

SYNOPSIS
Neuroendocrine tumors (NETs) are a diverse group of neoplasms that can arise in a variety of locations throughout the body and often metastasize early. A patient’s only chance for cure is surgical removal of the primary tumor and all associated metastases, although even when surgical cure is unlikely, patients can benefit from surgical debulking of their disease. A thorough preoperative workup will often require multiple clinical tests and imaging studies to locate the primary tumor, delineate the extent of the disease, and assess tumor functionality. This review will discuss the biomarkers important for the diagnosis of these unique tumors and the imaging modalities that are most helpful for surgical planning.
Keywords: pancreas neuro-endocrine tumors, carcinoid, octreoscan, gut hormones
INTRODUCTION
There has been a marked increase in the incidence of NETs over the past several decades, from approximately 1 case per 100,000 in 1973 to 5 cases per 100,000 in 2004.1 The reasons for this increase are unclear and could be due to increased environmental exposures, a greater understanding and awareness of these tumors, and the parallel, marked increased use of anatomic imaging studies over this period. Regardless of the cause, these tumors have gone from rare to commonplace, and clinicians need tools to help differentiate NETs from other neoplasms. Furthermore, 30% of patients with small bowel (SBNETs) and 64% of pancreatic NETs (PNETs) present with metastatic disease,1 and determining the primary NET site of origin is critical for guiding future surgical and medical therapy. This review will describe the different modalities commonly used in the diagnosis and follow-up of gatroenteropancreatic (GEP) NETs, including biochemical markers, gene expression tests, radiologic and nuclear medicine imaging.
BIOCHEMICAL MARKERS FOR GEP NETs
Approximately 50 years ago, Pearse proposed that all peptide-producing cells of the gut, pancreas, and to a lesser extent, the anterior pituitary gland, belonged to a larger system that shared similar chemical, ultrastructural, and functional characteristics.2 This was called the diffuse neuroendocrine cell system and Pearse held that all of these cells were of neural crest origin. GEP NETs were postulated to derive from a common endocrine progenitor termed amine precursor uptake and decarboxylation (APUD) cell. Neoplasms arising from this system are defined as epithelial neoplasms with predominant neuroendocrine differentiation.3 One property shared by these cells and their respective tumors is staining with neuroendocrine immunohistochemical (IHC) markers chromogranin A (CgA) and synaptophysin.4 Another is that approximately 80% of neuroendocrine tumors express the somatostatin subtype 2 receptors (SSTR2),5,6 allowing for the use of synthetic somatostatin (congeners) in the diagnosis and management of these neuroendocrine tumors.5–9 It has been suggested that the long latency period of NETs (up to 9 years for midgut carcinoids)10 may be related to the inhibitory and anti-proliferative action of native somatostatin and its congeners via membrane receptor coupling.5,7,8,11
NETs may occur throughout the body, including the lung (bronchial carcinoids), thyroid (medullary thyroid cancer), adrenal gland (pheochromocytoma), gastrointestinal (GI) tract (stomach, duodenum, jejunum and ileum, colon and rectum), pancreas, and the skin (Merkel cell carcinoma). This is not surprising since the cells of the diffuse neuroendocrine system have come to reside normally in these various organs and tissues. These tumors produce amines and peptides that can be exploited for diagnosis and followed for response to therapy (Table 1). These secreted substances may cause symptoms that give clues as to tumor location and are ideal markers to be selected for biochemical testing.10 This review will focus upon NETs of the gastroenteropancreatic (GEP) system, which may be functional (cause symptoms) or nonfunctional. The most frequently encountered GEP NETs are of the small bowel (SBNETs, or carcinoid tumors) and pancreas (PNETs), which account for approximately 70–75% of all tumors of the diffuse neuroendocrine system in humans.
Table 1.
Biochemical tests used for GEP NETs
| (Neuro) Peptide/Amine | Tumor | Value | Interfered with by |
|---|---|---|---|
| Urine 5-HIAA | GI NETs | Elevated in 88% midguts, 30% foreguts, rare in hindguts | Tryptophan-rich foods, caffeine, wine, several medications (see text) |
| Serotonin | GI NETs Some PNETs |
Elevated in 96% midgut, 43% foregut, 20% hindgut | Lithium, MAO inhibitors, morphine, methyldopa, reserpine |
| Chromogranin A | GI NETs PNETs |
80–90% midgut and foregut, most hindgut Useful to follow debulking, recurrence, progression |
Somatostatin analogues, PPIs, renal insufficiency, cirrhosis, CHF May also be elevated in HCC* and MTC♮ |
| Pancreastatin | GI NETs PNETs |
Elevated in 80% GI NETs Useful to follow debulking, recurrence, progression |
Renal insufficiency Medications affecting insulin levels |
| Neurokinin A | GI NETs | Elevated in 21–70% of midgut carcinoids Indicates poor prognosis if elevated |
Medications for hypertension, pain, and GI function |
| Gastrin | Gastrinoma | Elevated in 98% Should also have hyperchlorhydria, high basal acid ouput |
PPIs; atrophic gastritis/pernicious anemia, diabetic gastroparesis, gastric outlet obstruction, short bowel syndrome, retained antrum, H. pylori infection |
| Insulin | Insulinoma | Elevated in 98% Hypoglycemia with 72 h fast |
Exogenous recombinant insulin |
| Glucagon | Glucagonoma | Useful when syndrome is present | DM, acute burns and trauma, cirrhosis, renal failure, Cushing’s syndrome, bacteremia |
| VIP | VIPoma | Useful when syndrome is present | Recent radioisotope administration |
| Somatostatin | Somatastatinoma | Useful when syndrome is present | MTC, small cell lung cancer, pheochromocytoma |
| Pancreatic Polypeptide | PPoma | Good marker for nonfunctional PNETs and co-secreted with hormone in many functional PNETs | Other PNETS, nesidioblastosis, PP cell hyperplasia, renal dysfunction |
HCC: hepatocellular carcinoma
MTC: medullary thyroid carcinoma
Gastrointestinal NETs
