

Abstract
Genome instability and impaired DNA repair are hallmarks of carcinogenesis. The study was aimed at evaluating the DNA damage response in H2O2-treated lymphocytes using the alkaline comet assay in bladder cancer (BC) patients as compared to clinically healthy controls, elderly persons, and individuals with chronic inflammations. Polymorphism in DNA repair genes involved in nucleotide excision repair (NER) and base excision repair (BER) was studied using the PCR-RFLP method in the Belarusian population to elucidate the possible association of their variations with both bladder cancer risk and clinicopathological features of tumors. The increased level of H2O2-induced DNA damage and a higher proportion of individuals sensitive to oxidative stress were found among BC patients as compared to other groups under study. Heterozygosity in the XPD gene (codon 751) increased cancer risk: OR (95% CI) = 1.36 (1.03–1.81), p = 0.031. The frequency of the XPD 312Asn allele was significantly higher in T ≥ 2 high grade than in T ≥ 2 low grade tumors (p = 0.036); the ERCC6 1097Val/Val genotype was strongly associated with muscle-invasive tumors. Combinations of homozygous wild type alleles occurred with the increased frequency in patients with non-muscle-invasive tumors suggesting that the maintenance of normal DNA repair activity may prevent cancer progression.
1. Introduction
Oxidized DNA base lesions induced by environmental pollutants and endogenous metabolites lead to a variety of mutations and consequently to genetic instability, which is a hallmark of cancer [1, 2]. As shown in European populations, increased frequencies of chromosome aberrations and micronuclei are closely associated with cancer risk [3, 4]. When monitoring genomic alterations in the urothelial carcinomas in individual patients for a long time, the increased level of the mitotic recombination was found at the early carcinogenesis stage, and extensive genetic damage was accumulated during the evolution of the tumors [5]. When studying the cellular response to DNA damage in different types of cancer, activation of the ATM–Chk2–p53 signal pathway was observed in early human tumorigenesis [6] indirectly indicating accumulation of DNA lesions that, in turn, might be considered as the primary trait of upcoming genome instability and cell malignancy. Thus, the oxidatively induced DNA damage initiating genome instability is one of the principal factors of carcinogenesis, whereas the other one seems to be DNA repair deficiency or impairment.
The oxidatively damaged bases are predominantly removed via the BER pathway initiated with their excision by DNA glycosylases [7]. Among them, 8-oxo-guanine DNA glycosylase 1 (OGG1) is responsible for elimination of the highly mutagenic DNA lesion, 8-oxo-7,8-dihydroguanine (8-oxoGua). Another functionally important protein, X-ray repair cross-complementing protein 1 (XRCC1), interacts with DNA glycosylases, AP endonuclease-1 (APE-1), DNA polymerase β (POLβ), DNA ligase III (Lig III), poly (ADP-ribose) polymerase 1 (PARP-1), and polynucleotide kinase (PNK) at the damaged site, by modulating their activities and coordinating the subsequent enzymatic BER steps [8, 9]. The reduced BER activity has been newly discussed to trigger the development of sporadic cancers [10]. The performed proteomic analysis of BER deficient human cells has demonstrated that BER deficiency, leading to genome instability, results in dramatic changes in gene expression, resembling changes found in many cancers. These findings suggest that genetically unstable BER deficient cells may be a source of precancerous cells [10].
The majority of chemically induced DNA adducts are removed by the NER pathway that operates globally throughout the genome (global genome, GG NER) or during transcription (transcription coupled, TC NER); both subsets differ only in their initial recognition of the helix-distorting DNA damage [11, 12]. In this multistep repair process, DNA helicases unwind the double helix, thus opening access to the lesion site for other repair enzymes [13]. The XPD helicase, mutated in the cancer-prone xeroderma pigmentosum (XP), is part of the TFIIH complex that is essential for signaling events triggering transcription, cell cycle checkpoints, and DNA repair [14]. TC NER requires specific factors, including Cockayne syndrome (CS) protein B (CSB). The latter belongs to both the helicase superfamily 2 and to the SWI/SNF complex maintaining and remodeling chromatin structure [15, 16] and it acts at the crossroads of transcriptional networks [17, 18]. In the context of the present study, the recently reported data confirming involvement of NER-initiating proteins in the elimination of oxidatively generated DNA damage [19, 20] take on special significance.
We attempted to estimate the cellular response to oxidatively induced DNA damage and polymorphism in some DNA repair genes in bladder cancer. The frequencies of OGG1 Ser326Cys (rs1052133), XRCC1 Arg399Gln (rs25487), XPD Asp312Asn (rs1799793), and ERCC6 Met1097Val (rs2228526) polymorphisms have been recently determined in the bladder cancer (BC) patients as compared with clinically healthy residents of Belarus [21]. Our results indicated the association of the XPD 312Asp/Asn heterozygous genotype with an increased risk of bladder cancer, whereas the OGG1 326Ser/Cys heterozygous genotype has exhibited the protective effect. Here, genome integrity and stability was analyzed in peripheral blood lymphocytes using the comet assay. Besides, isolated DNA samples were genotyped for polymorphism of DNA repair genes involved in BER and NER to elucidate both the possible impact of some other genetic variations (XPD Lys751Gln and ERCC6 Gly399Asp) on bladder cancer susceptibility and the association of all six polymorphisms with clinicopathological parameters of tumors for evaluating their prognostic relevance.
2. Materials and Methods
2.1. Study Populations
The study included two independent experimental sets. In the first experimental set, the control group comprised 35 clinically healthy volunteers aged 22–63 years old who had no chronic and acute diseases and contacts with occupational hazards. They were recruited among residents of regions that were not affected by the Chernobyl fallout. Forty individuals with histologically verified bladder cancer (BC) were randomly selected among patients of the Department of Urology of N.N. Alexandrov National Cancer Centre of Belarus in 2011. The average age of BC patients was about 70 years; males and smokers amounted to 85% and 89% of the sample, respectively. Besides, the group of elderly people was represented by fifteen clinically healthy persons over 60 years; among them, 73% were males. Fifteen individuals comprised the group of chronic inflammatory diseases including chronic obstructive pulmonary disease (9 cases), chronic pyelonephritis (4 cases), and rheumatic disease and polyarthritis (2 cases); all of them were beyond the exacerbation phase. Their average age was about 50 years, and males amounted to 53% of the sample. The cellular response to oxidatively induced DNA damage was compared in these four groups using the comet assay.
In the second experimental set, the case group comprised 418 BC patients who were treated at the Department of Urology of N.N. Alexandrov National Cancer Centre of Belarus over 2011–2014. All urothelial carcinoma diagnoses were verified histologically after transurethral resection of tumors. The T stages were determined using the international Tumor-Node-Metastases (TNM) classification, and the grade of tumor tissue differentiation was established according to WHO classifications of 1973 and 2004 [22, 23]. Blood samples (3–5 mL) were collected by venal puncture by the qualified medical personnel in accordance with the Declaration of Helsinki (1964) [24]. The blood samples were accompanied with a demographic profile of patients and the clinicopathological description of tumors (Table 1).
Table 1.
