

Abstract
Our ability to build computational models that account for all known gene functions in a cell has increased dramatically. But why build whole-cell models, and how can they best be used? In this review we enumerate several areas in which whole-cell modeling can significantly impact research and technology.
Keywords: whole-cell modeling, model-driven discovery, predictive biology
Introduction
Whole-cell models, or computational models that account for the integrated function of every gene and molecule in a cell, have been described as “the ultimate goal” of systems biology, and “a grand challenge for the 21st century” (e.g., [1]). Although models of biological processes have been increasing in complexity and scope, until recently a number of significant challenges have prevented the construction of whole-cell models.
A recent study reported the construction of a whole-cell computational model for the bacterium Mycoplasma genitalium [2]. The approach combined diverse mathematical techniques from multiple fields to enable mechanistic modeling at multiple levels of abstraction in an integrated simulation. This approach enabled the simultaneous inclusion of thousands of heterogeneous experimental parameters in the model. The resulting whole-cell simulations captured a wide range of cellular behaviors, and suggested follow-on experiments that were validated experimentally [3].
A framework for whole-cell modeling has therefore been established, and other such models are currently underway. But why build whole-cell models, and how can they best be used? What applications can we look for in the future? Answering these questions forms the motivation for the current review (Figure 1).
Figure 1. Applications of whole-cell modeling to academia and industry.
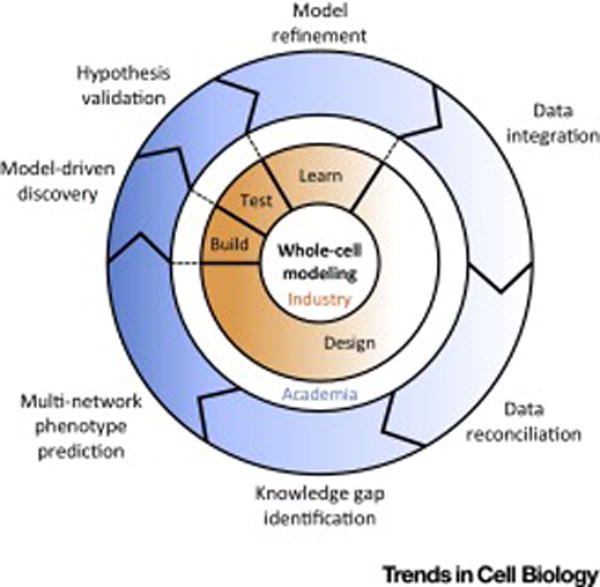
Heterogeneous data sets are integrated into a unified representation and are reconciled against each other in order to identify potential gaps in our current knowledge. Next, complex, multi-network phenotypes are simulated and predicted, and compared to data, which leads to model-driven discovery as hypotheses generated by the model are subject to experimental validation. This process is analogous to the “design-build-test-learn” cycle in industry (see dashed lines), where whole-cell models can provide a framework to accelerate the creation of genetically-modified organisms.
Five applications for whole-cell modeling
Integrate heterogeneous data sets
First, whole-cell models integrate heterogeneous data sets into a unified representation of our knowledge about a given organism. Biological datasets relevant to cell and molecular biology appear in many forms. They may be qualitative or quantitative, large or small, they depend on different technologies (gel electrophoresis, sequencing, fluorescence, etc.), and they represent varying aspects of cellular function. Although these datasets have been reported in such a way as to appear independent from one another (i.e., through publication in multiple journals at various points in time and from different labs), in fact they are deeply interconnected and are therefore most effectively considered as a whole.
Recognizing this, multiple groups have made an effort to generate massive datasets under one roof or collaboration, giving them the opportunity to consider all of these data together. For example, large-scale quantitative analyses have been made to understand Mycoplasma pneumoniae comprehensively [4], and similar analyses have been performed in other bacteria.
Computational efforts have focused on the challenge of data integration, which attempts to derive network structure and/or parameter values from multiple data sets. These approaches rely on the construction of networks that encompass key biological processes, and may incorporate combinations of probabilistic representations [5], machine learning-based algorithms [6], mechanistic constraint-based models [7], or large-scale ordinary differential equations [8].
Whole-cell modeling naturally leads to data integration because it attempts to incorporate all possible data pertaining to a particular cell type. The M. genitalium model accounted for ~1,700 parameters culled from the literature and was benchmarked or validated against such diverse data as metabolite concentration, flux, RNA expression, RNA decay, and chemical composition measurements, to name a few. A major advantage of whole-cell modeling is that these data are linked mechanistically in the model, through the simulated interaction of biological processes in the cell. This mechanistic linkage provides the most natural, intuitive interpretation of an integrated dataset.