The derivation of the term “carcinoid” (carcinoma-like, karzinoide) is credited to Oberndorfer, whose series of 6 cases published in 1907 identified what was thought to be a form of benign neoplasia.12,13 Carcinoid tumors of the small intestine account for approximately 55% of all adult neuroendocrine tumors,10 and 28–44% of all malignant tumors of the small bowel.14,15 Its incidence has increased 4-fold between 1973 and 2004 (from 2.1 to 9.3 cases per million), and it has transcended adenocarcinoma as the most common cancer type of the small bowel in 2000.15 The neuroendocrine cell giving rise to small bowel carcinoids of the jejunum and ileum is the Kulchitsky-enterochromaffin (EC) cell,16 which is a gut epithelial cell that contains secretory granules that store and release serotonin (5-hydroxytryptamine) and other peptides (such as CgA, synaptophysin, and substance P).4 They are actually derived from enterocyte stem cells, rather than from neural crest cells, as first proposed by Pearse.17
The majority of serotonin in the body (>90%) is produced in the GI tract, which is metabolized by monoamine oxidase (MAO) into its breakdown product 5-hydroxyindole acetic acid (5-HIAA) in the liver and lung, and then excreted into the urine. When SBNETs are metastatic to the liver, serotonin may not be metabolized prior to its release into the systemic circulation. Sustained serotonin elevation results in the carcinoid syndrome. Serotonin is normally released by EC cells in response to pressure (food), certain nutrients, and bacteria. It acts upon the neurons within the gut to stimulate peristalsis.18 Only 5–10% of persons with carcinoid tumors will have carcinoid syndrome, and 76% of these patients will have diarrhea, which may be secretory, hypermotile, malabsorptive, or obstructive.11 Facial flushing may affect 80% of persons with carcinoid syndrome and is usually episodic. It may be precipitated by catecholamine-driven emotion, excitability, exercise, decongestants, and eating. This symptom is generally mediated by kallikrein, bradykinins, substance P, histamines and other peptides, rather than serotonin. The episodic spikes of serotonin levels are often associated with hypotension, which worsens the catecholamine-driven serotonin release.11 Serotonin (and possibly the tachykinin, substance P) is associated with severe bronchial wheezing, which occurs in about 20% of patients with carcinoid syndrome. Cardiac fibrosis with right-sided valvular disease is seen in as many as 50% of patients.11,19 The primary mediator is thought to be serotonin, but substance P may contribute as well. Some patients may also develop pellagra, due to niacin depletion resulting from tryptophan being shunted to serotonin synthesis rather than nicotinic acid.
LABORATORY TESTS AND BIOMARKERS FOR GASTROINTESTINAL NETs
In the past, the gold standard for the biochemical diagnosis of carcinoid tumors was the measurement of the serotonin metabolite 5-HIAA in a 24-hour urine collection. This remains a useful test, with elevation found in 88% of carcinoid patients20, but can be falsely elevated by a variety of tryptophan-rich foods (cheese, bananas, kiwis, walnuts, tomatoes, pineapples, spinach, eggplant, avocados), wine, caffeine, and various medications (acetaminophen, monoamine oxidase inhibitors, isoniazid, phenothiazines, iodine, 5-fluorouracil). It is less commonly elevated in those with foregut tumors and hindgut tumors.21 It has a high sensitivity (approaching 100%), but low specificity (35%).22 Limitations of the test are its inconvenience for the patient and that it may be negative in those with low volume disease (such as a patient with a small bowel primary without nodal or liver metastases). Plasma assays for 5-HIAA are now available, and may become another useful tool in the management of these patients.23
The majority of serotonin in the blood is stored within platelets, and measurement of whole blood serotonin has been improved by performing liquid chromatography from platelet-rich plasma68. The plasma serotonin assay is now considered very reliable for the diagnosis of carcinoids when performed by CLIA-licensed and College of American Pathology (CAP)-approved commercial laboratories in the United States. This test a positive predictive value of 89% and negative predictive value of 93% of patients with midgut carcinoids, but is less accurate in those with foregut and hindgut carcinoids.21 It may not correlate as well with the tumor burden as other laboratory assays (e.g. chromogranin, pancreastatin) because platelets become saturated at high levels of serotonin. Excess serotonin remains unbound and continues to circulate in the blood.24 This assay can also be falsely elevated in patients taking lithium, MAO inhibitors, morphine, methyldopa, and reserpine.25
Chromogranin A is a 457 amino acid peptide that is widely distributed in endocrine and neuroendocrine tissues, is present in normal islet cells, and co-secreted from EC cells with serotonin. Its normal function is to promote formation of secretory granules and it serves as the precursor protein for several negative regulators of neuroendocrine cells (pancreastatin, vasostatin, catestatin). Serum CgA levels are considered one of the most useful markers for diagnosis and surveillance of patients with gastrointestinal (GI) NETs, including hindgut and foregut tumors, where 5-HIAA and serotonin levels are often within normal limits.26 The sensitivity depends on the specific assay used, but ranges from 67–93%, and the specificity from 85–96%.27 Levels of CgA may also be useful for determining prognosis, as patients with CgA >200 U/L have a lower median survival than those <200 U/L (2.1 vs. 7 years, respectively).28 Chromogranin A is also a useful marker for determining the efficacy of debulking procedures, disease recurrence, and progression.29,30 Unlike serotonin, CgA levels maintain a good relationship with overall tumor burden, even when circulating levels are high. Chromogranin A levels are increased by somatostatin analogues, use of proton pump inhibitors (but not H2 blockers), atrophic gastritis/pernicious anemia, and renal insufficiency.31
Pancreastatin is a 52 amino acid derivative of CgA. Its primary function is to decrease cellular glucose uptake.32 It is a useful marker for diagnosis, the effect of debulking, and for tumor progression.33 It is elevated in as many as 80% of patients with GI NETs.34 In contrast to the CgA assay, it is not affected by PPI or somatostatin analogue use. The pancreastatin assay does not cross-react with CgA.35 Pancreastatin is a useful marker for GI NET prognostication.34,36 and more accurately predicted patient outcome in SBNET and PNET patients than did serial measurements of CgA, serotonin, and neurokinin A (NKA) in one recent study.37 Patients with SBNETs and preoperative elevation of pancreastatin had a median progression free survival (PFS) of 1.7 years versus 6.5 years when this was normal. If pancreastatin normalized after surgery, PFS improved to 4.2 years (compared to 1.6 years if this remained high postoperatively).
