

Abstract
Recently we discovered that the central metabolite α-ketoglutarate (α-KG) extends lifespan in C. elegans through inhibition of ATP synthase and TOR signaling. Unexpectedly, here we find that (R)-2-hydroxyglutarate ((R)-2HG), an oncometabolite that interferes with various α-KG mediated processes, extends worm lifespan similarly. (R)-2HG accumulates in human cancers carrying neomorphic mutations in the isocitrate dehydrogenase (IDH) 1 and 2 genes. We show that, like α-KG, both (R)-2HG and (S)-2HG bind and inhibit ATP synthase, and inhibit mTOR signaling; these effects are mirrored in IDH1 mutant cells, suggesting a growth suppressive function of (R)-2HG. Consistently, inhibition of ATP synthase by 2-HG or α-KG in glioblastoma cells is sufficient for growth arrest and tumor cell killing under conditions of glucose limitation, such as when ketone bodies (instead of glucose) are supplied for energy. These findings inform therapeutic strategies and open avenues for investigating the roles of 2-HG and metabolites in biology and disease.
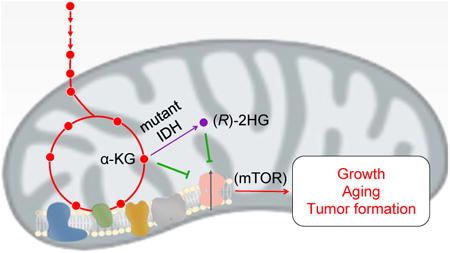
Graphical abstract
Introduction
Aberrant metabolism, long symbolic of inherited metabolic diseases, is now recognized as a hallmark of many other pathogenic conditions, including cancer (Warburg, 1956; Vander Heiden et al., 2009). Recently, we discovered that the common metabolite α-ketoglutarate (α-KG) increases the lifespan of adult C. elegans by inhibiting the highly conserved ATP synthase and the TOR pathway, mimicking dietary restriction in longevity (Chin et al., 2014). Furthermore, the observation that α-KG inhibits mTOR function in normal human cells implies a role for α-KG as an endogenous tumor suppressor metabolite (Chin et al., 2014). Known for its role in central carbon metabolism as a tricarboxylic acid (TCA) cycle intermediate, α-KG is universal to all cellular life. α-KG also serves as a co-substrate for a large family of dioxygenases with functions in cellular processes such as hypoxic response and epigenetic regulation. The identification of α-KG as a regulator of ATP synthase reveals a new mechanism for longevity regulation through metabolite signaling, and suggests that there likely exist other metabolites that play signaling roles in aging. Particularly, metabolites that are similar in structure to α-KG may also modify lifespan through interactions with ATP synthase, and the lifespan effects of metabolites may correlate with their involvement in human disease.
In the TCA cycle, α-KG is produced from isocitrate by isocitrate dehydrogenase (IDH). Catalytic arginine mutations in the IDH1 and IDH2 genes found in gliomas and acute myeloid leukemia (AML) result in neomorphic enzymes that instead convert α-KG to the structurally similar (R)-2HG, which accumulates to exceedingly high levels in these patients (Dang et al., 2009; Gross et al., 2010; Ward et al., 2010; Xu et al., 2011). (R)-2HG is now considered an oncometabolite, impairing epigenetic and hypoxic regulation through its binding to α-KG– dependent dioxygenases (Lu et al., 2012; Koivunen et al., 2012). The development of inhibitors of mutant IDH that normalize (R)-2HG levels is an attractive cancer therapeutic strategy (Wang et al., 2013; Rohle et al., 2013). Paradoxically however, brain cancer patients with the IDH mutations have longer median overall survival than patients without the mutations (Parsons et al., 2008; Yan et al., 2009; van den Bent et al., 2010), hinting at additional complexity in the biology of these cancers. (R)-2HG and (S)-2HG have also been found to accumulate in tissues of individuals with germline mutations in genes encoding the corresponding 2-HG dehydrogenases (Kranendijk et al., 2012; Steenweg et al., 2010). The resulting 2-HG aciduria is associated with neurological manifestations whose molecular mechanisms are unknown (Kranendijk et al., 2010). We set out to identify additional targets of 2-HG to elucidate the mechanisms underlying the seemingly disparate 2-HG–related phenotypes.
