

In the past several months, a remarkable transformation occurred in how the field regards lipoproteins, statins, and atherosclerosis. The straw – or tree trunk – that broke this stubborn camel’s back was the prospective, randomized, double-blinded IMPROVE-IT trial.1 This key study demonstrated that lowering plasma LDL concentrations in humans through the use of a non-statin – ezetimibe – not only reduced cardiovascular events, but did so to exactly the same extent as LDL lowering by statins. Preliminary data in humans suggest that the same is true of the new PCSK9 inhibitors, which are also non-statins.2, 3 Abundant prior data have told the same story for decades (reviewed in a recent AHA Council Statement4). For example, lowering plasma LDL concentrations surgically by partial ileal bypass, or by random Mendelian inheritance of key polymorphisms that lower plasma levels of LDL, or polymorphisms that lower levels of cholesterol- and triglyceride-rich apolipoprotein-B (apoB)-containing remnant lipoproteins, also reduce human atherosclerotic cardiovascular events.5, 6
Though a variety of pleiotropic effects are often attributed to statins, such as acting as anti-oxidants or anti-inflammatory agents, the bulk of the evidence supports that statins – and ezetimibe – reduce long-term human cardiovascular risk because they do one thing well. They lower plasma LDL levels.
Atherosclerosis has become as pathogenically simple as tuberculosis. What causes TB? Mycobacterium tuberculosis. What causes atherosclerosis? LDL and other cholesterol-rich, apoB-lipoproteins. Diabetes and smoking increase the risk of TB – and atherosclerosis – but cannot cause either disease on their own. Tuberculosis and atherosclerosis both involve extensive, and strikingly similar, host immune responses, including a persistent infiltrate of macrophages and T-cells, the development of foam cells, local induction of many of the same anti-emigration molecules that keep these cells in place,7, 8 and systemic elevations in so-called inflammatory markers, such as plasma C-reactive protein. But no primary immune derangement has ever been shown to cause TB in the absence of the bacillus – nor to cause atherosclerosis in the absence of abundant apoB-lipoproteins.
In this context, the interactions of LDL and other cholesterol-rich, apoB-lipoproteins with the vessel wall are now paramount to understanding how a normal artery becomes atherosclerotic, and how an existing atherosclerotic plaque worsens, stabilizes, or heals. The new study by Bartels, Christoffersen, et al. adds fresh insight by comparing the way established murine arterial plaques handle LDL during progression versus regression.9
Prior literature indicates that a key process initiates atherogenesis – namely, the subendothelial retention, or trapping, of plasma-derived apoB-lipoproteins, particularly LDL and remnants.10–12 In earliest atherogenesis, the affinity of specific domains of apoB to adhere directly to specific elements of the arterial matrix, particularly at branch points and other areas of non-laminar flow, drives lipoprotein retention.10–13 The retained lipoproteins become modified by arterial-wall enzymes and other processes to form a uniquely dangerous accumulation (Figure 1A). The resulting material provokes a series of strikingly maladaptive responses that include endothelial dysfunction and the recruitment and abnormal persistence of macrophages and T-cells.10–12, 14 Cellular and molecular programs that cause immune cells to remain in place may be adaptive for tuberculosis, an acid-fast bacillus that is not killed after phagocytosis and would therefore be spread throughout the body by emigrating macrophages. But retained and modified apoB-lipoproteins within the arterial wall inappropriately elicit many of the same evolutionarily conserved anti-emigration signals.7, 12, 15 The result is a crippling of the reticuloendothelial system, which otherwise has a huge capacity that could easily handle a few grams of intramural cholesterol and other debris.
Figure 1.