Neurokinin A is a tachykinin and bronchoconstrictor that represents an alternatively spliced isoform of substance P. One study examining 73 patients with midgut NETs (80% with metastases) found elevated levels in 70% and that levels appeared to correlate with metastatic tumor burden. Unfortunately, only a minority of patients had both pre and postoperative levels drawn.38 Diebold et al. demonstrated that in patients with metastatic midgut NETs (40% with liver metastases), serum NKA levels of <50 pg/ml correlated with improved 2 year survival (93% versus 49%) compared to those with more elevated levels. They suggested that when NKA levels normalized after surgery, patient outcomes improved, but survival statistics were not given.39 Sherman et al. did not find a correlation between preoperative NKA levels and PFS or OS in 52 midgut patients treated with surgery,37 and thus more data is needed to determine the prognostic value of NKA in midgut carcinoid patients.
CURRENT RECOMMENDATIONS FOR BIOCHEMICAL TESTING IN GASTROINTESTINAL NETS
The National Comprehensive Cancer Network (NCCN) guidelines for GI NETs suggest testing for CgA and collecting a 24 hour urine for 5-HIAA, but do not give specific recommendations for follow up.40. The European Neuroendocrine Tumor Society (ENETS) also recommends that the minimal testing performed for GI NETs should include serum CgA and urine 5-HIAA. They also recommend using these assays in follow up for tumor recurrence and progression. They further suggest that serotonin assays are insensitive and not recommended for either diagnosis or follow up, but state this biomarker’s utility may be improved using the platelet-based assays. They do not comment on pancreastatin and NKA (they suggest that further validation is warranted for newer markers), but do comment that neuron specific enolase should not be used.41 The North American Neuroendocrine Tumor Society (NANETS) again only mentions serum CgA and urinary 5-HIAA levels as potentially valuable for measuring response to therapy or progression. They suggest that 5-HIAA may be less useful in foregut (including PNETs) and hindgut NETs, as these tumors tend to not make high levels of serotonin.42 In patients with midgut and other GI NETs, it is our practice to measure to serum serotonin, CgA, pancreastatin, and less commonly NKA, preoperatively and at each follow up visit.10,33 These biomarkers are readily available and measureable by many CLIA-certified, ACP-sanctioned laboratories in the United States. Ideally, serial measurements should be from the same laboratory, recognizing that standards and quality control vary.
Pancreatic Neuroendocrine Tumors
Pancreatic neurodendocrine tumors, previously known as islet cell tumors, account for about 1–2% of all pancreatic neoplasms and for about 6% of all NETs.1,43 Their incidence has increased from 1.4 cases per million in 1973 to 3.0 in 2004.44 According to the National Cancer Database (NCBD), 85% were classified as nonfunctional, meaning there is no clinical syndrome associated with hormone excess. Because hormone levels are not collected in the database, and tumors classified as pathologically benign (like many insulinomas) are also not included, numbers derived from the NCDB may be overestimated.45 However, other recent studies suggest that PNETs are nonfunctional in 68–90% of cases.46
Human adult islet cells produce the hormones insulin, glucagon, somatostatin, vasoactive intestinal peptide (VIP), pancreatic polypeptide (PP), and serotonin; fetal islet cells can produce gastrin. Pancreatic NETs may secrete any of these hormones, and in addition, rare tumors may secrete adrenocorticotropic hormone (ACTH), Parathyroid hormone-related peptide (PTH-rP), calcitonin, and growth hormone releasing factor (GHRH).46 About 5% of patients with PNETs have an inherited predisposition, which includes members of multiple endocrine neoplasia type I (MEN1), von Hippel-Lindau (VHL), tuberous sclerosis (TS) and neurofibromatosis type I (NF1) families.47 Features of each of these more common PNET subtypes and how their biochemical diagnoses are made are covered in the sections that follow.
Gastrinomas
Gastrinomas, which cause the Zollinger-Ellison Syndrome (ZES), comprise about 15% of all functional PNETs,46 and are the most common PNET associated with MEN1. Approximately 90% of gastrinomas are found in the gastrinoma triangle, an area bordered by the confluence of the cystic and common duct superiorly, the pancreatic neck/body junction medially, and the second and third portions of the duodenum laterally.48 In patients with sporadic ZES, 50–88% of gastrinomas are duodenal in origin versus 70–100% of those in MEN1 patients. Approximately 22–35% of patients with pancreatic gastrinomas will have liver metastases. The mean size of these tumors is 3.8 cm.49
The predominant functional abnormality seen is inappropriately high circulating gastrin causing irreversible hyperchlorhydria (gastric acid overproduction) with subsequent typical or atypical ulcer formation, hemorrhage, and excess acid-induced malabsorptive diarrhea. The hallmarks of a gastrinoma are very elevated gastrin levels as well as gastric acid hypersecretion. Over 98% of gastrinoma patients have elevation of fasting gastrin, but this alone is nondiagnostic. The finding of hyperchlohyrdria and basal gastric acid output greater than 15 mEq/hr will help to confirm the diagnosis,49 and a gastric pH of less than 2 is also helpful.10,50 In the past, secretin infusion resulting in a paradoxical rise of gastrin was a useful way to make the diagnosis of marked hypergastrinemia,51 but is performed less commonly due to limited availability of this secretogogue.
There are other conditions associated with high levels of gastrin. These include atrophic gastritis/pernicious anemia, gastric outlet syndrome, retained gastric antrum, H. pylori infection, short bowel syndrome, and diabetic gastroparesis.49 Proton pump inhibitors will also cause significantly elevated gastrin through their significant suppression of gastric acid, with subsequent sustained hypergastrinemia and stimulation of CgA from gastric enterochromaffin-like (ECL) cells. Over time, ECL cell nodular hyperplasia can develop with secondary formation of small neuroendocrine tumors (usually less than 1 cm in size). Enterochromaffin cells in the stomach can also be stimulated by high gastrin and result in CgA elevation along with modest levels of serotonin.10 In MEN1 patients with ZES, hypercalcemia can increase gastrin levels and basal acid secretion. Parathyroidectomy can significantly improve this situation.52
Insulinoma
Insulinomas are the most common functional PNET, accounting for approximately 17% of cases.10,46 Patients are usually symptomatic with episodic hypoglycemia, although some patients can live for years with subclinical disease and have no overt symptoms, or will compensate for hypoglycemia with sugar ingestion. Up to 60% of all insulinomas are found in women, and the average age at diagnosis is 45 years.11 About 10% are associated with MEN1, and are second to gastrinomas in terms of MEN1-associated PNET frequency. Most are benign adenomas, but malignant insulinomas occur in approximately 10% of patients.