Results and Discussion
2-HG extends the lifespan of adult C. elegans
We demonstrated that α-KG promotes longevity through inhibition of ATP synthase (Chin et al., 2014). Given the structural similarity between α-KG and 2-HG (Figure 1A), and the association of 2-HG with cancer and neurological dysfunction, we asked whether 2-HG influences longevity. Surprisingly, both (R)-2HG and (S)-2HG increase the lifespan of C. elegans (Figure 1B-C). Notably, (R)-2HG, (S)-2HG, and α-KG interact distinctly with the α-KG dependent dioxygenases (Koivunen et al., 2012; Tarhonskaya et al., 2014). Thus, the similar effect of α-KG and (R)- and (S)-2-HG on lifespan points to a common mechanism that is independent of dioxygenases or any enantiomer-specific 2-HG effects (da Silva et al., 2002; Latini et al., 2005; Wajne et al., 2002; Chan et al., 2015). Since we identified the ATP synthase β subunit (ATP5B) as a target of α-KG (Chin et al., 2014), we asked whether 2-HG acts by a similar mechanism.
Figure 1. 2-HG extends the lifespan of adult C. elegans.

(A) Chemical structures of 2-hydroxyglutaric acid and α-ketoglutaric acid.
(B) (R)-2HG supplemented worms. Mean lifespan (days of adulthood) with vehicle treatment mveh = 14.0 (n = 112 animals tested); mα-KG = 20.7 (n = 114), P < 0.0001 (log-rank test); m(R)-2HG = 20.0 (n = 110), P < 0.0001 (log-rank test); mMet = 14.7 (n = 116), P = 0.4305 (log-rank test); mLeu = 13.2 (n = 110), P = 0.3307 (log-rank test).
(C) (S)-2HG supplemented worms. mveh = 15.7 (n = 85); mα-KG = 21.5 (n = 99), P < 0.0001 (log-rank test); m(S)-2HG = 20.7 (n = 87), P < 0.0001 (log-rank test).
All metabolites were given at a concentration of 8 mM. Number of independent experiments: 2.
ATP synthase is a molecular target of 2-HG
To determine whether 2-HG targets ATP5B, we first performed DARTS analysis (Lomenick et al., 2009) using U87 human glioblastoma cells. We found that both (R)-2HG and (S)-2HG bind to ATP5B (Figure 2A, and data not shown). Like α-KG, both 2-HG enantiomers inhibit ATP synthase (complex V) (Figure 2B-C and Figure S1A-C). This inhibition is specific as there is no inhibition by either enantiomer on other electron transport chain (ETC) complexes (Figure S1D-F) or ADP import into the mitochondria (Figure S1G). The inhibition of ATP synthase by 2-HG is also readily detected in live cells; treatment of U87 cells (wild-type IDH1/2) with membrane-permeable octyl esters of 2-HG or α-KG results in decreased cellular ATP content (Figure S2A) and decreased ATP/ADP ratio (Figure 2D and Figure S2B) under mitochondrial oxidative phosphorylation (OXPHOS) conditions, as with the ATP synthase inhibitor oligomycin (Figure S2A-B). As expected, both basal and ATP synthase-linked oxygen consumption rates (OCR) are decreased in the 2-HG treated cells (Figure 2E and Figure S2C-D), and lifespan increase by 2-HG is dependent on ATP synthase (Figure S2E).
Figure 2. 2-HG binds and inhibits ATP synthase.

(A) DARTS identifies ATP5B as a 2-HG binding protein. U87 cell extracts were used. Succinate serves as a negative control.