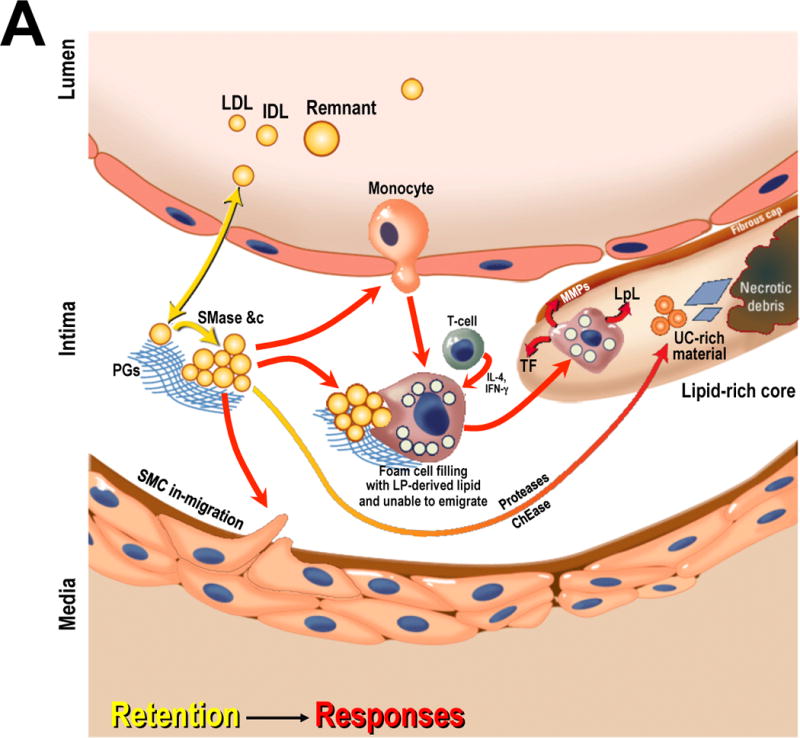
The central role of cholesterol-rich, apoB-lipoproteins in atherosclerosis initiation, progression, and regression.
Panel A: Initiation and progression of atherosclerosis. Arrows are color coded to indicate crucial mechanisms in the retention of cholesterol-rich apoB-lipoproteins within the arterial wall, which is the key initiating step in atherogenesis (yellow); and then maladaptive local responses to the retained and modified lipoproteins that lead to plaque growth and evolution (red).
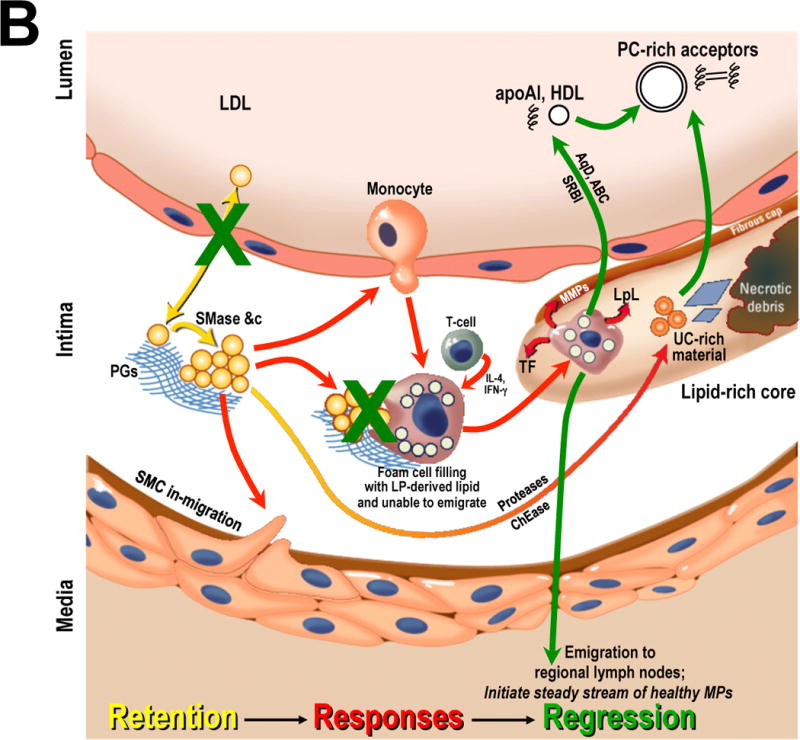
Panel B: Plaque stabilization, resolution of maladaptive sterile inflammation, and atherosclerosis regression. Drastic improvements in the plasma lipoprotein profile allow arterial healing. Shown schematically are vast reductions in circulating concentrations of apoB-lipoproteins combined with an increase in functional HDL and other putative acceptors of exchangeable material from the plaque. The green arrows indicate previously reported mechanisms for resolution of an established plaque. The two green Xs indicate new mechanisms found by Bartels, Christoffersen, et al. – namely, an improved endothelial barrier to LDL and a drop in the fractional degradation of LDL that enters the plaque during early regression.9
Abbreviations: ABC, ATP-binding cassette transporters; apoA-I, apolipoprotein A-I; AqD, aqueous diffusion; ChEase, cholesteryl esterase; IDL, intermediate-density lipoprotein; IFN-γ, interferon γ; IL-4, interleukin 4; LP, lipoprotein; LpL, lipoprotein lipase; MMPs, matrix metalloproteinases; MPs, macrophages; PC, phosphatidylcholine; PGs, proteoglycans; Remnant, cholesterol-rich apoB-containing remnant lipoprotein; SMase, secretory sphingomyelinase; SMC, smooth muscle cell; SRBI, scavenger receptor class B type I; TF, tissue factor; UC, unesterified cholesterol. Adapted from reference12 with permission.