The demonstration of Whipple’s triad, 1) symptoms of hypoglycemia after fasting, 2) low glucose when symptomatic, and 3) relief of symptoms with ingestion of glucose or food, is strongly suggestive of an insulinoma. Major symptoms may include headache, blurry vision, seizures, confusion, and even coma. Other commonly observed symptoms are due to peripheral catecholamine release, and include tremor, diaphoresis, and tachycardia. Approximately 98% of insulinoma patients will demonstrate inappropriate insulin secretion with symptomatic hypoglycemia within 72 hours, and therefore a supervised fast within a hospital setting has been recommended as the gold standard for diagnosis.19,53 Blood sugars should be measured every 4 hours until symptoms occur or blood glucose drops below 50 mg/dl, and then serum insulin, C-peptide, and glucose levels are drawn. A measurably low blood glucose, symptoms, and inappropriate elevation of insulin (usually greater than 6 uU/ml) or an insulin to glucose ratio of 0.3 or greater is highly suspicious for insulinoma. When proinsulin is cleaved by signal peptidase, C-peptide and insulin are formed, which are present in equal amounts in beta cells. If a patient has been administered exogenous insulin, C-peptide levels will be low, while these levels will be elevated (>200 pmol/L) in insulinoma.19 Some patients with PNETs and hypoglycemia may have elevated levels of proinsulin rather than insulin.54 For follow up of metastatic insulinoma, serial measurement of CgA and pancreastatin can be useful for assessing the extent of metastatic disease.19
VIPomas
Vasoactive intestinal polypeptide (VIP) secreting PNETs (VIPomas) were independently described by Priest and Alexander, and Verner and Morrison.55,56 Initially termed “pancreatic cholera,” and later, watery diarrhea, hypokalemia, achlorhydria (most often hypochlohydria) syndrome (WDHA), it is now known as watery diarrhea syndrome (WDS). VIP is a neuromodulator (not a hormone in the classical sense) which, in high sustained blood levels, acts as a powerful intestinal secretogogue, resulting in hypokalemia, metabolic acidosis, stool bicarbonate loss, and high volume alkalotic stool.11,57,58 VIPomas are an uncommon functional PNET, accounting for 1–2% of all functional PNETs.46,59 They can be seen in MEN1 patients even when other family members may have had gastrinomas or insulinomas.
The diagnosis of VIPoma is suspected in the setting of elevated plasma VIP and severe (often life threatening) watery diarrhea (usually greater than 1250 ccs/day) and profound hypokalemia. Initially, the severe diarrhea may be episodic (tumors may be nonautonomous). Flushing is seen in 20% of patients with WDS, also thought to be a direct action of VIP. Hypochlorhydria, not achlorhydria, is the seen in 80% of VIPoma/WDS patients.59 Both functional and nonfunctional biomarkers from certified commercial labs should be measured, to include VIP and PP.60,61 In the setting of metastasis, CgA and pancreastatin may be helpful to follow for progression and response to therapy. While the majority of VIPomas in adults arise from the pancreas, there are other nonpancreatic sources of VIP-secreting NETs, including pheochromocytoma, neuroblastoma, ganglioneuroma, bronchogenic carcinoma, and medullary thyroid carcinoma.10,60
Glucagonoma
Glucagonoma is a very rare functional tumor, accounting for 1% or less of all PNETs. The clinical manifestations of glucagonoma are very high circulating glucagon levels and a classic necrolytic migratory erythema skin rash, usually on the anterior lower extremity or perianal genital regions. It has come to be known as the “4D Syndrome” – dementia, diarrhea, deep vein thrombosis (DVT), and depression. Other clinical stigmata include a painful glossitis, weight loss (90%), mild type II diabetes mellitus (DM; 80%), low amino acid concentrations, and DVT (50%). The high circulating glucagon levels do not seem to be the cause of the dermopathy.10,57,61 Most glucagonomas are large at diagnosis, although they do not often present with classic symptoms. They are more often found in the pancreatic tail and have a very high rate of metastasis at the time of diagnosis. Like many of the functional PNETs, glucagonomas may also be seen in MEN1 patients.
The clinical diagnosis can be made by the finding of significant elevation of glucagon levels (>500 pg/ml), in the setting of symptoms listed above. Normal fasting levels are generally <150 pg/ml, and several conditions may cause mild elevations of glucagon (DM, acute burns and trauma, cirrhosis, renal failure, Cushing’s syndrome, and bacteremia). Pancreatic polypeptide and insulin levels may also be elevated in association with glucagonoma. As with other metastatic PNETs, serial CgA and pancreastatin levels may be helpful to monitor for progression.10
Somatostatinoma
Somatostatinomas are very rare tumors, accounting for <1% of PNETs.46 While most functional somatostatinomas are of PNET origin (60% of cases), duodenal, ampullary, and less commonly, jejunal somatostatinomas are also recognized. In PNET somatostatinomas, the excess native somatostatin causes hyperglycemia (75% present with DM type II), atony of the gallbladder (59% have gallbladder disease), hypochlorhydria and reduced gastric acid (>80%), steatorrhea and diarrhea (very common, from inhibition of prandial pancreatic enzyme release, bicarbonate, and reduced absorption of fats), and weight loss (possibly due to diarrhea and malabsorption, seen in about 33% of patients). Somatostatinomas are large, which may lead to destruction or loss of islet cells with reduced insulin production. Approximately 80% present with metastatic disease.10 The diagnosis is commonly made in retrospect, by IHC of the tumor, but if there is clinical suspicion, somatostatin levels should be measured. Patients with small cell and bronchogenic carcinomas of the lung have been described with elevated somatostatin levels, as well as up to 25% of patients with pheochromocytoma. Pancreatic polypeptide, CgA, and pancreastatin should also be followed, the latter two for monitoring progression.10
PPoma and nonfunctional tumors
PPomas are a group of nonfunctioning PNETs that comprise about 50% of all PNETs encountered. While PPomas are not recognized as functional PNETs, diarrhea has been associated with very high levels of PP.11 One recent report suggested an association of PPomas with DM, as 5 patients with DM and PPoma had improvement or resolution after resection.62 For the most part, the coassociation of elevated PP in PNETs making other hormones has maintained its value in the diagnosis and follow up of patients with both functional and nonfunctional PNETs.19,60,61 Therefore, PP is a good marker to test in all cases of suspected PNETs, in addition to the hormones suggested by a clinical syndrome, if present. Measurement of CgA and pancreastatin are also useful monitoring the effects of therapy and for progression.