(B) Inhibition of ATP synthase by 2-HG. 2-HG, released from octyl 2-HG (600 μM), decreases (P < 0.001) state 3, but not state 4o or 3u, respiration in mitochondria isolated from mouse liver. Octanol is used as vehicle. Oligo, oligomycin; FCCP, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone; AA, antimycin A.
(C) Inhibition of submitochondrial particle ATPase by 2-HG acid, but not by succinic acid. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; NS, P > 0.05. Oligo, oligomycin (32 μM).
(D) Decreased ATP/ADP ratio in U87 cells treated with octyl 2-HG but not octyl citrate, indicating specificity and excluding any effect involving octanol. *P < 0.05, ***P < 0.001; NS, P > 0.05.
(E) Decreased respiration as indicated by OCR (**P < 0.01) in octyl 2-HG treated U87 cells in glucose media. Octanol shows no effect on OCR compared to DMSO.
(A-E) Results were replicated in at least two independent experiments. (B-E) By unpaired t-test, two-tailed, two-sample unequal variance. Mean ± s.d. is plotted.
IDH1(R132H) mutant cells have decreased ATP content and mitochondrial respiration
At normal cellular concentrations of ∼200 μM (Gross et al., 2010), (R)-2HG is unlikely to cause significant inhibition of ATP synthase. However, in glioma patients with the IDH mutations where (R)-2HG accumulates to 10-100 times natural levels (Dang et al., 2009; Gross et al., 2010), inhibition of ATP synthase would be possible. To test this idea, we used U87 cells stably expressing IDH1(R132H), the most common IDH mutation in glioma (Yan et al., 2009). Similar to octyl (R)-2HG treated cells, the U87/IDH1(R132H) cells exhibit decreased ATP content and ATP/ADP ratio (Figure 3A), and OCR (Figure 3B) compared to isogenic IDH1(WT)-expressing U87 cells. Importantly, the decrease in respiration in the IDH1(R132H) cells is attributable to ATP synthase (complex V) inhibition. Whereas there is a clear difference in the basal respiration rates in U87/IDH1(WT) vs. U87/IDH1(R132H) cells, oligomycininsensitive respiration – which is independent of complex V – is not significantly different between IDH1(WT) and IDH1(R132H) cells (Figure 3C). Furthermore, complex V knockdown using ATP5B RNAi normalizes the respiration difference between IDH1(R132H) and IDH1(WT) cells (Figure 3D). Consistently, the difference in ATP content of U87/IDH1(WT) and U87/IDH1(R132H) cells is diminished upon treatment with octyl (R)-2HG (Figure 3E). Similar results were obtained in HCT 116 IDH1(R132H/+) cells (Figure S3A-B). In addition, mitochondrial membrane potential in IDH1 mutant cells is higher than that in IDH1 wildtype cells (Figure 3F and Figure S3C), consistent with inhibition of complex V (Johnson et al., 1981). In contrast, inhibition of ETC complex I, III, or IV each causes dissipation of mitochondrial membrane potential (Johnson et al., 1981).
Figure 3. Inhibition of ATP synthase in IDH1(R132H) cells.

(A) Decreased ATP levels and ATP/ADP ratio in U87/IDH1(R132H) cells (****P < 0.0001).
(B) Decreased respiration in U87/IDH1(R132H) cells (**P = 0.0037).
(C-D) Decreased respiration in U87/IDH1(R132H) cells is complex V-dependent (**P < 0.01, ***P < 0.001; NS, P > 0.05).
(E) Decreased ATP content in U87/IDH1(R132H) cells is attributable to (R)-2HG (****P < 0.0001; NS, P > 0.05).
(F) Increased mitochondrial membrane potential in U87/IDH1(R132H) cells normalized to cell number (****P < 0.0001).
(G) 2-HG accumulation in U87/IDH1(R132H) cells (***P = 0.0003).