In atherosclerosis, persistent macrophages secrete lipases that perversely accelerate further retention and modification of apoB-lipoproteins within the developing plaque. Many of these cells die but their carcasses fail to undergo normal disposal by phagocytosis (efferocytosis), leading to necrotic core formation and further harmful immune activation.14 Persistent, living macrophages in an atheroma also release proteases, which weaken the overlying fibrous cap, and tissue factor, which ensures vigorous clot formation upon plaque rupture – and hence the risk of arterial occlusion (Figure 1A, reviewed in12, 15).
What about the other direction? How can an existing atherosclerotic plaque stabilize or heal? Over a half-century of studies on experimental atherosclerosis in animals, including non-human primates, indicates that essentially all features of advanced atherosclerotic plaques can reverse, including seemingly permanent changes such as necrosis and extracellular lipid accumulations, including crystalline material.15 The key is drastic improvements in the plasma lipoprotein profile, most commonly, vast reductions in total cholesterol or LDL. Plaque stabilization and shrinkage have been documented in humans as well, and we and others are convinced that these effects will become easier to achieve clinically with new, more potent LDL-lowering agents.
Studies on the cellular infiltrate in experimental atherosclerotic plaques indicate that regression is not merely a rewinding of progression, but instead a resolution response that involves emigration of the maladaptive macrophage infiltrate, followed by the initiation of a stream of healthy, normally functioning phagocytes that clear retained apoB-lipoproteins, apoptotic cells, necrotic debris, and other components of advanced plaques (Figure 1B and references7, 15, 16).
Nevertheless, there has been a conspicuous gap in our knowledge of the interactions of LDL and other cholesterol-rich, apoB-lipoproteins with the vessel wall during plaque regression. As far as we know, Bartels, Christoffersen, et al. are the first to address this crucial issue.9 Their experimental system was LDL receptor-negative mice fed on a high-cholesterol diet for approximately 4–5 months, to produce sustained elevations in plasma LDL concentrations and the formation of atherosclerotic plaques in the aorta. Then, weekly doses of an anti-Apob antisense oligonucleotide (ASO) corrected the hypercholesterolemia. Concentrations of LDL and VLDL in plasma fell to nearly undetectable levels within 2–3 days of the first administration of the anti-Apob ASO. Control animals received a partially mismatched ASO and remained hypercholesterolemic. In an impressive effort, the authors assessed key morphologic, genetic, and metabolic properties of the aortic plaques just one week or four weeks after beginning ASO treatments.
The experimental system has several noteworthy features that strengthen the study. First, the primary atherogenic lipoprotein was LDL, which remains a key disease-causing lipoprotein in humans and the major successful therapeutic target for atherosclerosis. Many studies of atherosclerosis progression and regression use the apoE-deficient mouse, but its major lipoprotein is a highly abnormal particle called ß-VLDL that enters the arterial wall and becomes retained there, but can be handled somewhat differently from LDL. Second, the atherosclerotic plaques in the Bartels/Christoffersen study exhibited necrosis and fibrosis, i.e., advanced well beyond just fatty streaks. Third, the animals remained on the high-cholesterol diet even during administration of the ASOs, despite the ability of high-cholesterol diets to provoke sterile inflammation.17 Fourth, plasma HDL cholesterol concentrations, though readily detectable, did not rise during the regression period in animals treated with the anti-Apob ASO. Other studies in mice indicate that a fall in plasma apoB-lipoprotein levels, combined with a rise in functional HDL, produces the most rapid regression of established plaques.18 Although morphologic regression of atheromata in the current study in four weeks may seem fast, the rate is leisurely compared with that seen in other models and fits with this prior work. As noted below, the absence of morphologic resolution at one week simplifies interpretation of the study. Fifth, the initial time point after ASO administration is listed as one week, but because the fall in plasma LDL concentration was not instantaneous, it may be more like 4–5 days.