The demographic features of BC patients and clinicopathological parameters of tumors.
| Features | Patients | |
|---|---|---|
| n | Frequency % | |
| Gender | ||
| Males | 344 | 82.3 |
| Females | 74 | 17.7 |
| Age (years) | ||
| Min | 31 | |
| Max | 93 | |
| Mean ± SD | 66.7 ± 10.9 | |
| Median | 67 | |
| Smoking | ||
| Smokers | 283 | 67.7 |
| Nonsmokers | 117 | 28.0 |
| Not specified | 18 | 4.3 |
| Tumor stages | ||
| TIS | 1 | 0.2 |
| Ta | 91 | 21.8 |
| T1 | 198 | 47.4 |
| T2 | 72 | 17.2 |
| T3 | 27 | 6.5 |
| T4 | 27 | 6.5 |
| Not specified | 2 | 0.4 |
| Tumor grades | ||
| 1973 | ||
| CIS | 1 | 0.2 |
| G1 | 139 | 33.3 |
| G2 | 186 | 44.5 |
| G3 | 86 | 20.6 |
| Not specified | 6 | 1.4 |
| 2004 | ||
| PUNLMP | 11 | 2.6 |
| CIS | 1 | 0.2 |
| Low | 241 | 57.7 |
| High | 156 | 37.3 |
| Not specified | 9 | 2.2 |
| Recurrence | ||
| No | 268 | 64.1 |
| Yes | 150 | 35.9 |
370 individuals were randomly recruited as controls among healthy volunteers involved in blood donation at the Republic Research and Production Center for Transfusiology and Medical Biotechnologies (Minsk) and elderly people who were observed at the Department of Gerontology and Geriatrics at the Belarusian Medical Academy of Postgraduate Education. Individuals from both control subgroups had no positive cancer history or acute diseases and should be considered the population-based controls. The control population was predominantly represented by males (68.7%). Like the BC patients, noncancer individuals were between 31 and 94 years old, with the average age of 64.5 ± 13.5 years as opposed to 66.7 ± 10.9 years in the case group. The controls were matched to the cases by the recruitment period, the ethnic origin (both were predominantly Belarusians or other Eastern Slavs), and age. However, they differed from each other in the smoking status, since smokers amounted to 31% among controls and to 68% among patients. It should be mentioned that the same control population was used in the previous work in order to study the possible impact of OGG1 Ser326Cys (rs1052133), XRCC1 Arg399Gln (rs25487), XPD Asp312Asn (rs1799793), and ERCC6 Met1097Val (rs2228526) polymorphisms on susceptibility to bladder cancer [21]. Herein, the control population was used for the similar purpose concerning XPD Lys751Gln (rs13181) and ERCC6 Gly399Asp (rs2228528) polymorphisms, while the next steps of the study were carried out using the enlarged case group stratified into several categories depending on tumor stages and grades.
Informed consent was obtained from each participant included in the study before the collection of blood samples. All participants were interviewed to complete a questionnaire covering medical, residential, and occupational history as well as age, gender, and the tobacco smoking status. The smoking status was summarized as “smokers” (combining current smokers and ex-smokers) or “nonsmokers” (including never smoking persons).
2.2. Estimation of Genome Integrity in Freshly Isolated Lymphocytes Using the Comet Assay
The approach for evaluation of genome integrity in order to diagnose genome instability in isolated lymphocytes was earlier described in detail [25–27]. In this investigation, 2-3 mL peripheral blood was collected into the heparinized Vacutainer tubes and kept at 4°C for no longer than 2 h. Lymphocytes were isolated from whole blood samples by centrifugation over 2.5 mL Histopaque at 1500 rpm for 30 min. Then lymphocytes were washed twice with RPMI 1640, suspended in cold PBS, and exposed to hydrogen peroxide (100 μM H2O2) at 4°C for 1 min, followed by washing with cold PBS. Intact and treated cells were incubated in RPMI 1640 with 10% fetal bovine serum (FBS) during a 3-h period at 37°C. Their viability was traditionally evaluated with the trypan blue exclusion test and usually varied in the range of 96–98%.
All the reagents and procedures were used according to the admitted protocol of the alkaline comet assay (single cell gel electrophoresis) [28]. Briefly, procedures included slide preparation, lysis of cell membranes for DNA elution by keeping the slides in the cold lysing solution (2.5 M NaCl, 10 mM Na2EDTA, 10 mM Tris, 1% Triton-X100, pH 10) for 1 h, DNA unwinding in fresh electrophoresis buffer (1 mM Na2EDTA, 300 mM NaOH) for 20 min, and horizontal electrophoresis for 20 min at 1 V/cm, 300 mA and pH > 13. After electrophoresis, slides were washed twice for 5 min with 0.4 M Tris buffer (pH 7.5) for neutralization and fixed in ice-cold 96% ethyl alcohol for 10 min. After staining with ethidium bromide, slides were analyzed with a fluorescence microscope Olympus BX-50. Visual estimation of DNA damage in arbitrary unites (a.u.) was carried out according to published recommendations [29]. Two slides were prepared for each point of analysis, and at least 100 cells were scored per each of two replicate slides by one researcher that provided the concordance between the results. The levels of DNA damage were calculated as average values.
Basal DNA damage was determined after 180 min incubation of intact lymphocytes in RPMI 1640 with 10% FBS. The initial level of oxidatively induced DNA damage was estimated immediately after mutagenic treatment, and the residual level of DNA damage was measured 180 min after exposure. To estimate DNA repair kinetics, samples of H2O2-treated lymphocytes were collected at 0, 30, 60, and 180 min of their incubation.
2.3. Genotyping
DNA for genotyping procedures was extracted using the traditional phenol-chloroform technique. Single nucleotide polymorphisms (SNPs) in some DNA repair genes were determined by the PCR-RFLP method under conditions used in the previous work [21]. In addition to polymorphisms OGG1 Ser326Cys, XRCC1 Arg399Gln, XPD Asp312Asn, and ERCC6 Met1097Val, XPD Lys751Gln (rs13181) and ERCC6 Gly399Asp (rs2228528) were analyzed in the present study. These polymorphisms were detected at conditions described elsewhere [30, 31]. The PCR products were digested with restriction enzymes, electrophoresed through 2.5% agarose gels containing ethidium bromide, and visualized under UV light. DNA repair genes and corresponding genotypes are shown in Table 2.
Table 2.
Characteristics of allelic variants and some conditions for their detection.
| Gene polymorphisms | Primer sequences | Restriction enzyme | PCR products (bp) |
|---|---|---|---|
| Nucleotide excision repair | |||
|
| |||
| ERCC2/XPD Asp312Asn rs1799793 | (F) 5′-CTG TTG GTG GGT GCC CGT ATC TGT TGG TCT-3′ (R) 5′-TAA TAT CGG GGC TCA CCC TGC AGC ACT TCC T-3′ |
StyI | Asp/Asp: 507 + 244; Asp/Asn: 507 + 474 + 244 + 33; Asn/Asn: 474 + 244 + 33 |
|
| |||
| ERCC2/XPD Lys751Gln rs13181 | (F) 5′-GCC CGC TCT GGA TTA TAC G-3′ (R) 5′-CTA TCA TCT CCT GGC CCC C-3′ |
Pst I | Lys/Lys: 290 + 146; Lys/Gln: 290 + 127 + 146; Gln/Gln: 227 + 146 |
|
| |||
| ERCC6/CSB Met1097Val rs2228526 | (F) 5′-CCT GCT T CT AAC ATA TCT GT-3′ (R) 5′-AAT CAC TGA CAA CTC TTC TG-3′ |
Nla III | Met/Met: 123 + 78; Met/Val: 201 + 123 + 78; Val/Val: 201 |
|
| |||
| ERCC6/CSB Gly399Asp rs2228528 | (F) 5′-TGA AGA GTC TGA GTA TTT CC-3′ (R) 5′-ATC TTC ATC TCC ATC ATC TC-3′ |
RsaI | Gly/Gly: 180 + 91; Gly/Asp: 271 + 180 + 91; Asp/Asp: 271 |
|
| |||
| Base excision repair | |||
|
| |||
|
XRCC1
Arg399Gln rs25487 |
(F) 5′-GGA CTG TCA CCG CAT GCG TCG G-3′ (R) 5′-GGC TGG GAC CAC CTG TGT T-3′ |
MspI | Arg/Arg: 115 + 34; Arg/Gln: 149 + 115 + 34; Gln/Gln: 149 |
|
| |||
|
OGG1
Ser326Cys rs1052133 |
(F) 5′-CTG TTC AGT GCC GAC CTG CGC CGA-3′ (R) 5′-ATC TTG TTG TGC AAA CTG AC-3′ |
MboI | Ser/Ser: 224 + 23; Ser/Cys: 247 + 224 + 23; Cys/Cys: 247 |
2.4. Statistical Analysis
Pearson's χ 2 test (or Fisher's exact test when necessary) was used to verify the significance of differences between the groups of BC patients and controls as well as between groups of different tumor stage and grade categories in genotype/allele frequencies. Student's t-test was used for comparison of the groups by age and other continuous variables, including the DNA damage levels. Nonparametric Mann-Whitney U test was also used in the latter case. DNA repair efficiency (RE) was calculated as percentage of DNA lesions eliminated at consequent time points relative to their initial level. The DNA repair rate in different groups was compared by the coefficients of linear regression (β) [25–27].