(H) Metabolic profile of octyl (R)-2HG treated U87 cells (***P < 0.001, *P = 0.0435). It is possible that the flux rate has changed without affecting the absolute abundance of the intermediates.
(I) Model of metabolite signaling through ATP synthase inhibition.
(A-H) By unpaired t-test, two-tailed, two-sample unequal variance. Mean ± s.d. is plotted. Results were replicated in at least two independent experiments.
The intracellular (R)-2HG levels are ∼20-100 fold higher in U87 and HCT 116 cells expressing IDH1(R132H) than in control cells (Figure 3G and Figure S3D). The elevated (R)-2HG levels are comparable to those found in cells treated with octyl (R)-2HG (Figure 3H), and levels reported for IDH1-mutant tumor samples (Dang et al., 2009; Gross et al., 2010; Reitman et al., 2011). The detection of similar (R)-2HG levels in tumors as in the octyl (R)-2HG treated cells suggests that the tumor cells likely experience reduced ATP synthase and mitochondrial respiration, raising potential prognostic or therapeutic implications (see below).
2-HG accumulation does not alter the levels of common metabolites
The metabolite 2-HG is linked to the TCA cycle and related amino acid metabolic pathways (Figure 3I). To explore potential metabolic changes upon octyl 2-HG treatment, we measured metabolite levels in octyl 2-HG treated cells cultured in 1,2-13C-glucose-containing medium by LC-MS. As expected, 2-HG accumulates 20-100 fold after octyl 2-HG treatment (Figure 3H and Figure S3E). There is no dramatic change (< 2-fold) in TCA cycle metabolites or related amino acids (Figure 3H and Figure S3E). As expected, the bulk of the increased 2-HG came from the hydrolysis of exogenously provided octyl 2-HG as indicated by the unlabeled M+0 isotopomer (Figure S3F; data not shown for octyl (S)-2HG treatment). There is also no major change (< 2-fold) in labeled TCA cycle intermediates and related amino acids (Figure S3G). Similarly, treatment with octyl α-KG causes an increase in α-KG levels without other substantial changes in the metabolic profile (Figure S3H). The steady-state metabolic profiles observed in 2-HG (or α-KG) treated cells support the notion that the bioenergetic shift results from the direct inhibition of ATP synthase by 2-HG (or α-KG) rather than secondary effects (Figure 3I).
IDH1(R132H) mutant cells exhibit intrinsic vulnerability to glucose limitation
As the end component of the mitochondrial ETC, ATP synthase is a major source of cellular energy and the sole site for OXPHOS (Walker, 2013). When glycolysis is inhibited, such as under conditions of glucose insufficiency, cells are forced to rely on mitochondrial respiration as a source of ATP. The inherent inhibition of ATP synthase and mitochondrial respiration in mutant IDH1 cancer cells thus suggests a potential Achilles' heel for these cancers. Supporting this idea, when cultured in glucose-free, galactose-containing medium to ensure that respiration is the primary source of energy, IDH1(R132H) cells exhibit drastically decreased cell viability (Figure 4A and Figure S4A). These results indicate a particular sensitivity of IDH1(R132H) mutant cells to the deprivation of glucose. The mutant cell line is not sensitive to FBS deprivation (data not shown), indicating that its increased vulnerability to glucose starvation is specific. This vulnerability to glucose starvation is also evident in U87 cells treated with octyl α-KG or octyl 2-HG (Figure 4B-D and Figure S4B) and in ATP5B knockdown cells (Figure 4E). These findings raise the possibility that cancer cells with the IDH1(R132H) mutation (and the concomitant ATP synthase/mitochondrial respiration defect) may also be particularly sensitive to nutrient conditions analogous to glucose limitation.
Figure 4. Inherent vulnerability, or the loss of cell viability, characteristic of cells with ATP5B knockdown, 2-HG accumulation, or IDH mutations.

(A) U87/IDH1(R132H) cells have increased vulnerability to glucose starvation (***P < 0.001).