The four-week time point after initiation of anti-Apob ASO injections – a non-statin – proves that this was an environment for plaque regression and repair: foam cell content decreased, collagen increased, and gene expression patterns clearly improved by then. All of these changes are consistent with previously reported features of atherosclerosis regression (green arrows in Figure 1B; see also references7, 15, 16). But the one-week time point is the more mechanistically fascinating. At one week, neither morphologic changes – content of macrophages, foam cells, necrotic material, or even the size of the total pool of arterial-wall LDL – nor changes in gene expression – including mRNAs for allegedly key cytokines and scavenger receptors – were evident. Yet at one week, the two major findings of this study were already apparent. Endothelial permeability for LDL entry into the plaques decreased, and the fractional degradation of LDL that still made it into the plaques fell dramatically (green Xs in Figure 1B).
How can we explain an improved endothelial barrier to LDL after rapid correction of hypercholesterolemia? Alterations in endothelial permeability, though apparently not essential to lesion development, may play a contributory role, but only if some of the infiltrated apoB-lipoproteins become retained. For example, early prelesional accumulation of apoB-lipoproteins within the arterial wall is focally concentrated in sites that are known to be prone to the later development of atheromata, particularly in areas near non-laminar flow, but the rates of LDL entry into prelesional susceptible versus prelesional resistant sites are not different.10–13 Surprisingly, we still do not completely understand how LDL crosses the endothelium into or out of the arterial wall, nor do we understand what makes the local endothelium more permeable to LDL once a plaque develops underneath. Bartels, Christoffersen, et al. ruled out a non-specific process by showing that permeability to Evans blue dye does not change during that crucial first week of anti-Apob ASO treatment. Caveolin-1 – and by implication unesterified cholesterol in the membrane – might be involved: knock-out of this abundant endothelial protein impedes LDL from crossing into the arterial wall and slows atherogenesis in hypercholesterolemic mice.19, 20
A crucial question remains: what pool of LDL might be acting on the endothelium to alter permeability to apoB-lipoproteins during progression or regression of plaques? In the current study, the endothelial barrier might have improved after anti-Apob ASO treatment because there was less LDL in the plasma flowing past these cells so that, say, the endothelial plasma membrane might become less enriched in unesterified cholesterol through aqueous diffusion. Or the key effect of anti-Apob ASO treatment could have been less LDL to pass through the endothelium, a process that itself might induce cellular changes. There would have been less newly retained and aggregated LDL under the endothelium to release fatty acids, modified phospholipids, sterols, ceramides, and other potentially biologically active molecules. Anti-Apob ASO treatment decreased the amount of newly degraded LDL, which can be another source of harmful products within the plaque.21 There could also be a pool of LDL that induces rapidly reversible alterations in other elements of the plaque that were not assessed but communicate with the endothelium.
It should be experimentally feasible to distinguish roles for these different pools of LDL – and the distinct mechanisms by which they could act on the arterial endothelium. For example, would high plasma concentrations of LDL made from mutated apoB100 that lacks its domains for binding arterial matrix – a previous technical triumph from the Borén laboratory11 – still alter endothelial function? Or LDL in the presence of antibodies that block its binding to arterial matrix and hence atherogenesis?22 Or LDL after knock-out or overexpression of specific arterial-wall enzymes that are known to accelerate apoB-lipoprotein retention, aggregation, and the release of lipolytic products?10, 23, 24 Would direct injection of LDL into the subendothelial cushion impair endothelial function? To our knowledge, this last approach has not been tried since the 1930s.25
One clue comes from prior studies showing that intramural retention and aggregation of apoB-lipoproteins occur within mere minutes to hours after the onset of hypercholesterolemia, long before the earliest known signs of endothelial dysfunction. The most straightforward conclusion is that early alterations in endothelial function cannot be a cause, and may be a consequence, of the initial retention and modification of apoB-lipoproteins within the arterial wall.10 We speculate that newly retained and modified LDL may be the most pathogenic pool during regression as well – and that shrinkage of the freshest pool of retained material, as opposed to the total pool of arterial-wall LDL, allows the overlying endothelium to recover in a few days. As Bartels, Christoffersen, et al. note, the shapes of the kinetic curves suggest a lag in labeled LDL entry, especially after treatment with the anti-Apob ASO; future studies should confirm and explain this intriguing pattern.