When genotyping the DNA samples for DNA repair gene polymorphisms, the statistical significance for deviation from Hardy-Weinberg equilibrium was determined using χ 2 test. p ≤ 0.05 values were considered significant. The relative risk was estimated as odds ratio (OR) with 95% confidence intervals (CI).
3. Results
3.1. Genome Integrity In Isolated Lymphocytes after Oxidative Stress In Vitro
To estimate adequately the cellular response to oxidized DNA damage in bladder cancer, it was compared among several groups and first of all between the cases and controls (Table 3, Figure 1). The results indicated the absence of statistically significant differences between the levels of basal DNA damage in the control and case groups, whereas the levels of H2O2-induced DNA damage in BC patients exceeded those in healthy volunteers during the whole period of observations. The greatest differences were observed immediately after lymphocyte mutagenic exposure. Nevertheless, the slopes of repair kinetics closely resembled each other (the insert in Figure 1), and, being compared by means of regression analysis, these data revealed no differences between two groups with respect to the DNA repair velocity. DNA repair efficiency was also equal in lymphocytes from patients and controls (Table 3), suggesting that induced DNA damage was eliminated in a similar manner in both groups.
Table 3.
The cellular response to the oxidative stress in vitro in the case group as compared to controls.
| Features under study | Exposure, time of lymphocyte incubation (min) | BC patients (n = 40) |
Controls/healthy donors (n = 35) |
|---|---|---|---|
| Basal DNA damage | Intact lymphocytes | ||
| 180 | 11.4 ± 1.0 | 9.2 ± 0.8 | |
|
| |||
| Oxidatively induced DNA damage | Exposure to H2O2 | ||
| 0 | 117.1 ± 7.1 | 85.6 ± 4.4a | |
| 30 | 41.8 ± 3.8 | 31.7 ± 3.2a | |
| 60 | 29.9 ± 2.7 | 22.8 ± 2.1a | |
| 180 | 17.7 ± 1.6 | 13.3 ± 1.1a | |
|
| |||
| DNA repair efficiency | 30 | 65.2 | 63.0 |
| 60 | 74.5 | 73.4 | |
| 180 | 84.3 | 84.4 | |
aSignificant differences are observed between the levels of H2O2-induced DNA damage in BC patients and healthy controls (p = 0.00035, 0.045, 0.037, and 0.026 at 0, 30, 60, and 180 min after mutagenic exposure according to two sided Student's t-test, and 0.01 < p < 0.05 according to the nonparametric Mann-Whitney U test).
Figure 1.
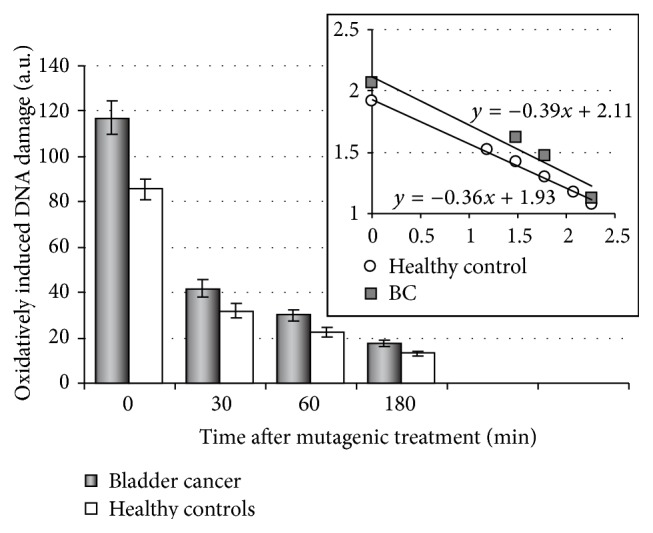
The oxidatively induced DNA damage and DNA repair kinetics in isolated lymphocytes from BC patients as compared to healthy donors. The case group included 40 BC patients; the control group comprised 35 clinically healthy donors. The insert reflects the DNA repair kinetics on a logarithmic scale. The coefficients of linear regression in the groups of patients (β = −0.39) and controls (β = −0.36) are approximately equal.
Then the cellular response to H2O2 exposure in BC patients was compared with that in elderly persons and individuals with chronic inflammatory diseases (Table 4). The significant differences were found between all the groups with respect to the initial levels of oxidatively induced DNA damage, with the highest level in the BC patients indicating increased cellular sensitivity to oxidative stress in bladder cancer.
Table 4.
The cellular response to DNA damage in the groups of BC patients, elderly people, and individuals with chronic inflammatory diseases.
| Features under study | BC patients (n = 40) |
Individuals older than 60 years (n = 15) |
Individuals with chronic inflammations (n = 15) |
|---|---|---|---|
| Average age (mean ± SE) |
69.55 ± 1.57a | 62.8 ± 0.74 | 48.87 ± 2.86 |
| Sex ratio females/males (% of males) |
6/34 (85) | 4/11 (73.33) | 7/8 (53.33) |
| Smokers/nonsmokers (% of smokers) |
5/35 (89) | 6/9 (60) | 2/13 (13.33) |
| Basal DNA damage (a.u.) at 180 min | 11.38 ± 1.09 | 9.93 ± 2.61 | 6.0 ± 1.32 |
| H2O2-induced DNA damage (a.u.) at 0 min | 117.13 ± 7.01b | 89.33 ± 11.55 | 92.47 ± 7.97 |
| Residual level of H2O2-induced DNA damage (a.u.) at 180 min | 17.7 ± 1.59 | 18.2 ± 4.21 | 12.77 ± 3.03 |
| DNA repair efficiency for 30 min incubation | 65.24 ± 2.08 | 69.91 ± 3.82 | 66.23 ± 4.03 |
| DNA repair efficiency for 180 min incubation | 84.25 ± 1.33 | 81.27 ± 3.27 | 84.72 ± 3.22 |
aSignificant differences concerning age were revealed between BC patients and elderly persons (p = 0.0004) and between BC patients and individuals with chronic inflammatory diseases (p = 0.0001).
bSignificant differences concerning the initial level of H2O2-induced DNA damage were observed between BC patients and elderly persons (p = 0.05) and between those and individuals with inflammations (p = 0.027).
In another approach, the frequency of sensitive individuals (with an enhanced DNA damage response) was estimated in the same groups using the earlier established reference intervals for all the parameters under study in the control population of 172 residents of Belarus [32]. In brief, the normal lymphocyte response to DNA damage was determined due to calculating 10th and 90th percentiles for levels of basal and exogenous DNA damage as well as for DNA repair efficiency measured at certain time points after mutagenic exposure. The marginal values were determined as follows: 15 a.u. for basal DNA damage, 110 a.u. for the initial level, 25 a.u. for the residual level of H2O2-induced DNA damage, and 70% for DNA repair efficiency by the end of cell incubation. Subjects with the levels of DNA damage exceeding these values as well as with DNA repair efficiency, which is lower than the normal parameter, were attributed to the group of “sensitive” individuals. It is seen from Table 5 that half of the BC patients sample manifested the increased sensitivity of lymphocytes to H2O2 immediately after treatment as opposed to 14.3% in the control group and 26.7% among elderly persons and individuals with chronic inflammations, respectively. Both approaches have demonstrated that the cellular responses to oxidatively induced DNA damage in bladder cancer strongly differed from those in healthy donors and to a lesser degree in aging and inflammations. Consequently, the increased initial level of H2O2-induced DNA damage in isolated lymphocytes might serve as a potential biomarker of genome instability predisposing to cancer.
Table 5.
The frequency of individuals with increased lymphocyte sensitivity to DNA damage in various study groups.
| Study groups | Proportion of sensitive subjects (%) with respect to | |||
|---|---|---|---|---|
| Basal DNA damage | H2O2-induced DNA damage | Total | ||
| Initial level | Residual level | |||
| BC patients (n = 40) | 17.5 | 50a | 10 | 62.5b |
| Individuals older than 60 years (n = 15) | 20.0 | 26.67 | 26.67 | 46.67 |
| Individuals with chronic inflammatory diseases (n = 15) | 6.67 | 26.67 | 6.67 | 46.67 |
| Controls (n = 35) | 8.57 | 14.29 | 5.71 | 40.0 |
aSignificant differences were revealed between all the groups by criterion χ 2 (p = 0.009) and between the case group and controls (p = 0.001).
bSignificant differences were observed between BC patients and controls (p = 0.05).