(B-D) Octyl α-KG or octyl 2-HG treated U87 cells exhibit decreased viability upon glucose starvation (****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05).
(E) ATP5B knockdown inhibits U87 cell growth (***P = 0.0004).
(A-E) Cells were cultured in galactose medium.
(F) HCT 116 IDH1(R132H/+) cells exhibit increased vulnerability to glucose-free medium supplemented with (R)-3-hydroxybutyrate (***P < 0.001).
(G-I) U87 cells with ATP5B knockdown or octyl esters of α-KG or 2-HG treatment and HCT 116 IDH1(R132H/+) cells exhibit decreased mTOR Complex 1 activity in glucose-free, galactose-containing medium. All lanes in (I) are on the same blot; space indicates position of unnecessary lanes that were digitally removed. Octanol has no effect on mTOR activity.
(A-F) By unpaired t-test, two-tailed, two-sample unequal variance. Mean ± s.d. is plotted. (A-I) Results were replicated in at least two independent experiments.
In complex organisms, glucose limitation can occur as a consequence of ketosis, wherein cells use ketone bodies (instead of glucose) for energy. Ketosis is naturally induced upon prolonged starvation (or fasting), during which cells derive energy from fat reservoirs while sparing protein in muscle and other tissues from catabolism. Ketosis can also be induced by feeding a low-carbohydrate-high-fat “ketogenic diet”, which has shown benefits against cancer (Stafford et al., 2010). One reason for this may be that tumor cells largely depend on glucose for growth and survival. Since metabolism of ketone bodies depends entirely on OXPHOS, one prediction is that inhibiting ATP synthase (or other ETC components) in cancer cells would confer a survival disadvantage if ketone bodies were the only source of energy. Since U87 cells are unable to utilize ketone bodies for energy, we determined the effect of ketogenic conditions using HCT 116 cells expressing mutant IDH1. When cultured in glucose-free medium containing the ketone body (R)-3-hydroxybutyrate, IDH1 mutant HCT 116 cells showed a profound decrease in viability compared to the parental cells (Figure 4F), confirming the suspected metabolic weakness of IDH mutant cells. These results further support our discovery that (R)-2HG accumulation in mutant IDH cancer cells results in ATP synthase inhibition, and also suggest novel metabolic therapeutic strategies in cancer treatment.
Decreased mTOR signaling and cell growth by 2-HG
Inhibition of ATP synthase leads to decreased TOR signaling in mammalian cells, worms, and flies (Chin et al., 2014; Sun et al., 2014). We found that ATP5B knockdown (Figure 4G), treatment with octyl esters of 2-HG (Figure 4H), and IDH1(R132H) mutation (Figure 4I) all decrease the phosphorylation of mTOR complex 1 substrates. This effect occurs initially (4 h) in an AMPK-independent manner, with 2-HG decreasing mTOR signaling without significantly altering AMPK activity; however, prolonged exposure to 2-HG (24 h) also activates AMPK (Figure S4C), consistent with the idea that mTOR itself may directly sense ATP (Dennis et al., 2001) in addition to responding to AMP levels through crosstalk with AMPK (Shaw, 2009; Inoki et al., 2012).
TOR is a major regulator of cell growth (Blagosklonny and Hall, 2009). Consistent with the decreased TOR signaling, we observed growth inhibition in ATP5B knockdown cells (Figure S4D), in cells treated with octyl α-KG or octyl 2-HG, (Figure S4E-G), and in IDH1(R132H) expressing cells (Figure S4H-I). ATP5B RNAi normalizes the growth difference between IDH1(R132H) and IDH1(WT) cells (Figure S4J). Growth inhibition by 2-HG (and by α-KG) is also observed in WI-38 normal human diploid fibroblasts, in immortalized non-malignant HEK 293 cells, and in other cancer cell lines tested (Figure S4K-N). These results suggest that when present in excess, 2-HG acts as a growth inhibitory metabolite across cell types. Further work is warranted to test whether the growth inhibitory effect of (R)-2HG underlies the longer median overall survival of glioma patients with IDH mutations.
