotow H, Murray EJ. Ro 09-2210 exhibits potent anti-proliferative effects on activated T cells by selectively blocking MKK activity. Biochemistry. 1998;37:9579–85. doi: 10.1021/bi972914c. [DOI] [PubMed] [Google Scholar]
- 62.Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–6. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 63.Delaney AM, Printen JA, Chen H, Fauman EB, Dudley DT. Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol Cell Biol. 2002;22:7593–602. doi: 10.1128/MCB.22.21.7593-7602.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allen LF, Sebolt-Leopold J, Meyer MB. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK) Semin Oncol. 2003;30:105–16. doi: 10.1053/j.seminoncol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 65.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–47. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 66.Smith CK, Windsor WT. Thermodynamics of nucleotide and non-ATP-competitive inhibitor binding to MEK1 by circular dichroism and isothermal titration calorimetry. Biochemistry. 2007;46:1358–67. doi: 10.1021/bi061893w. [DOI] [PubMed] [Google Scholar]
- 67.Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008;18:6501–4. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]
- 68.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 71.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6:2209–19. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 72.Huynh H, Soo KC, Chow PK, Tran E. Targeted inhibition of the extracellular signal-regulated kinase kinase pathway with AZD6244 (ARRY-142886) in the treatment of hepatocellular carcinoma. Mol Cancer Ther. 2007;6:138–46. doi: 10.1158/1535-7163.MCT-06-0436. [DOI] [PubMed] [Google Scholar]
- 73.Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 74.Farley J, Brady WE, Vathipadiekal V, Lankes HA, Coleman R, Morgan MA, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14:134–40. doi: 10.1016/S1470-2045(12)70572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011;29:2357–63. doi: 10.1200/JCO.2010.33.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Friday BB, Yu C, Dy GK, Smith PD, Wang L, Thibodeau SN, et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res. 2008;68:6145–53. doi: 10.1158/0008-5472.CAN-08-1430. [DOI] [PubMed] [Google Scholar]
- 77.Wabnitz PA, Mitchell D, Wabnitz DA. In vitro and in vivo metabolism of the anti-cancer agent CI-1040, a MEK inhibitor, in rat, monkey, and human. Pharm Res. 2004;21:1670–9. doi: 10.1023/b:pham.0000041464.27579.d0. [DOI] [PubMed] [Google Scholar]
- 78.Rice KD, Aay N, Anand NK, Blazey CM, Bowles OJ, Bussenius J, et al. Novel Carboxamide-Based Allosteric MEK Inhibitors: Discovery and Optimization Efforts toward XL518 (GDC-0973) ACS Med Chem Lett. 2012;3:416–21. doi: 10.1021/ml300049d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature. 2013;501:232–6. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- 80.Iverson C, Larson G, Lai C, Yeh LT, Dadson C, Weingarten P, et al. RDEA119/BAY 869766: a potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009;69:6839–47. doi: 10.1158/0008-5472.CAN-09-0679. [DOI] [PubMed] [Google Scholar]
- 81.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 82.Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–81. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- 83.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–9. doi: 10.1016/S1470-2045(12)70269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alcala AM, Flaherty KT. BRAF inhibitors for the treatment of metastatic melanoma: clinical trials and mechanisms of resistance. Clin Cancer Res. 2012;18:33–9. doi: 10.1158/1078-0432.CCR-11-0997. [DOI] [PubMed] [Google Scholar]
- 86.Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014;50:2072–81. doi: 10.1016/j.ejca.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 87.Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA, 3rd, Patnaik A, Jones SF, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol. 2015;26:58–64. doi: 10.1093/annonc/mdu482. [DOI] [PubMed] [Google Scholar]
- 88.Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol. 2015;75:183–9. doi: 10.1007/s00280-014-2615-5. [DOI] [PubMed] [Google Scholar]
- 89.Isshiki Y, Kohchi Y, Iikura H, Matsubara Y, Asoh K, Murata T, et al. Design and synthesis of novel allosteric MEK inhibitor CH4987655 as an orally available anticancer agent. Bioorg Med Chem Lett. 2011;21:1795–801. doi: 10.1016/j.bmcl.2011.01.062. [DOI] [PubMed] [Google Scholar]