3.2. Association of DNA Repair Gene Polymorphisms with Bladder Cancer Risk and Clinicopathological Characteristics of Tumors
Polymorphism in some DNA repair genes has been recently reported to affect susceptibility to bladder cancer in Belarus [21]. Herein, the results of genotyping for XPD Lys751Gln (rs13181) and ERCC6 Gly399Asp (rs2228528) polymorphisms are added (Table 6). The ERCC6 Gly399Asp polymorphisms were found to be neutral unlike the XPD polymorphisms. In the latter case, cancer risk was mainly associated with the XPD 751Lys/Gln heterozygous genotype (OR (95% CI) = 1.36 (1.03–1.81) (p = 0.031)), which indicated that heterozygosity in this codon predisposes to tumorigenesis as it was earlier noticed for the XPD codon 312.
Table 6.
Distribution of allelic variants of some DNA repair genes in the group of BC patients as compared to controls.
| Genotypes/variant alleles | BC cases | Controls | p | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| ERCC2/XPD Lys751Gln (rs13181) | |||||
| Lys/Lys | 120 | 29.2 | 132 | 36.2a | 0.039 |
| Lys/Gln | 212 | 51.6 | 160 | 43.8b | 0.031 |
| Gln/Gln | 79 | 19.2 | 73 | 20.0 | >0.05 |
| Lys/Gln + Gln/Gln | 291 | 70.8 | 233 | 63.8c | 0.039 |
| Gln | 370/822 | 45.0 | 306/730 | 41.9 | >0.05 |
| ERCC6/CSB Gly399Asp (rs2228528) | |||||
| Gly/Gly | 283 | 68.0 | 259 | 71.0 | >0.05 |
| Gly/Asp | 121 | 29.1 | 101 | 27.7 | >0.05 |
| Asp/Asp | 12 | 2.9 | 5 | 1.4 | >0.05 |
| Gly/Asp + Asp/Asp | 133 | 32.0 | 106 | 29.0 | >0.05 |
| Asp | 145/832 | 17.4 | 111/730 | 15.2 | >0.05 |
The genotypic distribution is in accordance with Hardy-Weinberg equilibrium in the control and case groups: χ 2 = 3.63 and 0.72 (p = 0.06 and 0.39) for ERCC2/XPD Lys751Gln polymorphism; χ 2 = 1.95 and 0.05 (p = 0.16 and 0.83) for ERCC6/CSB Gly399Asp polymorphism.
aOR [95% CI] = 0.73 [0.54–0.98], p = 0.039; bOR [95% CI] = 1.36 [1.03–1.81], p = 0.031; cOR [95% CI] = 1.37 [1.02–1.86], p = 0.039. OR values describe a homozygous wild type genotype of the XPD gene as a protective factor, whereas the heterozygous genotype and sum of genotypes containing a variant allele seem to be risk factors for developing bladder cancer.
The comparison of the genotype distribution depending on the tumor stages and tumor tissue differentiation (Table 7) revealed lack of differences, except for the ERCC6 Met1097Val polymorphism (rs2228526). The frequency of the ERCC6 1097Val/Val genotype was significantly increased in muscle-invasive tumors as compared to non-muscle-invasive ones (p = 0.0045), and the similar trend concerned the Val allele frequency, which was almost doubled in patients with T2 tumors as compared to Ta neoplasms (37.5% and 20.9%, resp.; p = 0.0009). These data suggested that the carriers of the ERCC6 1097Val allele, predominantly in the homozygous state, have a higher probability of developing advanced cancer, what is also indicated by the odds ratio: OR (95% CI) = 2.86 (1.36–6.05) (p = 0.0061) for the Val/Val genotype.
Table 7.
Distribution of genotypes of some DNA repair genes in non-muscle-invasive (Ta/T1) and muscle-invasive (T ≥ 2) tumors depending on their differentiation.
| DNA repair gene polymorphisms | Genotype frequency (%) depending on the tumor stages and grades | ||||||
|---|---|---|---|---|---|---|---|
| Ta/T1 | T ≥ 2 | G1 | G2 | G3 | low | high | |
| OGG1 326 rs1052133 | n = 288 | n = 126 | n = 135 | n = 185 | n = 86 | n = 240 | n = 156 |
| Ser/Ser | 67.0 | 69.0 | 71.1 | 65.9 | 67.4 | 67.5 | 69.2 |
| Ser/Cys | 28.8 | 27.0 | 24.4 | 30.8 | 27.9 | 28.8 | 26.3 |
| Cys/Cys | 4.2 | 4.0 | 4.4 | 3.2 | 4.7 | 3.8 | 4.5 |
|
| |||||||
| XRCC1 399 rs25487 | n = 288 | n = 126 | n = 135 | n = 186 | n = 86 | n = 241 | n = 156 |
| Arg/Arg | 39.6 | 44.4 | 41.5 | 38.2 | 46.5 | 39.0 | 44.2 |
| Arg/Gln | 49.0 | 44.4 | 48.9 | 48.4 | 43.0 | 48.5 | 44.9 |
| Gln/Gln | 11.5 | 11.1 | 9.6 | 13.4 | 10.5 | 12.5 | 10.9 |
|
| |||||||
| XPD 312 rs1799793 | n = 288 | n = 126 | n = 135 | n = 185 | n = 86 | n = 240 | n = 156 |
| Asp/Asp | 29.2 | 32.5 | 24.4 | 35.7 | 30.2 | 31.3 | 30.8 |
| Asp/Asn | 54.5 | 49.2 | 62.2 | 44.9 | 52.3 | 53.7 | 49.3 |
| Asn/Asn | 16.3 | 18.3 | 13.3 | 19.5 | 17.5 | 15.0 | 19.9 |
|
| |||||||
| XPD 751 rs13181 | n = 280 | n = 124 | n = 131 | n = 181 | n = 84 | n = 234 | n = 152 |
| Lys/Lys | 28.9 | 30.6 | 28.2 | 30.4 | 31.0 | 31.2 | 28.3 |
| Lys/Gln | 53.2 | 51.6 | 57.3 | 49.2 | 51.2 | 52.1 | 52.0 |
| Gln/Gln | 17.9 | 17.7 | 14.5 | 20.4 | 17.8 | 16.7 | 19.7 |
|
| |||||||
| ERCC6 1097 rs2228526 | n = 289 | n = 126 | n = 135 | n = 186 | n = 86 | n = 241 | n = 156 |
| Met/Met | 50.9 | 46.8 | 57.0 | 45.2 | 47.7 | 48.1 | 49.3 |
| Met/Val | 44.3 | 40.5 | 37.8 | 47.3 | 43.0 | 45.6 | 41.7 |
| Val/Val | 4.8 | 12.7a | 5.2 | 7.5 | 9.3 | 6.2 | 9.0 |
|
| |||||||
| ERCC6 399 rs2228528 | n = 283 | n = 126 | n = 133 | n = 183 | n = 85 | n = 236 | n = 155 |
| Gly/Gly | 67.8 | 67.5 | 65.4 | 69.4 | 67.0 | 65.7 | 69.7 |
| Gly/Asp | 29.0 | 30.2 | 31.6 | 27.3 | 30.6 | 31.4 | 27.7 |
| Asp/Asp | 3.2 | 2.4 | 3.0 | 3.3 | 2.4 | 2.9 | 2.6 |
aSignificant differences were observed between Ta/T1 and T ≥ 2 tumors with respect to frequencies of the homozygous ERCC6 1097 Val/Val genotype according to χ 2 test (p = 0.0045).