The vast majority of nonfunctional PNETs are diagnosed as a result of nonspecific abdominal pain or symptoms of obstruction of the pancreatic or bile duct. Because of this, nonfunctional PNETs tend to be larger when detected (5.9 cm), they have a higher rate of metastases (60%), and poorer prognosis (5 year survival of 33%) 44
CURRENT RECOMMENDATIONS FOR BIOCHEMICAL TESTING IN PNETS
The NCNN recommends checking PP, CgA, calcitonin, PTH-rP, and GHRH for generic PNETs. If the patient has a recognizable syndrome they recommend checking specific hormone levels. When insulinoma is suspected, then insulin, proinsulin, and C-peptide should be checked, and consideration should be given to a 72 hour fast. Serum VIP levels should be checked if one suspects VIPoma, serum glucagon for those with glucagonoma symptoms, and basal or stimulated gastrin for suspected gastrinoma patients.40 ENETs suggests checking CgA in cases of nonfunctional PNETs, with PP being more uncertain, except in MEN1 patients. Further tests are indicated if the patient demonstrates symptoms.63 The recommendations from NANETs are similar, although given in more detail for functional tumors. For nonfunctional tumors, they recommend CgA and PP.52
BIOMOLECULAR DIAGNOSTICS IN NETs
WREN Assay
Modlin et al. set out to identify a genetic signature for NETs that could be tested from peripheral blood samples that might be useful for diagnosis, assessment of tumor burden, and response to therapy.64 They retrieved data from tissue-based microarrays from normal tissues, and from 9 primary GEP NETs and 6 GEP NET metastases. They identified a group of genes that showed elevated expression in GEP NETs, and then tested these genes in peripheral blood samples as a training set (67 normal samples, 63 GEP NETs). The validation set included 92 normal samples and 143 GEP NETs. They selected 75 genes for further study by qPCR (21 from tissue-based results, 32 from blood-based, and 22 from the literature), which was then further reduced to a 51 gene panel. In PNETs, 79% of samples were accurately identified using the PCR test, as were 88% of GI NETs; the sensitivity was 90% and specificity was 94% for GI NETs, and 80% and 94%, respectively, for PNETs. In comparison to serum CgA levels from 81 GEP NET patients and 95 controls, the PCR-based test outperformed the biomarker (CgA sensitivity 32%, accuracy 60%), even in patients where CgA was low. The authors concluded that their panel could identify GEP NETs regardless of primary tumor site or metastasis, which could be useful for screening and potentially response to therapy. This group is actively recruiting patients to determine how well it might perform under these circumstances.
Biotheranostics Test
A 92-gene molecular assay (CancerTYPE ID, bioTheranostics, Inc.) was developed for determining the site of unknown primary tumors using qPCR from paraffin-embedded biopsy specimens. In a trial examining 790 tumors comprising 28 different tumor types and 50 subtypes, it was found to have an 87% sensitivity, >98% specificity, and a positive predictive value of 61–100%.65 This test was later applied specifically to 75 NETs (12 GI, 22 pulmonary, 10 pancreas, 10 pheochromocytoma, 11 medullary thyroid carcinoma, and 10 Merkel cell carcinomas), of which 59% were metastases and 41% were primary tumors. This panel correctly classified the tumors as a NET in 74/75 cases. The 4 genes that were most important for making this distinction were ELAVL4, CADPS, RGS17, and KCNJ11. Fifteen additional genes were used for further subtyping of NETs, which was accurate in 71/75 (95%) of cases.66 One shortcoming of this study is the inability of this test to determine the GI NET subtype—the test does not identify the site of the primary tumor (small bowel versus duodenal versus rectal), only that the tumor is a GI NET. Still, the test performed well overall, and shows promise in differentiation of lung, pancreatic, and GI NET primaries from tissue samples of metastases, allowing for more tailored therapy for patients.
Gene Expression Classifiers and IHC to Differentiate SBNETs from PNETs
Sherman et al. evaluated the expression of a panel of genes by qPCR in primary tumors and metastases from 61 patients with SBNETs and 25 with PNETs. They were able to refine this panel down to 4 genes in the G-protein coupled receptor pathway (BRS3, OPRK1, OXTR, and SCTR), and in 136 metastases accurately predicted the origin from SBNETs in 94/97 cases (97%) and 34/39 PNETs (87%). The algorithm made primary predictions in 122 cases using qPCR of just BRS3 and OPKR1, though when one of the genes had undetectable expression (14 cases), the results for OXTR and/or SCTR were used to come up with a prediction.67 Maxwell et al. compared the results of this gene expression classifier (GEC) to an IHC algorithm that employed CDX2, PAX6, and Islet1 in first tier staining, followed by IHC for PR, PDX1, NESP55, and PrAP if the first step stains were equivocal. The IHC algorithm was correct in determining the site of origin in 23/27 (85%) of SBNET metastases, and 10/10 (100%) PNET metastases. Comparison of these results revealed improved performance of the GEC for determining SBNET primaries and of IHC for PNET primaries. Although the overall accuracy was 94% for the GEC and 89% for IHC, they concluded that this IHC algorithm should be used first because of its widespread availability, and that GEC be reserved for cases of indeterminate IHC results.68 The ability to differentiate SBNET from PNET primaries from a biopsy of a metastatic liver tumor could aid in surgical exploration, and selection of therapy for these patients with metastatic disease, such as Everolimus, Sunitinib, or chemotherapy in patients with PNETs.