Summary
We demonstrate that, similar to α-KG, both enantiomers of 2-HG bind and inhibit ATP synthase and extend the lifespan of C. elegans. Inhibition of ATP synthase by these related metabolites decreases mitochondrial respiration and mTOR signaling. Both 2-HG and α-KG exhibit broad growth inhibitory effects and reduce cancer cell viability in glucose restricted conditions. It is now recognized that (R)-2HG, which accumulates in IDH mutant cancers, facilitates oncogenic transformation. Little is known, however, about how (R)-2HG modifies the phenotype of IDH mutant gliomas once the tumors are formed. Our findings suggest that in addition to interfering with various α-KG binding factors with importance in cancer, (R)-2HG also acts – through inhibition of ATP synthase and mTOR signaling downstream – to decrease tumor cell growth and viability. The latter property may contribute to the improved prognosis of IDH mutant glioma patients. This idea is consistent with emerging findings that inhibition of ETC complex I in cancer could be an effective therapeutic strategy (Wheaton et al., 2014; Birsoy et al., 2014). Together, these findings highlight a hopeful approach to cancer prevention and treatment by targeting certain aging pathways through metabolic modulation.
The effects of excess 2-HG are likely to be context dependent. Whereas its growth inhibitory effects may be beneficial in cancer, in 2-HG aciduria, the inhibition of ATP synthase and resulting impaired mitochondrial function could contribute to neurological dysfunction (da Silva et al., 2002; Wajne et al., 2002; Kolker et al., 2002; Latini et al., 2005). Thus, the identification of ATP synthase as a target of both 2-HG enantiomers provides a congruent molecular basis for 2-HG associated cancer and neurological disorders. We postulate that altered mitochondrial energy metabolism may contribute to the inverse susceptibility to cancer and neurodegenerative diseases (e.g., Parkinson's Disease). Finally, our findings raise the possibility that nutrient and/or metabolic intervention per se, such as diets that lower reliance on glucose, and/or approaches that perturb cellular energy metabolism (e.g., by targeting OXPHOS), may benefit glioma patients. Such approaches may be particularly valuable for improving the survival of glioma patients without IDH mutations, who otherwise have no means to inherently curb mitochondrial respiration, and for cancer prevention and treatment in general.
Experimental Procedures
Lifespan analysis
Lifespan experiments were performed as described (Chin et al., 2014). Lifespan assays were conducted at 20 °C on solid nematode growth media (NGM). L4 or young adult animals were picked onto NGM assay plates containing D-2-HG (Sigma, H8378), L-2-HG (Sigma, 90790), α-KG (Sigma, K1128), or vehicle control (H2O). Assay plates were seeded with OP50. For RNAi experiments, NGM assay plates also contained 1 mM isopropyl-b-D-thiogalactoside (IPTG) and 50 μg/mL ampicillin, and were seeded with the appropriate RNAi feeding clone (Thermo Scientific/OpenBiosystems). The C. elegans TOR (let-363) RNAi clone was obtained from Joseph Avruch (MGH/Harvard). To assess the survival of the worms, the animals were prodded with a platinum wire every 2–3 days, and those that failed to respond were scored as dead. Worms that ruptured, bagged, or crawled off the plates were censored. Lifespan data were analysed using GraphPad Prism; P values were calculated using the log-rank (Mantel–Cox) test unless stated otherwise.
Target identification using drug affinity responsive target stability (DARTS)
DARTS was performed as described (Lomenick et al., 2009).
Measurement of mitochondrial respiration
Mitochondrial respiration was analyzed using isolated mitochondria (Brand and Nicholls, 2011).
Cell growth and viability assays
Cells were seeded in 12-well plates and after overnight incubation were treated with indicated concentrations of each compound. After harvesting, cells were stained with Acridine Orange (AO) and DAPI. Cell number and viability were measured based on AO and DAPI fluorescence measured by NC3000 (ChemoMetec) following the manufacturer's instructions.