The distribution of genotypes/alleles for polymorphisms of DNA repair genes did not depend on tumor grades. However, the analysis of genetic variations in papillary neoplasms of low malignant potential (PNLMP) as compared with high grade or poorly differentiated cancer revealed some peculiarities concerning ERCC6 Met1097Val and OGG1 Ser326Cys polymorphisms (Figure 2). The frequencies of homozygous wild type genotype of ERCC6 gene and heterozygous genotype of the OGG1 gene were significantly increased in LMP tumors, whereas the genotypes containing at least one variant allele of the ERCC6 gene occurred more often in patients with G3 or high grade urothelial carcinomas. Thus, neoplasms of low malignant potential were distinct from others with respect to genotype distribution of both the OGG1 Ser326Cys and the ERCC6 Met1097Val polymorphisms.
Figure 2.
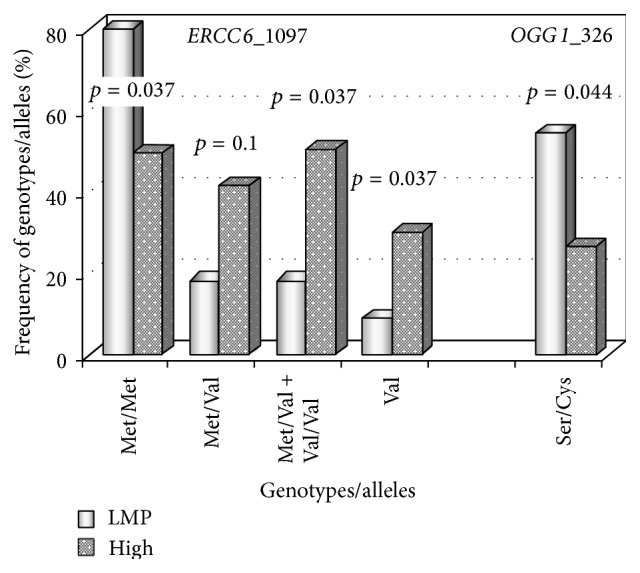
Distribution of some genotypes/alleles in patients with papillary urothelial neoplasms of low malignant potential (LMP, 11 samples) as compared to high grade carcinomas (high, 156 samples). Four coupled bars correspond to ERCC6 Met1097Val polymorphisms. The frequencies of the Met/Met genotype are 81.8% and 49.4% in LMN and high grade tumors, whereas the frequencies of the Met/Val + Val/Val genotypes are 18.2% and 50.6% in the same types of urothelial carcinomas, respectively. The last coupled bars reflect the frequencies of the OGG1 (codon 326) heterozygous genotype, which are 54.5% in LMP neoplasms and 26.3% in high grade tumors.
When dividing tumors into four categories (Ta/T1 low, Ta/T1 high, T ≥ 2 low, and T ≥ 2 high), evident differences were found only in muscle-invasive carcinomas, with the homozygous wild type genotype of the XPD gene (codon 312) being associated with low grade cancer, whereas the frequencies of genotypes containing a variant Asn allele were significantly increased in high grade neoplasms (Figure 3(a)). Two other polymorphisms (XPD Lys751Gln and ERCC6 Gly399Asp) showed similar trends, but the differences between T ≥ 2 low grade tumors and T ≥ 2 high grade carcinomas were not statistically confirmed (Figure 3(b)).
Figure 3.

Distribution of some genotypes/alleles of DNA repair genes in non-muscle-invasive and muscle-invasive tumors depending on their grades. (a) The frequencies of XPD Asp312Asn polymorphisms in Ta/T1 low grade tumors (227 samples) as compared to Ta/T1 high grade neoplasms (59 samples) as well as in T ≥ 2 low grade tumors (24 samples) as opposed to T ≥ 2 high grade neoplasms (97 samples). In the latter case, the frequencies of the Asp/Asp, Asn/Asn, and Asn/Asn+Asp/Asn genotypes and the Asn alleles were as follows: 50% and 28.9% (p = 0.049), 8.3% and 20.6% (p = 0.16), 50% and 71.1% (p = 0.049), and 29.2% and 45.9% (p = 0.036) in T ≥ 2 low grade and T ≥ 2 high grade tumors, respectively. (b) The frequencies of XPD (codon 751) and ERCC6 (codon 399) polymorphisms in T ≥ 2 low grade tumors (24 samples for each polymorphisms) as compared to T ≥ 2 high grade neoplasms (95 and 97 samples, resp.).
Based on the assumption that the wild type of DNA excision repair genes may provide elimination of mutagenic/carcinogenic DNA lesions thus promoting both decrease in cancer risk and inhibition of tumor expansion/malignancy, the frequencies of combined homozygous wild type alleles were estimated depending on T stages. The combination involving Ser/Ser, Arg/Arg, Asp/Asp, Lys/Lys, Met/Met, and Gly/Gly genotypes of OGG1 (codon 326), XRCC1 (codon 399), XPD (codons 312 and 751), and ERCC6 (codons 1097 and 399), respectively, was a rare event occurring only in Ta/T1 tumors (2.1%). As shown in Figure 4(a), the total frequency of combinations represented by any five homozygous wild type genotypes was significantly higher in non-muscle-invasive carcinomas (18.7%) as compared to advanced tumors (7.9%) and together with the former combination containing all six wild type homozygotes they achieved 20.8% in Ta/T1 neoplasms as opposed to 7.9% in T ≥ 2 tumors (Figure 4(b)). Accordingly, homozygosity for the wild type alleles of the DNA repair genes under study seemed to prevent tumor expansion. The distribution of these combinations did not depend on tumor grades.
Figure 4.

Distribution of wild type homozygous combinations in non-muscle-invasive and muscle-invasive urothelial carcinomas. 6 hmz correspond to combination of all six wild type homozygotes; 5 hmz correspond to total combinations containing any five wild type homozygotes. The Ta/T1 group was represented by 289 samples, whereas the T ≥ 2 group consisted of 120 samples.
4. Discussion
Before discussing the data, it should be noted that evaluation of bladder cancer risk implies a case-control study, but the comparison of the demographic profiles of BC patients and controls (Section 2.1 and Table 1) shows the significant differences between groups concerning tobacco smoking status that is likely to limit interpretation of the results. On the other hand, the random selection of sizeable populations, which are matched by the recruitment period, age and ethnicity, allows a higher frequency of smokers among BC patients to be considered as a disease-specific feature in support of findings that bladder cancer is an age-, gender-, and smoking-related disease [33, 34]. It should also be mentioned that the percentage of smokers in the control population reflects the situation with tobacco consumption in Belarus, and the gene-smoking relationship in bladder cancer has been previously characterized [21, 35]. A close association of bladder cancer with age and a tobacco smoking habit suggests that oxidative stress contributes to its development.
The comet assay used in the first experimental set remains a widespread and efficient tool in biomonitoring studies [36, 37]. Our approach resembles a challenge assay, which is based on detecting chromosome breakage and has developed for revealing exposure-induced DNA repair deficiency as a functional biomarker of cancer risk [38]. In our studies, basal and exogenous DNA lesions were identified as potential biomarkers of genome destabilization, and their levels as well as DNA repair kinetics after oxidative stress in vitro were measured as average group values and individually. Using this approach, we diagnosed and specified genome instability in lymphocytes of patients with some genetic disorders and occupationally exposed subjects [25–27]. Taking into account conflicting data on the relationship between age and the yield of DNA damage in the comet assay [36, 39], the age differences, in particular between groups of BC patients and individuals with chronic inflammations (Table 4), might be a limitation of the present study. However, lack of such correlation for basal and H2O2-induced DNA damage [27] confirms the reliability of our observations.
Herein, the significantly increased levels of H2O2-induced DNA damage were found in lymphocytes of BC patients as compared to controls with the pronounced effect immediately after mutagenic treatment due to dysfunction of antioxidant defense and disturbance of redox homeostasis [40, 41] rather than a reduced DNA repair rate or efficiency (Figure 1 and Table 3). However, reactive oxygen species (ROS) are known to contribute to inflammations [42, 43], aging, and related diseases [44, 45], whereas inflammations, resulting from and triggering ROS production, forego and accompany carcinogenesis [46–48]. Therefore, it was reasonable for comparing the cellular response to oxidative stress in different conditions. The initial levels of H2O2-induced DNA damage as well as the proportion of individuals with increased cellular sensitivity to hydrogen peroxide in the group of BC patients exceeded those among elderly persons and subjects with chronic inflammatory diseases (Tables 4 and 5). The results seemed to demonstrate an essential role of the abnormal cellular response to oxidatively generated DNA damage in bladder cancer.