Identify limits of our knowledge
Next, whole-cell models identify the limits of our current knowledge for a given biological system. With all the data that is generated for a particular cell or organism, there remains a dramatic gap between what is known and what remains to be discovered. To completely “solve” a cell would ideally involve rigorous and coordinated statistical design of experiments to comprehensively identify the main and interaction effects in a given network. Anything short of this will inevitably lead to both under-explored areas of the network, which are essentially gaps in our knowledge, as well as controversy when published experimental results exhibit seemingly inconsistent results in the absence of sufficient network context.
Whole-cell models can diagnose both under-explored areas and areas of seeming inconsistency in a network. With regard to finding gaps in the model, model predictions were compared to experimental observations in a single-gene disruption library of M. genitalium, producing a detailed map of model-experiment comparisons for all 525 genes in the chromosome [3]. This map highlights poorly understood cellular functions gene-by-gene to suggest possibly fruitful areas of inquiry. Others have found ways to identify and fill in metabolic network gaps automatically (e.g., [9]).
In terms of areas of high inconsistency in the network, the totality of existing data for an organism is rarely self-consistent and reproducible. For example, kinetic parameter measurements for a particular enzyme can vary over orders of magnitude [3] – not to mention the variability that can occur when values are obtained from different types of measurements. This can be due to variation in laboratory techniques, but it also often reflects limitations in our understanding of the system. In the above case of kinetic parameter variation, for example, a simple Michaelis-Menten representation may not adequately describe a more complex underlying phenomenon. Constructing mathematical models that integrate massive amounts of data forces these inconsistencies to the foreground and requires them to be resolved, whether on the experimental or the modeling side, or both.
Predict complex, multi-network phenotypes
The most compelling application of whole-cell modeling to date has been the ability to identify and even elucidate emergent behaviors that cross-traditional network boundaries. In the origins of this field, a minimal cell model was described which could predict changes in cell composition, cell size, cell shape, and the timing of chromosome synthesis in response to environmental changes [10]. More recently, the complete whole-cell model of M. genitalium has demonstrated the potential to reveal complex phenomena that are difficult or prohibitive to investigate experimentally in the context of the entire cell, including: the instantaneous protein chromosomal occupancy as well as the temporal dynamics and interactions of every DNA-binding protein at the genomic scale at single-cell resolution; a novel, emergent control on the duration of the cell cycle; quantitative assessments of cellular energetics and the synthesis dynamics of the high-energy intermediates; and the “molecular pathologies” of single-gene disruption strains [2]. Another novel approach to simulating whole-cell behaviors in E. coli, based on spatial stochastic simulations, was used to quantify variation in how individual cells in a population express a set of genes in response to an environmental signal [11]. All of these predictions remain to be experimentally validated, and may even motivate the development of novel measurement technologies in the future. Nevertheless, in each case, the biological insights generated in these studies would have been impossible to identify or quantify without a comprehensive model.
Suggest future experiments that may lead to new knowledge
The early promise of systems biology was that mathematical modeling could be used to suggest new experiments through testable predictions. One effective implementation of this idea occurs as large sets of computer simulations are directly compared to experimental outcomes. For any given complex experiment, whole-cell models can be used to produce a corresponding simulation, resulting in two sets of data, one computational and one experimental. These two data sets can be directly compared to determine how well the model describes experimental observations. The areas in which model and experiment agree validate the model, but the discrepancies between predictions and observations hold the true value, as each discrepancy represents a high-probability opportunity for a discovery. Such a premise has been implemented in the context of genome-scale metabolic modeling, leading to new functional assignments for genes [12].
Whole-cell models broaden the scope and increase the diagnostic power of such studies. For example, M. genitalium model simulations were compared to experimentally measured growth rates to determine discrepancies. The outcome of this comparison led to the prediction of specific kinetic parameters for three enzymes, which were then all successfully validated experimentally [3]. We find it striking that simply knowing the growth rates of certain disruption strains was sufficient to constrain kinetic parameter values for specific proteins; this observation highlights the value of an approach in which all of the biological processes are connected.