We are on somewhat firmer ground when trying to understand the drop in the fractional degradation of LDL that entered the plaque during early regression. Again, the authors ruled out a non-specific effect: phagocytic and pinocytic capacities of the cells in the plaques did not measurably decrease during the first week of anti-Apob ASO treatment. At that initial time point, the lack of any significant change in macrophage expression of mRNAs for classical scavenger receptors, mentioned above, is consistent with prior work showing that these molecules are unnecessary for full foam cell formation in murine atherosclerosis.26
Instead, the authors’ data suggest less degradation of retained LDL because there was less modification of these particles within the arterial wall, inferred in part from a decrease in overall protein nitrosylation within plaques after one week of anti-Apob ASO treatment. One possibility would be lower local levels of enzymes, such as the secretory sphingomyelinase, that promote apoB-lipoprotein retention and aggregation. Calmer endothelium secretes less secretory sphingomyelinase basolaterally.23 Moreover, less cholesterol entry into nearby macrophages would decrease their production of damage-associated molecular patterns that activate endothelium.27 An additional option would be decreased expression of known receptors on macrophages for uptake of enzymatically aggregated LDL, such as LRP128 and the syndecan-4 heparan sulfate proteoglycan.24 But these are only hypotheses. The mechanistic chain – from rapid correction of atherogenic hypercholesterolemia, to these or other molecular effects, to a sharp reduction in intramural LDL degradation – remains to be discovered.
Most importantly, the rapid decreases in permeability to LDL and in the fractional degradation of LDL that enters the plaque may turn out to be crucial for subsequent plaque stabilization, resolution of maladaptive sterile inflammation, and atherosclerosis regression. The role of these effects in altering the balance between pro- and anti-emigration signals for immune cells within the plaque,7 production and stability of lipid pro-resolving mediators such as resolvins and lipoxins,14 and, as noted above, the generation of harmful byproducts from modification and degradation of retained LDL, will be fruitful areas for study.
Overall, this work provides novel information about endothelial permeability to LDL and then intramural degradation of newly entered LDL during a crucial early time point in a model of atherosclerosis regression. These are completely new findings, in an area of considerable scientific and clinical interest, particularly as stronger LDL-lowering medicines – statins and non-statins – have become approved for clinical use.
Acknowledgments
Sources of funding
The authors acknowledge support from NIH-USA grants HL084312 (EAF), HL075662 and HL107497 (IT), and HL38956 (KJW). Support is also acknowledged from the Swedish Heart-Lung Foundation (Hjärt-Lungfonden), the Ruth and Yonatan Ben-Avraham Fund, and the American Heart Association (KJW).
Footnotes
Disclosure
The authors declare no competing interests.
Contributor Information
Kevin Jon Williams, Email: kjwilliams@temple.edu, Kevin-Jon.Williams@wlab.gu.se.
Ira Tabas, Email: iat1@columbia.edu.
Edward A. Fisher, Email: Edward.Fisher@nyumc.org.