Metabolic profile analysis
Cells were cultured for 24 h, rinsed with PBS, and medium containing 1,2-13C-glucose (1 g/L) added. After 24 h culture, cells were rinsed with ice-cold 150 mM NH4AcO (pH 7.3) followed by addition of 400 μL cold methanol and 400 μL cold water. Cells were scraped off, transferred to an Eppendorf tube, and 10 nmol norvaline as well as 400 μL chloroform added to each sample. For the metabolite extraction, samples were vortexed for 5 min on ice, spun down, and the aqueous layer transferred into a glass vial and dried. Metabolites were resuspended in 70% ACN, and 5 μL sample loaded onto a Phenomenex Luna 3u NH2 100A (150 × 2.0 mm) column. The chromatographic separation was performed on an UltiMate 3000RSLC (Thermo Scientific) with mobile phases A (5 mM NH4AcO, pH 9.9) and B (ACN) and a flow rate of 300 μL/ min. The gradient ran from 15% A to 95% A over 18 min, 9 min isocratic at 95% A, and re-equilibration for 7 min. Metabolite detection was achieved with a Thermo Scientific Q Exactive mass spectrometer run in polarity switching mode (+3.0 kV / -2.25 kV). TraceFinder 3.1 (Thermo Scientific) was used to quantify metabolites as area under the curve using retention time and accurate mass measurements (≤ 3 ppm). Relative amounts of metabolites were calculated by summing up all isotopomers of a given metabolite and were normalized to internal standard and cell number. Natural occurring 13C was accounted for as described in (Yuan et al., 2008).
Statistical analyses
All experiments were repeated at least two times with identical or similar results. Data represent biological replicates. Appropriate statistical tests were used for every figure. Mean ± s.d. is plotted in all figures.
Supplementary Material
Highlights.
2-HG, like α-KG, inhibits ATP synthase and extends the lifespan of C. elegans.
IDH1(R132H) mutant cells have reduced ATP content, respiration, and mTOR signaling.
IDH1(R132H) mutant cells exhibit intrinsic vulnerability to glucose limitation.
ATP synthase is a target of 2-HG's growth suppressive activity in IDH mutant cells.
Acknowledgments
We thank Chris Walsh for insightful suggestions and advice. Supported by the Oppenheimer Program, NIH grants R01 AT006889 and P01 HL028481, and UCLA Jonsson Cancer Center Foundation and National Center for Advancing Translational Sciences UCLA Clinical and Translational Science Institute (CTSI) Grant UL1TR000124. X.F. is a recipient of the China Scholarship Council Scholarship. R.M.C. is a postdoctoral fellow supported by the UCLA Tumor Immunology Training Program (NIH T32 CA009120). M.Y.P. was a trainee of the UCLA C&MB training grant (NIH T32 GM007185).
Footnotes
Please see the Supplemental Information for detailed experimental procedures.