Provide a framework for the safe and effective design of genetically-modified organisms
The advent of rapid and inexpensive DNA synthesis is leading an era of largely or even completely synthetic organisms. Just as computer-aided design (CAD) and other modeling-based predictive tools have transformed other engineering disciplines, we expect “Bio-CAD” tools to play a major role in the field of synthetic biology.
Network modeling approaches have facilitated the rational engineering and perturbation of biological systems in academia [13–14], and are beginning to be applied in industry as well. In the biotechnology sector, companies such as the Emerald Cloud Lab and Transcriptic seek to automate the entire engineering cycle - and with it, the model-driven design process. In parallel, Ginkgo Bioworks and SGI-DNA offer services, which greatly facilitate the design of novel organisms. Zymergen, Genomatica and Amyris use modeling to inform their strain designs. As whole-cell modeling develops, we expect it to become part of the expanding toolkit for this new industry.
Despite these advances, synthetic biology still lacks many predictive tools needed to enable efficient design. Computational whole-cell models can provide a useful guiding framework that could eventually transform current genome engineering into a more precise and predictive discipline [15].
Concluding remarks
As advances in computational methods related to whole-cell modeling are developed, the number of applications will increase dramatically. New approaches will be required to model more complex cells, such as pathogenic microbes or mammalian cells, and eventually even multicellular systems, tissues or ecosystems. These and other advances will enable whole-cell modeling to realize its potential: to serve as a foundational platform for interpreting complex behaviors and facilitating discovery across a host of medical, research and biotechnological applications.
Acknowledgments
We gratefully acknowledge the Stanford Center for Systems Biology (NIH P50GM107615), as well as an NIH Pioneer Award (5DP1LM01150-05) and an Allen Distinguished Investigator Award to M.W.C., and a Dean’s Postdoctoral Fellowship of the Stanford School of Medicine to J.C. We also thank N. Ruggero and M. Paull for their valuable feedback on this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tomita M. Whole–cell simulation: a grand challenge of the 21st century. Trends Biotechnol. 2001;19:205–10. doi: 10.1016/s0167-7799(01)01636-5. [DOI] [PubMed] [Google Scholar]
- 2.Karr JR, et al. A whole-cell computational model predicts phenotype from genotype. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanghvi JC, et al. Accelerated discovery via a whole–cell model. Nat Methods. 2013;10:1192–5. doi: 10.1038/nmeth.2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wodke JA, et al. MyMpn: a database for the systems biology model organism Mycoplasma pneumoniae. Nucleic Acids Res. 2015;43:D618–23. doi: 10.1093/nar/gku1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandrasekaran S, Price ND. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2010;107:17845–17850. doi: 10.1073/pnas.1005139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrera J, et al. Computational design of genomic transcriptional networks with adaptation to varying environments. Proc Natl Acad Sci USA. 2012;109:15277–15282. doi: 10.1073/pnas.1200030109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, et al. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science. 2009;325:1544–9. doi: 10.1126/science.1174671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrera J, et al. An integrative, multi-scale, genome-wide model reveals the phenotypic landscape of Escherichia coli. Mol Syst Biol. 2014;10:735. doi: 10.15252/msb.20145108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganter M, et al. Predicting network functions with nested patterns. Nat Commun. 2014;5:3006. doi: 10.1038/ncomms4006. [DOI] [PubMed] [Google Scholar]
- 10.Domach MM, et al. Computer model for glucose-limited growth of a single cell of Escherichia coli B/r-A. Biotechnol. Bioeng. 1984;26:1140. doi: 10.1002/bit.260260925. [DOI] [PubMed] [Google Scholar]
- 11.Roberts E, et al. Noise contributions in an inducible genetic switch: a whole-cell simulation study. PLoS Comput Biol. 2011;7:e1002010. doi: 10.1371/journal.pcbi.1002010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed JL, et al. Systems approach to refining genome annotation. Proc Natl Acad Sci USA. 2006;103:17480–17484. doi: 10.1073/pnas.0603364103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung YK, et al. Metabolic engineering of Escherichia coli for the production of polylactic acid and its copolymers. Biotechnol Bioeng. 2010;105:161–171. doi: 10.1002/bit.22548. [DOI] [PubMed] [Google Scholar]
- 14.Cahan P, et al. CellNet: network biology applied to stem cell engineering. Cell. 2014;158:903–915. doi: 10.1016/j.cell.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purcell O, et al. Towards a whole-cell modeling approach for synthetic biology. Chaos. 2013;23:025112. doi: 10.1063/1.4811182. [DOI] [PMC free article] [PubMed] [Google Scholar]