References
- 1.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM, for the IMPROVE-IT Investigators Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 2.Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJ, for the ODYSSEY LONG TERM Investigators Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 3.Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA. Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators ular events. N Engl J Med. 2015;372:1500–9. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- 4.Hegele RA, Gidding SS, Ginsberg HN, McPherson R, Raal FJ, Rader DJ, Robinson JG, Welty FK. Nonstatin low-density lipoprotein–lowering therapy and cardiovascular risk reduction—statement from ATVB Council. Arterioscler Thromb Vasc Biol. 2015 doi: 10.1161/ATVBAHA.115.306442. (ePub ahed of print 16 Sept) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Holm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, König IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schafer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36. doi: 10.1016/j.jacc.2012.08.1026. [DOI] [PubMed] [Google Scholar]
- 7.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bracho-Sanchez E, Ramkhelawon B, Desvignes L, Moore K, Ernst J. Induction of neural guidance molecule expression in macrophages and dendritic cells by Mycobacterium tuberculosis. J Immunol. 2013;190:55.9. (Meeting Abstract Supplement) [Google Scholar]
- 9.Bartels ED, Christoffersen C, Lindholm MW, Nielsen LB. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ Res. 2015;117:xxx–xxx. doi: 10.1161/CIRCRESAHA.115.307182. [in this issue] [DOI] [PubMed] [Google Scholar]
- 10.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL, Borén J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. doi: 10.1038/nature00804. [DOI] [PubMed] [Google Scholar]
- 12.Williams KJ, Tabas I. Lipoprotein retention – and clues for atheroma regression. Arterioscler Thromb Vasc Biol. 2005;25:1536–1540. doi: 10.1161/01.ATV.0000174795.62387.d3. [DOI] [PubMed] [Google Scholar]
- 13.Steffensen LB, Mortensen MB, Kjolby M, Hagensen MK, Oxvig C, Bentzon JF. Disturbed laminar blood flow vastly augments lipoprotein retention in the artery wall: a key mechanism distinguishing susceptible from resistant sites. Arterioscler Thromb Vasc Biol. 2015;35:1928–35. doi: 10.1161/ATVBAHA.115.305874. [DOI] [PubMed] [Google Scholar]
- 14.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5:91–102. doi: 10.1038/ncpcardio1086. [DOI] [PubMed] [Google Scholar]
- 16.Daoud AS, Jarmolych J, Augustyn JM, Fritz KE. Sequential morphologic studies of regression of advanced atherosclerosis. Arch Pathol Lab Med. 1981;105:233–9. [PubMed] [Google Scholar]
- 17.Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A, 3rd, Kirk EA, O’Brien KD, Chait A. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:685–91. doi: 10.1161/ATVBAHA.107.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–71. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frank PG, Lisanti MP. Caveolin-1 and caveolae in atherosclerosis: differential roles in fatty streak formation and neointimal hyperplasia. Curr Opin Lipidol. 2004;15:523–9. doi: 10.1097/00041433-200410000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Fernández-Hernando C, Yu J, Suárez Y, Rahner C, Dávalos A, Lasunción MA, Sessa WC. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009;10:48–54. doi: 10.1016/j.cmet.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ketelhuth DF, Rios FJ, Wang Y, Liu H, Johansson ME, Fredrikson GN, Hedin U, Gidlund M, Nilsson J, Hansson GK, Yan ZQ. Identification of a danger-associated peptide from apolipoprotein B100 (ApoBDS-1) that triggers innate proatherogenic responses. Circulation. 2011;124:2433–2443. doi: 10.1161/CIRCULATIONAHA.111.051599. [DOI] [PubMed] [Google Scholar]
- 22.Brito V, Mellal K, Portelance SG, Pérez A, Soto Y, deBlois D, Ong H, Marleau S, Vázquez AM. Induction of anti-anti-idiotype antibodies against sulfated glycosaminoglycans reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2012;32:2847–54. doi: 10.1161/ATVBAHA.112.300444. [DOI] [PubMed] [Google Scholar]
- 23.Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, Tabas I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase. Implications for early atherogenesis and ceramide-mediated cell signaling. J Biol Chem. 1998;273:4081–4088. doi: 10.1074/jbc.273.7.4081. [DOI] [PubMed] [Google Scholar]
- 24.Boyanovsky BB, Shridas P, Simons M, van der Westhuyzen DR, Webb NR. Syndecan-4 mediates macrophage uptake of group V secretory phospholipase A2-modified low density lipoprotein. J Lipid Res. 2009;50:641–650. doi: 10.1194/jlr.M800450-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christianson OO. Observations of lesions produced in arteries of dogs by injection of lipids. Lipids injected: human fat, fatty acids, soaps and cholesterol. Arch Pathol. 1939;27:1011–1020. [Google Scholar]
- 26.Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu ML, Scalia R, Mehta JL, Williams KJ. Cholesterol-induced membrane microvesicles as novel carriers of damage-associated molecular patterns: mechanisms of formation, action, and detoxification. Arterioscler Thromb Vasc Biol. 2012;32:2113–2121. doi: 10.1161/ATVBAHA.112.255471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakr SW, Eddy RJ, Barth H, Wang F, Greenberg S, Maxfield FR, Tabas I. The uptake and degradation of matrix-bound lipoproteins by macrophages require an intact actin cytoskeleton, Rho family GTPases, and myosin ATPase activity. J Biol Chem. 2001;276:37649–58. doi: 10.1074/jbc.M105129200. [DOI] [PubMed] [Google Scholar]