Author Contributions: The majority of experiments were designed and performed by X.F.; lifespan assays by R.M.C.; DARTS by H.H.; mitochondrial respiration study design and analyses by L.V. and K.R.; enzyme inhibition assays by R.M.C.; compound syntheses by G.D., Y.X. and M.E.J.; metabolomic profiling and analysis by X.F. and D.B.; S.L., L.T., D.A.N., M.Y.P., F.F., C.C., R.M.P., M.A.T., A.L., K.F.F., M.J., S.G.C., T.F.C., T.G.G., D.B., H.R.C., M.E.J., L.V., and K.R. provided guidance, specialized reagents, and expertise. X.F., K.R. and J.H. wrote the paper. X.F., R.M.C. and J.H. analyzed data. All authors discussed the results and contributed to aspects of preparing the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Birsoy K, Possemato R, Lorbeer FK, Bayraktar EC, Thiru P, Yucel B, Wang T, Chen WW, Clish CB, Sabatini DM. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging Albany (NY) 2009;1:357–362. doi: 10.18632/aging.100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F, Medeiros BC, Tyvoll DA, Majeti R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21:178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;509:397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva CG, Ribeiro CA, Leipnitz G, Dutra-Filho CS, Wyse AA, Wannmacher CM, Sarkis JJ, Jakobs C, Wajner M. Inhibition of cytochrome c oxidase activity in rat cerebral cortex and human skeletal muscle by D-2-hydroxyglutaric acid in vitro. Biochim Biophys Acta. 2002;1586:81–91. doi: 10.1016/s0925-4439(01)00088-6. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science. 2001;294:1102–1105. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Walsh ML, Bockus BJ, Chen LB. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J Cell Biol. 1981;88:526–535. doi: 10.1083/jcb.88.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolker S, Pawlak V, Ahlemeyer B, Okun JG, Horster F, Mayatepek E, Krieglstein J, Hoffmann GF, Kohr G. NMDA receptor activation and respiratory chain complex V inhibition contribute to neurodegeneration in d-2-hydroxyglutaric aciduria. Eur J Neurosci. 2002;16:21–28. doi: 10.1046/j.1460-9568.2002.02055.x. [DOI] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, Salomons GS, Van der Knaap MS, Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571–587. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranendijk M, Struys EA, van Schaftingen E, Gibson KM, Kanhai WA, van der Knaap MS, Amiel J, Buist NR, Das AM, de Klerk JB, et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science. 2010;330:336. doi: 10.1126/science.1192632. [DOI] [PubMed] [Google Scholar]
- Latini A, da Silva CG, Ferreira GC, Schuck PF, Scussiato K, Sarkis JJ, Dutra Filho CS, Wyse AT, Wannmacher CM, Wajner M. Mitochondrial energy\metabolism is markedly impaired by D-2-hydroxyglutaric acid in rat tissues. Mol Genet Metab. 2005;86:188–199. doi: 10.1016/j.ymgme.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford P, Abdelwahab MG, Kim do Y, Preul MC, Rho JM, Scheck AC. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr Metab (Lond) 2010;7:74. doi: 10.1186/1743-7075-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenweg ME, Jakobs C, Errami A, van Dooren SJ, Adeva Bartolome MT, Aerssens P, Augoustides-Savvapoulou P, Baric I, Baumann M, Bonafe L, et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotype-phenotype study. Hum Mutat. 2010;31:380–390. doi: 10.1002/humu.21197. [DOI] [PubMed] [Google Scholar]
- Sun X, Wheeler CT, Yolitz J, Laslo M, Alberico T, Sun Y, Song Q, Zou S. A Mitochondrial ATP Synthase Subunit Interacts with TOR Signaling to Modulate Protein Homeostasis and Lifespan in Drosophila. Cell Rep. 2014;8:1781–1792. doi: 10.1016/j.celrep.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarhonskaya H, Rydzik AM, Leung IK, Loik ND, Chan MC, Kawamura A, McCullagh JS, Claridge TD, Flashman E, Schofield CJ. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun. 2014;5:3423. doi: 10.1038/ncomms4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, Frenay M, Tijssen CC, Lacombe D, Idbaih A, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res. 2010;16:1597–1604. doi: 10.1158/1078-0432.CCR-09-2902. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajne M, Vargas CR, Funayama C, Fernandez A, Elias ML, Goodman SI, Jakobs C, van der Knaap MS. D-2-Hydroxyglutaric aciduria in a patient with a severe clinical phenotype and unusual MRI findings. J Inherit Metab Dis. 2002;25:28–34. doi: 10.1023/a:1015165212965. [DOI] [PubMed] [Google Scholar]
- Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans. 2013;41:1–16. doi: 10.1042/BST20110773. [DOI] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Bennett BD, Rabinowitz JD. Kinetic flux profiling for quantitation of cellular metabolic fluxes. Nat Protoc. 2008;3:1328–1340. doi: 10.1038/nprot.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.