Among various underlying mechanisms, mutations or polymorphisms in genes responsible for antioxidant defense, redox regulation, and oxidatively damaged base repair are currently studied. Our second experimental set was focused on the latter mechanism, and it would be interesting to discuss involvement of DNA helicases in removing oxidized DNA lesions. An inability to repair oxidatively generated damage accumulating in the brain was hypothesized to cause the neurological degeneration in xeroderma pigmentosum [49]. As recently reported, the neurodegeneration in Cockayne syndrome is associated with ROS-induced damage in the mitochondria, independent of nuclear transcription coupled repair [50]. Moreover, CSB protein appears to behave as an electron scavenger in the mitochondria whose absence leads to increased levels of ROS in CSB-mutated cells [50]. The CSA and CSB proteins, in addition to their basic role in TC NER, can participate in BER directly by interaction with BER proteins and indirectly by modulating gene expression [51]. Using high performance liquid chromatography coupled to electrochemical detection (HPLC-EC) to measure the genomic 8-oxoGua levels in mouse NER- or BER-deficient embryo fibroblasts [20] as well as the immunofluorescence method to detect binding of CSB and XPC to oxidative lesions in different nuclear compartments in fibroblast cell lines derived from patients [19], the experimental evidence for a direct involvement of some XP and CS gene products in repair of oxidatively induced damage has been provided. In spite of the fact that the OGG1 DNA glycosylase dominates in 8-oxoGua repair, NER (XPC and XPA) and transcription-coupled repair proteins (CSB and CSA) are similar but are minor contributors [19].
In our studies, the OGG1 (codon 326) heterozygous genotype decreased bladder cancer risk, especially in smokers with OR = 0.55 (0.34–0.89) (p = 0.014) [21] and prevented high grade tumors as compared to neoplasms of low malignant potential (Figure 2). Our findings, at least with respect to cancer predisposition, are in line with some other data [52, 53]. The ERCC6/CSB 1097Val/Val genotype enhanced susceptibility to advanced (T ≥ 2) urothelial carcinoma (Table 7), and the ERCC2/XPD 312Asn allele seemed to promote tumor malignancy, since its frequency was increased in patients with T ≥ 2 high grade tumors as compared to T ≥ 2 low grade neoplasms (Figure 3(a)). The effects associated with impaired activity of CSB and XPD proteins might be mediated by accumulation of ROS, which act as the second messengers in intracellular signaling cascades inducing and maintaining the oncogenic phenotype of cancer cells [54, 55].
It is generally accepted that the “driver” mutations in a few key genes trigger certain (sometimes alternative) pathways of cancer pathogenesis. In bladder cancer, the mutations in FGFR3 gene are strongly associated with superficial tumors, whereas mutations in TP53 gene lead to muscle-invasive cancer [56]. However, the molecular analysis of tumor tissue samples from the same Belarusian patients has shown that about 30% among them are of the wild type genotype with respect to both genes suggesting multiple genetic origins of urothelial carcinomas [57]. The current molecular-genetic analysis of bladder cancer includes the whole genome sequencing, detection of genome-wide gene expression profiles, studies of DNA repair and replication processes, the immune and inflammatory responses, and other common hallmarks of human cancers [58–62]. These investigations are aimed at revealing novel molecular markers with high predictive and prognostic relevance. In the context of our study, the results concerning a set of mutations, which were not earlier recognized as significant events for bladder cancer, are of great interest. Specifically, among 32 identified genes, there was the NER ERCC2/XPD gene, and its fifteen of sixteen genetic variations were represented by deleterious missense mutations with dominant negative effects [63].
In spite of the fact that genetic variations in excision repair genes are not attributed to driver mutations in bladder cancer [62], they may modulate susceptibility to cancer initiation and cancer progression. For example, genome-wide association studies (GWAS) have identified more than 300 validated associations between genetic variants and risk of approximately 70 common diseases [64]. The functions of genes identified as relevant for bladder cancer focus on detoxification of carcinogens, maintenance of DNA integrity, control of the cell cycle, and apoptosis. Our data indicate both the accumulation of oxidatively induced DNA damage and impact of modified XPD and CSB proteins on risk and clinical course of bladder cancer. It is typical that all known SNPs are associated with bladder cancer with odds ratios lower than 1.5; however, when interacting, they may collectively result in a substantial cancer risk [64]. Combinations of the homozygous wild type alleles are expected to exert a reverse effect. Indeed, the combined wild type homozygotes for some DNA repair genes reduced susceptibility to bladder cancer [35] and even prevented tumor expansion (Figure 4(b)).
The impact of excision repair gene polymorphisms on susceptibility to different cancers, including urothelial carcinoma, has been widely discussed in literature [21, 35]. Their associations with clinicopathological parameters of tumors are still poorly understood, although there are intriguing findings indicating the dual effects of DNA repair gene polymorphisms with respect to bladder cancer risk/recurrences/progression and clinical outcomes. Improved overall and disease-specific survivalas well as decreased mortality risk of BC patients after chemotherapy and radiotherapy was observed in carriers of variant allelesof the XPC gene [65] and the XPD 751Gln allele combined with the XPC 939Gln allele [66]. The clinical outcomes were also affected by a series of XRCC1 polymorphisms [67] and by the OGG1 326 Cys/Cys genotype [68]. Hence, so-called “risky” genotypes/alleles of some DNA repair genes decreased tumor resistance to radiation or chemical treatment, thereby improving clinical outcomes. At the same time, the “risky” ERCC6 1097Val allele increased the frequency of urothelial carcinoma recurrences [69] and the XRCC1 399 A/A (Gln/Gln) genotype greatly reduced recurrence free survival of BCG treated patients [70]. The higher frequency of muscle-invasive tumors was observed in carriers of XRCC1 194 СT+TT genotypes as compared to the wild type CC genotype [70] and in carriers of the mutated APE1 148Glu allele [71]. Our results indicating the association of some polymorphic variants of ERCC6/CSB and ERCC2/XPD genes with advanced bladder cancer (T ≥ 2 as compared to Ta/T1 tumors or T ≥ 2 high as opposed to T ≥ 2 low grade carcinomas) fit into an overall picture, but the problem needs to be further explored to confirm some regularities arising from our own and literature data.
5. Conclusion
Using the alkaline comet assay, the increased level of H2O2-induced DNA damage was found in isolated lymphocytes of BC patients as compared to healthy controls, elderly people, and individuals with chronic inflammatory diseases. The proportion of individuals with the enhanced cellular response to oxidative stress was also significantly higher among BC patients than among healthy subjects. These results showed that accumulation of oxidatively induced DNA damage might serve as a potential biomarker of genome instability predisposing to cancer.