- 90.Ishii N, Harada N, Joseph EW, Ohara K, Miura T, Sakamoto H, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 2013;73:4050–60. doi: 10.1158/0008-5472.CAN-12-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hamidi H, Lu M, Chau K, Anderson L, Fejzo M, Ginther C, et al. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br J Cancer. 2014;111:1788–801. doi: 10.1038/bjc.2014.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, Kluger HM. MEK targeting in N-RAS mutated metastatic melanoma. Mol Cancer. 2014;13:45. doi: 10.1186/1476-4598-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–56. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 94.Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S, et al. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. Br J Haematol. 2010;149:537–49. doi: 10.1111/j.1365-2141.2010.08127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoon J, Koo KH, Choi KY. MEK1/2 inhibitors AS703026 and AZD6244 may be potential therapies for KRAS mutated colorectal cancer that is resistant to EGFR monoclonal antibody therapy. Cancer Res. 2011;71:445–53. doi: 10.1158/0008-5472.CAN-10-3058. [DOI] [PubMed] [Google Scholar]
- 96.Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T, et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett. 2011;21:1315–9. doi: 10.1016/j.bmcl.2011.01.071. [DOI] [PubMed] [Google Scholar]
- 97.von Euw E, Atefi M, Attar N, Chu C, Zachariah S, Burgess BL, et al. Antitumor effects of the investigational selective MEK inhibitor TAK733 against cutaneous and uveal melanoma cell lines. Mol Cancer. 2012;11:22. doi: 10.1186/1476-4598-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choo EF, Belvin M, Chan J, Hoeflich K, Orr C, Robarge K, et al. Preclinical disposition and pharmacokinetics-pharmacodynamic modeling of biomarker response and tumour growth inhibition in xenograft mouse models of G-573, a MEK inhibitor. Xenobiotica. 2010;40:751–62. doi: 10.3109/00498254.2010.514365. [DOI] [PubMed] [Google Scholar]
- 99.Narita Y, Okamoto K, Kawada MI, Takase K, Minoshima Y, Kodama K, et al. Novel ATP-competitive MEK inhibitor E6201 is effective against vemurafenib-resistant melanoma harboring the MEK1-C121S mutation in a preclinical model. Mol Cancer Ther. 2014;13:823–32. doi: 10.1158/1535-7163.MCT-13-0667. [DOI] [PubMed] [Google Scholar]
- 100.Goto M, Chow J, Muramoto K, Chiba K, Yamamoto S, Fujita M, et al. E6201 [(3S,4R,5Z,8S,9S,11E)-14-(ethylamino)-8, 9,16-trihydroxy-3,4-dimethyl-3,4,9,19-tetrahydro-1H-2-benzoxacyclotetradecine-1,7 (8H)-dione], a novel kinase inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK)-1 and MEK kinase-1: in vitro characterization of its anti-inflammatory and antihyperproliferative activities. J Pharmacol Exp Ther. 2009;331:485–95. doi: 10.1124/jpet.109.156554. [DOI] [PubMed] [Google Scholar]
- 101.Byron SA, Loch DC, Wellens CL, Wortmann A, Wu J, Wang J, et al. Sensitivity to the MEK inhibitor E6201 in melanoma cells is associated with mutant BRAF and wildtype PTEN status. Mol Cancer. 2012;11:75. doi: 10.1186/1476-4598-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cohen RB, Aamdal S, Nyakas M, Cavallin M, Green D, Learoyd M, et al. A phase I dose-finding, safety and tolerability study of AZD8330 in patients with advanced malignancies. Eur J Cancer. 2013;49:1521–9. doi: 10.1016/j.ejca.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 104.Miller CR, Oliver KE, Farley JH. MEK1/2 inhibitors in the treatment of gynecologic malignancies. Gynecol Oncol. 2014;133:128–37. doi: 10.1016/j.ygyno.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 105.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Signal. 2010;3:ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B, et al. Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B-RafV600E mutation. Cancer Res. 2011;71:5535–45. doi: 10.1158/0008-5472.CAN-10-4351. [DOI] [PubMed] [Google Scholar]
- 107.Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–6. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dai B, Meng J, Peyton M, Girard L, Bornmann WG, Ji L, et al. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011;71:3658–68. doi: 10.1158/0008-5472.CAN-10-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11:1143–54. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- 110.Goetz EM, Ghandi M, Treacy DJ, Wagle N, Garraway LA. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014;74:7079–89. doi: 10.1158/0008-5472.CAN-14-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 112.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–82. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]