IMAGING TESTS FOR DIAGNOSIS AND STAGING OF GEP NETs
Gastroenteropancreatic neuroendocrine tumors are rare, and typically indolent neoplasms that metastasize early. Surgical resection of the primary tumor, regional nodal disease and distant metastases is the best chance for cure, symptomatic relief, and/or long-term survival.69,70 Thus, it is recommended that patients with GEP NETs of any stage be considered for surgery, especially when the primary tumor can be excised and 70–90% of their metastatic burden can be debulked.70–72
Preoperative imaging is crucial in determining resectability, and the ideal study will identify the primary tumor, define its relationship to surrounding organs and vessels, and detect distant metastases.73 Conventional imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), ultrasound (US), or endoscopy are generally employed to define anatomic relationships, whereas somatostatin receptor imaging and positron emission scanning (PET) are used to determine functionality and scan for distant disease. Each of these modalities will be reviewed and their role in the preoperative workup of GEP NETs discussed.
Computed tomography
A CT scan is usually the first study ordered in the evaluation of a suspected GEP NET. Whether the primary resides within the pancreas, small bowel, colon or rectum, a multiphase study should be obtained with both intravenous (IV) contrast, as these tumors and their metastases are most often hypervascular and are usually identified in the early arterial phase of a triple phase scan (Figure 1).74,75 The later portal venous or delayed phases may detect hypovascular GEP NETs. Diseased mesenteric lymph nodes often develop calcifications, which can help identify them on CT.76 Oral contrast is also helpful, either with conventional radiopaque enteral contrast, or more recently, using negative enteral contrast (methylcellulose for CT enteroclysis, or simply water or polyethylene glycol) which may help to highlight small bowel lesions better.77
Figure 1. CT scan of a patient with a PNET and numerous hepatic metastases.

(a) Early arterial phase demonstrating multiple hypervascular enhancing hepatic metastases. Arrow indicates a large metastasis with a necrotic center. (b) Venous phase. In this later phase, contrast has washed out of the hepatic metastases and only the necrotic core of the metasasis indicated by the arrow in (a) can be seen as clear evidence of hepatic disease.
Primary GEP NETs are detected by CT approximately 73% of the time, though rates vary widely (39–94%) depending on the study and subset of NETs examined.78 Study sensitivity is affected by image acquisition protocols, tumor size, location, and contrast with surrounding tissue.75 SBNETs tend to be small and multifocal, and may be missed in approximately 50% of scans.79 PNETs are more easily detected, with reported rates of approximately 70% for all PNETs and 80–100% when the primary is greater than 2 cm in size.43,80–82 In cases where the location of the primary tumor is unknown, detection rates are much lower (approximately 35%)50, though inability to detect the primary on imaging should not preclude surgical exploration in the majority of patients, as a recent single-institution study demonstrated that 90% of unknown lesions could be identified intraoperatively.83 Detection rates for hepatic metastases and soft tissue metastases are approximately 80%.78,79,84
Magnetic resonance imaging
MRI is superior to CT in detailing hepatic metastases and the pancreatic ductal system, and is useful in patients with renal failure or an allergy to iodinated contrast (Figure 2).85–87 It is often unnecessary in the workup of GEP NETs, however, as a CT scan is commonly obtained and sufficient for surgical planning. This study should also be obtained with IV gadolinium contrast, and as with CT, most lesions enhance (are hyperintense) on arterial phases.82 In a study comparing MRI, CT and OctreoScan, MRI detected hepatic metastases with 95.2% sensitivity, compared to sensitivities of 78.5% for CT and 49.3% for OctreoScan.86 We find MRI helpful when planning hepatic debulking, as it generally defines lesions more clearly than CT and also detects hepatic lesions that CT frequently misses. It is not as useful for examining the mesentery and small intestine.
Figure 2.

MRI demonstrating numerous enhancing hepatic metastases on a T2 weighted image.
Ultrasound
The primary role of conventional US in the preoperative workup of GEP NETs is assessment of hepatic tumor burden. Its sensitivity for detecting primary GEP NETs is only 36%.84 For hepatic metastases, its sensitivity (88%)84 is less than contrast-enhanced CT or MRI, though the addition of microbubble contrast agents can improve this to nearly 100%. These small gas bubbles oscillate up to hundreds of meters per second in the blood, perfusing the tumors and enhancing their reflectivity.88 This is an alternative to standard imaging in patients who cannot tolerate contrast.89 Ultrasound is the intraoperative tool of choice to localize hepatic metastases for ablation, or to examine the pancreas for small tumors that were undetectable on CT or MRI.
Endoscopy
Endoscopy is a useful study to localize primary foregut and hindgut GEP NETs and can be used to obtain a tissue diagnosis. In some cases, small (< 1 cm) intraluminal tumors that do not invade beyond the mucosa can be treated during this procedure with snare removal.75,90 It is often combined with ultrasound (endoscopic ultrasound, EUS), which may aid in both primary tumor detection and local staging. In PNETs, EUS locates primaries with 93% sensitivity and 95% specificity. For suspected duodenal NETs, it is less sensitive, with detection rates of 45–60%.91 In cases of where the location of the primary is unknown but the small bowel is suspected, either double balloon enteroscopy (which may need to be done from below and above) or video capsule endoscopy can be employed. Unfortunately, double balloon enteroscopy has a detection rate of only 33% in this context, and capsule endoscopy will uncover only 45% of small bowel primaries.92,93
Though it is rarely used as a stand-alone procedure, EUS may add useful information during surgical planning. In a small series of 14 patients with PNETs, EUS was compared to CT to determine whether EUS could be helpful for surgical decision making. In 36% of cases, EUS altered the surgical plan by either identifying a solitary PNET or additional multifocal PNETs that were missed by CT.94 EUS has the added benefit of being able to detect tumors less than 2 cm in size, which may by missed by CT or MRI.91