Some excision repair gene polymorphisms modified the susceptibility to bladder cancer and were associated with clinicopathological parameters of tumors. Polymorphisms in XPD gene (codons 312 and 751) increased cancer risk, and at the same time the variant XPD 312Asn allele was significantly associated with muscle-invasive high grade tumors. Polymorphisms in ERCC6 gene (codon 1097), especially the Val/Val homozygous genotype, occurred with the higher frequency in muscle-invasive tumors as compared to non-muscle-invasive ones, and polymorphic variants in the XPD (codon 751) and ERCC6 (codon 399) genes manifested the trends resembling effects of the XPD Asp312Asn polymorphisms with respect to T ≥ 2 high grade tumors as compared to T ≥ 2 low grade carcinomas. Interestingly, the combinations of homozygous wild type genotypes were associated with non-muscle-invasive tumors and their frequency was more than twice lower in T ≥ 2 carcinomas suggesting that the maintenance of normal DNA repair activity, specifically of some XP and CS gene products, seems to inhibit cancer initiation and/or cancer progression. Based on the literature data, one can assume that their positive effects, at least in part, are mediated through elimination of mutagenic/carcinogenic oxidatively induced DNA damage.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Streffer C. Strong association between cancer and genomic instability. Radiation and Environmental Biophysics. 2010;49(2):125–131. doi: 10.1007/s00411-009-0258-4. [DOI] [PubMed] [Google Scholar]
- 2.Dizdaroglu M. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Letters. 2012;327(1-2):26–47. doi: 10.1016/j.canlet.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Bonassi S., Norppa H., Ceppi M., et al. Chromosomal aberration frequency in lymphocytes predicts the risk of cancer: results from a pooled cohort study of 22 358 subjects in 11 countries. Carcinogenesis. 2008;29(6):1178–1183. doi: 10.1093/carcin/bgn075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonassi S., El-Zein R., Bolognesi C., Fenech M. Micronuclei frequency in peripheral blood lymphocytes and cancer risk: evidence from human studies. Mutagenesis. 2011;26(1):93–100. doi: 10.1093/mutage/geq075. [DOI] [PubMed] [Google Scholar]
- 5.van Tilborg A. A. G., de Vries A., de Bont M., Groenfeld L. E., van der Kwast T. H., Zwarthoff E. C. Molecular evolution of multiple recurrent cancers of the bladder. Human Molecular Genetics. 2000;9(20):2973–2980. doi: 10.1093/hmg/9.20.2973. [DOI] [PubMed] [Google Scholar]
- 6.Bartkova J., Hořejší Z., Koed K., et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 7.Hazra T. K., Das A., Das S., Choudhury S., Kow Y. W., Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair. 2007;6(4):470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campalans A., Marsin S., Nakabeppu Y., O'Connor T. R., Boiteux S., Radicella J. P. XRCC1 interactions with multiple DNA glycosylases: a model for its recruitment to base excision repair. DNA Repair. 2005;4(7):826–835. doi: 10.1016/j.dnarep.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 9.Berquist B. R., Singh D. K., Fan J., et al. Functional capacity of XRCC1 protein variants identified in DNA repair-deficient Chinese hamster ovary cell lines and the human population. Nucleic Acids Research. 2010;38(15):5023–5035. doi: 10.1093/nar/gkq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markkanen E., Fischer R., Ledentcova M., Kessler B. M., Dianov G. L. Cells deficient in base-excision repair reveal cancer hallmarks originating from adjustments to genetic instability. Nucleic Acids Research. 2015;43(7):3667–3679. doi: 10.1093/nar/gkv222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoeijmakers J. H. J. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 12.Shuck S. C., Short E. A., Turchi J. J. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Research. 2008;18(1):64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuper J., Kisker C. DNA helicases in NER, BER, and MMR. Advances in Experimental Medicine and Biology. 2013;767:203–224. doi: 10.1007/978-1-4614-5037-5-10. [DOI] [PubMed] [Google Scholar]
- 14.Fuss J. O., Tainer J. A. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair. 2011;10(7):697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newman J. C., Bailey A. D., Weiner A. M. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(25):9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lake R. J., Fan Y. H. Structure, function and regulation of CSB: a multi-talented gymnast. Mechanisms of Ageing and Development. 2013;134(5-6):202–211. doi: 10.1016/j.mad.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fousteri M., Mullenders L. H. F. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Research. 2008;18(1):73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 18.Vélez-Cruz R., Egly J.-M. Cockayne syndrome group B (CSB) protein: at the crossroads of transcriptional networks. Mechanisms of Ageing and Development. 2013;134(5-6):234–242. doi: 10.1016/j.mad.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Menoni H., Hoeijmakers J. H. J., Vermeulen W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. Journal of Cell Biology. 2012;199(7):1037–1046. doi: 10.1083/jcb.201205149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parlanti E., D'Errico M., Degan P., et al. The cross talk between pathways in the repair of 8-oxo-7,8-dihydroguanine in mouse and human cells. Free Radical Biology and Medicine. 2012;53(11):2171–2177. doi: 10.1016/j.freeradbiomed.2012.08.593. [DOI] [PubMed] [Google Scholar]
- 21.Ramaniuk V. P., Nikitchenko N. V., Savina N. V., et al. Polymorphism of DNA repair genes OGG1, XRCC1, XPD and ERCC6 in bladder cancer in Belarus. Biomarkers. 2014;19(6):509–516. doi: 10.3109/1354750x.2014.943291. [DOI] [PubMed] [Google Scholar]
- 22.Babjuk M., Oosterlinck W., Sylvester R., et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder, the 2011 update. European Urology. 2011;59(6):997–1008. doi: 10.1016/j.eururo.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Stenzl A., Cowan N. C., De Santis M., et al. The updated EAU guidelines on muscle-invasive and metastatic bladder cancer. European Urology. 2009;55(4):815–825. doi: 10.1016/j.eururo.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 24.World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. Proceedings of the 18th WMA General Assembly; June 1964; Helsinki, Finland. http://www.wma.net/en/30publications/10policies/b3/index.html. [Google Scholar]
- 25.Savina N. V., Smal M. P., Kuzhir T. D., et al. Chromosomal instability at the 7q11.23 region impacts on DNA-damage response in lymphocytes from Williams-Beuren syndrome patients. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2011;724(1-2):46–51. doi: 10.1016/j.mrgentox.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Savina N. V., Smal M. P., Kuzhir T. D., et al. Biomarkers for genome instability in some genetic disorders: a pilot study. Biomarkers. 2012;17(3):201–208. doi: 10.3109/1354750x.2011.651157. [DOI] [PubMed] [Google Scholar]
- 27.Savina N. V., Smal M. P., Kuzhir T. D., Ershova-Pavlova A. A., Goncharova R. I. DNA-damage response associated with occupational exposure, age and chronic inflammation in workers in the automotive industry. Mutation Research—Genetic Toxicology and Environmental Mutagenesis. 2012;748(1-2):21–28. doi: 10.1016/j.mrgentox.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Tice R. R., Agurell E., Anderson D., et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environmental and Molecular Mutagenesis. 2000;35(3):206–221. doi: 10.1002/(sici)1098-2280(2000)35:3lt;206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 29.Collins A. R., Fleming I. M., Gedik C. M. In vitro repair of oxidative and ultraviolet-induced DNA damage in supercoiled nucleoid DNA by human cell extract. Biochimica et Biophysica Acta. 1994;1219(3):724–727. doi: 10.1016/0167-4781(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 30.Spitz M. R., Wu X., Wang Y., et al. Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients. Cancer Research. 2001;61(4):1354–1357. [PubMed] [Google Scholar]
- 31.Chang C.-H., Chiu C.-F., Wang H.-C., et al. Significant association of ERCC6 single nucleotide polymorphisms with bladder cancer susceptibility in Taiwan. Anticancer Research. 2009;29(12):5121–5124. [PubMed] [Google Scholar]
- 32.Kuzhir T. D., Savina N. V., Ershova-Pavlova A. A., et al. Assessment of the genome integrity in lymphocytes of people engaged in industry. Doklady of the National Academy of Sciences of Belarus. 2012;56(4):72–76. (Rus). [Google Scholar]
- 33.Janković S., Radosavljević V. Risk factors for bladder cancer. Tumori. 2007;93(1):4–12. doi: 10.1177/030089160709300102. [DOI] [PubMed] [Google Scholar]