18FDG positron emission scanning
18-fluoro-deoxy-glucose PET (FDG PET) is a functional imaging study that is used to detect a variety of tumor types. It has limited utility for GEP NETs, especially in the preoperative setting, as most of these tumors are of low or intermediate grade and metabolically inactive, and thus do not to take up 18FDG well.95 High-grade GEP NETs tend to be metabolically active and are most likely to have uptake on 18FDG-PET. In this context, uptake identifies lesions likely to have more rapid progression.96,97 A recent study found shorter overall survival (15 months) in GEP NET patients that had a maximum standardized uptake value (SUVmax) of 4.5 or greater. Patients with tumors having lower uptake (i.e. SUVmax < 4.5) had an overall survival of 120 months.98
Somatostatin receptor imaging
Somatostatin is an endogenous peptide that inhibits cellular proliferation and secretion when it binds to one of five types of somatostatin receptor (SSTR1 – SSTR5). These G-protein coupled receptors are normally expressed by neuroendocrine cells in a wide variety of tissue types including the brain, pituitary, pancreas, thyroid, spleen, adrenal glands, large and small intestine, kidney, peripheral nervous system, immune cells and the vasculature.99–101 SSTR2 is the most highly expressed SSTR subtype on the majority of well differentiated NETs, and is the primary receptor for somatostatin-based imaging and treatment.99,102
There are two types of somatostatin receptor-based imaging available. Both can be used in the diagnosis and surveillance of GEP NET patients, and also to select candidates for peptide receptor radionuclide therapy (PRRT).103 Additionally, each uses analogues of endogenous somatostatin to bind to SSTR, as the short half-life of the native peptide precludes its use for this purpose.102 The most common type of somatostatin-based imaging is a scintigraphic study called the OctreoScan (Mallinckrodt, St. Louis, MO), which uses the radiotracer 111In-DPTA-D-Phe-1-octreotide, and binds mainly to SSTR2 but also to SSTR5.101 Recently, a PET scan has been developed that uses the positron emitter 68Ga to label a variety of somatostatin analogues, which then bind to a variety of SSTR subtypes. The most common of these labeled analogues are 68Ga-DOTATOC, 68Ga-DOTANOC, and 68Ga-DOTATATE. Each have slightly different affinities for the SSTR subtypes, though this does not translate into variable clinical efficacy.87,104
Octreoscan
The octreoscan is a nuclear medicine study that is available in a large number of centers worldwide and is probably the most commonly used imaging study used in the diagnosis and surveillance of NETs. Patient preparation includes transition to short acting octreotide four to six weeks prior to image acquisition to minimize the drug’s interference with the imaging ligand, and voiding immediately prior to the study. In cases of suspected GEP NETs, it may be beneficial to have the patient use an over-the-counter laxative the night prior to the study to minimize the accumulation of isotope in the lumen of the bowel.105 The patient is then scanned four and 24 hours after IV injection of the analogue.101
In its basic form, ligand-receptor binding produces a dark spot on the full body planar image (Figure 3).105 Since 1999, however, this scintigraphic image is usually fused with single photon emission computed tomography (SPECT) and CT to increase its diagnostic accuracy (Figure 4). The addition of SPECT allows for the scintigraphic image to be displayed as tomographic slices, which minimizes the interference physiologic ligand uptake has on NET detection. Fusion with CT increases the anatomic definition of the study.106–108 A study comparing octreoscan-SPECT/CT to planar octreoscan showed that fusing the images positively impacted patient care and altered management decisions in 15% of cases.109
Figure 3. Planar octreoscan demonstrating a primary PNET (arrow).

Physiologic uptake is seen in the liver, spleen and bladder. (a) Image acquired at 4 h. (b) Image acquired at 24 h.
Figure 4. Octreoscan fused with SPECT/CT.

This axial image depicts the same PNET (arrow) as is seen in the planar images in Figure 3. Physiologic uptake is seen in the spleen and kidneys.
Even when fused with SPECT/CT, the anatomic resolution of the octreoscan is insufficient as the only study used for surgical planning. The primary purpose of this functional imaging is to specifically identify tumors as NETs based on their expression of SSTR2. It is also used as an adjunct to CT or MRI to detect distant metastases and localize primary GEP NETs when the primary tumor site is unknown. The Octreoscan’s sensitivity for detection of hepatic metastases ranges from 49–91%.79,84,86,110,111 In comparison to 68Ga-DOTATOC, Octreoscan is more likely to miss small lymph nodes and peritoneal metastases, as well as bone metastases.112 In known primary tumors, Octreoscan has a sensitivity of 80% and specificity of > 95%. The study’s ability to detect primary tumors seems to be related to tumor size (<>2 cm), rather than just SSTR2 expression by the tumor.113 Its detection rate for unknown primary tumors has been reported to be 24–39%.84,114
68Ga-PET
Introduction of 68Ga-labeled radioligands to somatostatin receptor-based imaging have enhanced the sensitivity and utility of this type of functional imaging study. The advantages of these radioligands (68Ga-DOTATOC, 68Ga-DOTANOC, and 68Ga-DOTATATE) over the ligand used in the Octreoscan (111In-DPTA-D-Phe-1-octreotide) are the ease and lower cost at which they can be synthesized, enhanced patient convenience (as the image is acquired one hour post-contract injection), and ability to quantify lesion uptake of the ligand. This modality better aids preoperative planning as it can resolve imaged structures to within millimeters. The more precise spatial resolution is due to the fact that it measures the radiation of two photons coincidentally. In comparison, SPECT can only achieve a 1 cm limit of detection and measures the gamma radiation of only one photon directly.108 Similar to octreoscan-SPECT/CT, 68Ga-PET images are fused with CT to further improve their anatomic specificity (Figure 5).
Figure 5.
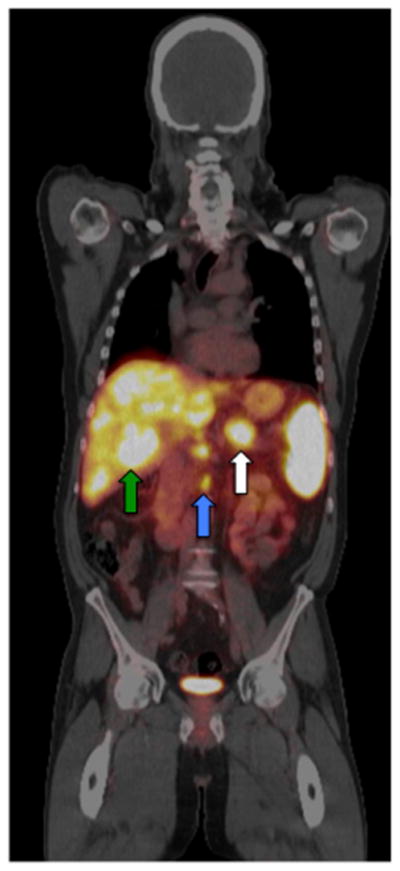
68Ga-DOTATOC PET/CT of a patient with innumerable hepatic metastases (green arrow), lymph node metastases (blue arrow) and a primary PNET (white arrow). Physiologic uptake is seen in the spleen.