- 34.Franekova M., Halasova E., Bukovska E., Luptak J., Dobrota D. Gene polymorphisms in bladder cancer. Urologic Oncology: Seminars and Original Investigations. 2008;26(1):1–8. doi: 10.1016/j.urolonc.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Ramaniuk V. P., Nikitchenko N. V., Savina N. V., Kuzhir T. D., Goncharova R. I. Polymorphism of excision repair genes XPD, XRCC1, and hOGG1 in the population of the republic of Belarus and its impact on carcinogenesis. Russian Journal of Genetics: Applied Research. 2015;5(2):141–154. doi: 10.1134/s2079059715020094. [DOI] [Google Scholar]
- 36.Møller P. Genotoxicity of environmental agents assessed by the alkaline comet assay. Basic and Clinical Pharmacology and Toxicology. 2005;96(1):1–42. doi: 10.1111/j.1742-7843.2005.pto960101.x. [DOI] [PubMed] [Google Scholar]
- 37.Dusinska M., Collins A. R. The comet assay in human biomonitoring: gene-environment interactions. Mutagenesis. 2008;23(3):191–205. doi: 10.1093/mutage/gen007. [DOI] [PubMed] [Google Scholar]
- 38.Au W. W., Giri A. K., Ruchirawat M. Challenge assay: a functional biomarker for exposure-induced DNA repair deficiency and for risk of cancer. International Journal of Hygiene and Environmental Health. 2010;213(1):32–39. doi: 10.1016/j.ijheh.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Piperakis S. M., Kontogianni K., Karanastasi G., Iakovidou-Kritsi Z., Piperakis M. M. The use of comet assay in measuring DNA damage and repair efficiency in child, adult, and old age populations. Cell Biology and Toxicology. 2009;25(1):65–71. doi: 10.1007/s10565-007-9046-6. [DOI] [PubMed] [Google Scholar]
- 40.Klaunig J. E., Kamendulis L. M., Hocevar B. A. Oxidative stress and oxidative damage in carcinogenesis. Toxicologic Pathology. 2010;38(1):96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 41.Dawane J. S., Pandit V. A. Understanding redox homeostasis and its role in cancer. Journal of Clinical and Diagnostic Research. 2012;6(10):1796–1802. doi: 10.7860/JCDR/2012/4947/2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C., Zhou H.-M. The role of manganese superoxide dismutase in inflammation defense. Enzyme Research. 2011;2011:6. doi: 10.4061/2011/387176.387176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffman S., Nolin J., McMillan D., Wouters E., Janssen-Heininger Y., Reynaert N. Thiol redox chemistry: role of protein cysteine oxidation and altered redox homeostasis in allergic inflammation and asthma. The Journal of Cellular Biochemistry. 2015;116(6):884–892. doi: 10.1002/jcb.25017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rybka J., Kupczyk D., Kędziora-Kornatowska K., et al. Age-related changes in an antioxidant defense system in elderly patients with essential hypertension compared with healthy controls. Redox Report. 2011;16(2):71–77. doi: 10.1179/174329211x13002357050897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haider S., Saleem S., Perveen T., et al. Age-related learning and memory deficits in rats: role of altered brain neurotransmitters, acetylcholinesterase activity and changes in antioxidant defense system. Age. 2014;36(3):1291–1302. doi: 10.1007/s11357-014-9653-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartsch H., Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbeck's Archives of Surgery. 2006;391(5):499–510. doi: 10.1007/s00423-006-0073-1. [DOI] [PubMed] [Google Scholar]
- 47.Kundu J. K., Surh Y.-J. Inflammation: gearing the journey to cancer. Mutation Research/Reviews in Mutation Research. 2008;659(1-2):15–30. doi: 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Ohnishi S., Ma N., Thanan R., et al. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxidative Medicine and Cellular Longevity. 2013;2013:9. doi: 10.1155/2013/387014.387014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lehmann A. R. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes and Development. 2001;15(1):15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- 50.Cleaver J. E., Brennan-Minnella A. M., Swanson R. A., et al. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(37):13487–13492. doi: 10.1073/pnas.1414135111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khobta A., Epe B. Repair of oxidatively generated DNA damage in Cockayne syndrome. Mechanisms of Ageing and Development. 2013;134(5-6):253–260. doi: 10.1016/j.mad.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 52.De Ruyck K., Szaumkessel M., De Rudder I., et al. Polymorphisms in base-excision repair and nucleotide-excision repair genes in relation to lung cancer risk. Mutation Research. 2007;631(2):101–110. doi: 10.1016/j.mrgentox.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Huang M., Dinney C. P., Lin X., Lin J., Grossman H. B., Wu X. High-order interactions among genetic variants in DNA base excision repair pathway genes and smoking in bladder cancer susceptibility. Cancer Epidemiology Biomarkers and Prevention. 2007;16(1):84–91. doi: 10.1158/1055-9965.epi-06-0712. [DOI] [PubMed] [Google Scholar]
- 54.Storz P. Reactive oxygen species in tumor progression. Frontiers in Bioscience. 2005;10(2):1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 55.Liou G.-Y., Storz P. Reactive oxygen species in cancer. Free Radical Research. 2010;44(5):479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Rhijn B. W. G., van der Kwast T. H., Vis A. N., et al. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Research. 2004;64(6):1911–1914. doi: 10.1158/0008-5472.can-03-2421. [DOI] [PubMed] [Google Scholar]
- 57.Smal M. P., Rolevich A. I., Polyakov S. L., Krasny S. A., Goncharova R. I. FGFR3 and TP53 mutations in a prospective cohort of Belarusian bladder cancer patients. Experimental Oncology. 2014;36(4):246–251. [PubMed] [Google Scholar]
- 58.Lindgren D., Frigyesi A., Gudjonsson S., et al. Combined gene expression and genomic profiling define two intrinsic molecular subtypes of urothelial carcinoma and gene signatures for molecular grading and outcome. Cancer Research. 2010;70(9):3463–3472. doi: 10.1158/0008-5472.CAN-09-4213. [DOI] [PubMed] [Google Scholar]
- 59.Li X., Chen J., Hu X., et al. Comparative mRNA and microrna expression profiling of three genitourinary cancers reveals common hallmarks and cancer-specific molecular events. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0022570.e22570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo G., Sun X., Chen C., et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nature Genetics. 2013;45(12):1459–1463. doi: 10.1038/ng.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morrison C. D., Liu P., Woloszynska-Read A., et al. Whole-genome sequencing identifies genomic heterogeneity at a nucleotide and chromosomal level in bladder cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(6):E672–E681. doi: 10.1073/pnas.1313580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cazier J. B., Rao S. R., McLean C. M., et al. Whole-genome sequencing of bladder cancers reveals somatic CDKN1A mutations and clinicopathological associations with mutation burden. Nature Communications. 2014;5:p. 3756. doi: 10.1038/ncomms4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Golka K., Selinski S., Lehmann M.-L., et al. Genetic variants in urinary bladder cancer: Collective power of the ‘wimp SNPs’. Archives of Toxicology. 2011;85(6):539–554. doi: 10.1007/s00204-011-0676-3. [DOI] [PubMed] [Google Scholar]
- 65.Sasaki M., Sakano S., Okayama N., et al. DNA repair gene polymorphisms may be associated with prognosis of upper urinary tract transitional cell carcinoma. Neoplasia. 2008;10(3):255–265. doi: 10.1593/neo.07982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sanyal S., De Verdier P. J., Steineck G., et al. Polymorphisms in XPD, XPC and the risk of death in patients with urinary bladder neoplasms. Acta Oncologica. 2007;46(1):31–41. doi: 10.1080/02841860600812693. [DOI] [PubMed] [Google Scholar]
- 67.Sacerdote C., Guarrera S., Ricceri F., et al. Polymorphisms in the XRCC1 gene modify survival of bladder cancer patients treated with chemotherapy. International Journal of Cancer. 2013;133(8):2004–2009. doi: 10.1002/ijc.28186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ha Y.-S., Yan C., Kim I. Y., Yun S.-J., Moon S.-K., Kim W.-J. Tissue hOGG1 genotype predicts bladder cancer prognosis: a novel approach using a peptide nucleic acid clamping method. Annals of Surgical Oncology. 2011;18(6):1775–1781. doi: 10.1245/s10434-010-1500-7. [DOI] [PubMed] [Google Scholar]
- 69.Gu J., Zhao H., Dinney C. P., et al. Nucleotide excision repair gene polymorphisms and recurrence after treatment for superficial bladder cancer. Clinical Cancer Research. 2005;11(4):1408–1415. doi: 10.1158/1078-0432.CCR-04-1101. [DOI] [PubMed] [Google Scholar]
- 70.Mittal R. D., Singh R., Manchanda P. K., et al. XRCC1 codon 399 mutant allele: a risk factor for recurrence of urothelial bladder carcinoma in patients on BCG immunotherapy. Cancer Biology and Therapy. 2008;7(5):645–650. doi: 10.4161/cbt.7.5.5763. [DOI] [PubMed] [Google Scholar]
- 71.Narter K. F., Ergen A., Agaçhan B., Görmüs U., Timirci Ö., Isbir T. Bladder cancer and polymorphisms of DNA repair genes (XRCC1, XRCC3, XPD, XPG, APE1, hOGGl) Anticancer Research. 2009;29(4):1389–1394. [PubMed] [Google Scholar]