One of the largest series (n = 109) comparing 68Ga-PET to conventional imaging found that 68Ga-DOTANOC PET/CT had a sensitivity of 78.3% and specificity of 92.5% for primary GEP-NETs and 97.4% sensitivity and 100% specificity for metastases. In both cases, this was a significant improvement compared to CT, MRI or US. Further, patient management was altered for 19% of patients on the basis of the 68Ga-DOTANOC PET/CT results. For 5.5% of patients, the primary tumor was detected by 68Ga-DOTANOC PET/CT but missed by other imaging modalities, allowing them to undergo surgical resection. In 6.4% of patients, 68Ga-DOTANOC PET/CT demonstrated new resectable lesions, aiding preoperative planning and potentially improving postoperative outcome. Unnecessary surgery was avoided in 3.6% of patients with evidence of widespread disease on 68Ga-DOTANOC PET/CT.115
Despite the excellent image quality and sensitivity of 68Ga-PET/CT, its expense and limited availability make it unlikely to usurp contrast-enhanced CT as the most useful preoperative imaging study. However, it is very helpful in cases where CT or MRI fail to locate the primary tumor and often uncovers metastases missed by other modalities. In one study, 68Ga-DOTANOC PET/CT found the primary site in 59% of patients with advanced disease but an unknown primary. CT was only able to detect the primary site in 20%.116 Buchmann et al. compared 68Ga-DOTATOC to octreoscan-SPECT/CT and found that 68Ga-DOTATOC detected more than 279 lesions in their group of 27 patients with histologically-proven NETs, whereas octreoscan-SPECT/CT only detected 157. When the number of liver metastases detected by each modality were compared, the concordance rate (lesions detected by both modalities) was only 66%. In lymph node metastases, the concordance rate was 40.1%. In both cases, 68Ga-DOTATOC proved to be the superior somatostatin-based imaging study to delineate the extent of patient disease.117
Achieving good surgical outcomes requires prudent, meticulous planning. Imaging studies are a crucial part of this process and are required to identify the primary tumor site and the extent of metastatic disease, as well as determine the resectability of the disease. The most practical initial study for GEP-NETs is a contrast-enhanced CT scan as it is fast, available at most centers, and excellent in its anatomic detail. For patients that are unable to tolerate iodinated contrast, MRI is a good alternative anatomic study. MRI is also very helpful defining the extent of hepatic disease in patients selected to undergo hepatic debulking procedures. If extra-abdominal metastases are suspected or require further investigation, 68Ga-PET will likely provide the best supplemental information to the surgeon, though it will take time for this modality to gain FDA approval and dissemination throughout the United States. Thus, in cases where 68Ga-PET is unavailable, the octreoscan remains the most helpful NET-specific modality to identify tumors specifically as NETs and to detect metastases. In PNETs, gastric and duodenal NETs, EUS is an excellent adjunct to CT and gives good information regarding primary tumor location, multiplicity, invasion and likely lymph node involvement. It can also be used to obtain a tissue diagnosis. For GI NETs, endoscopy should be used sparingly, given its moderate sensitivity for locating primary tumors and inability to detect distant disease.
CONCLUSIONS
The increasing incidence of NETs over the past decades, and specifically those of gastroenteropancreatic origin, pose several challenges for the clinician. Since a high percentage of patients present with distant disease, one of the difficulties has been in the early identification of these tumors. Increasing recognition of the signs and symptoms characteristic of the specific clinical syndromes associated with functional tumors will promote screening using appropriate NET biochemical markers in the blood, and imaging tests to define the locations of primary tumors. Conversely, the frequent incidental finding of a suspicious lesion on anatomic imaging should lead to appropriate serum testing for functional or nonfunctional NETs, as well as possibly somatostatin-based imaging tests. In metastatic lesions, biopsy samples can now be used to identify the site of unknown primary using qPCR-based tests, which may enhance their discovery and the selection of appropriate surgical or medical therapy.
KEY POINTS.
Many NETs secrete substances that can cause symptoms, but also aid biochemical diagnosis and localization of the primary tumor.
There are many foods and medications that can interfere with biomarker assays.
In cases where a PNET is suspected, PP, CgA, calcitonin, PTH-rP, and GHRH should be drawn during the patient’s initial visit.
When a GI NET is suspected, CgA and serotonin levels should be obtained.
Molecular testing may be used to identify an unknown metastasis as a NET and can be more accurate than traditional histologic procedures (IHC) in differentiating between primary tumor sites.
Acknowledgments
Dr. Maxwell’s work is supported by NIH 5T32#CA148062-05.
Key Abbreviation
- NET
neuroendocrine tumors
- PNET
pancreas neuroendocrine tumors
- SBNET
small bowel neuroendocrine tumors
- PP
pancreatic polypeptide
- CgA
chromogranin A
- GHRH
growth hormone releasing hormone
- PTHrP
parathyroid hormone related peptide
- APUD
amine precursor uptake and decarboxylation
- MAO
monoamine oxidase
- 5-HIAA
5-hydroxy indole acetic acid
- GEP
gastroentericpancreatic
- NKA
neurokinase A
- NCCN
National Comprehensive Cancer Network
- ENETS
European Neuroendocrine Tumor Society
- NANETS
North American Neuroendocrine Tumor Society
- SSTR1-5
somatostatin receptor subtypes 1 to 5
- NCDB
National Cancer Database
- ZES
Zollinger Ellison Syndrome
- VHL
von Hippel Lindau
- MEN-1
Multiple endocrine neoplasia type 1
- NF-1
Neurofibromatosis type 1
- TS
tuberous sclerosis
- WDHA
watery diarrhea, hypokalemia, achlorhydria
- WDS
watery diarrhea syndrome
- PRRT
peptide receptor radionuclide therapy
Footnotes
Drs. O’Dorisio and Howe have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 2.Pearse AGE. Common cytochemical properties of cells producing polypeptide hormone, with particular reference to calcitonin and the thyroid C cells. Vet Rec. 1966;79:587–590. doi: 10.1136/vr.79.21.587. [DOI] [PubMed] [Google Scholar]
- 3.Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The Pathologic Classification of Neuroendocrine Tumors: A Review of Nomenclature, Grading, and Staging Systems. Pancreas. 2010;39:707–712. doi: 10.1097/MPA.0b013e3181ec124e. [DOI] [PubMed] [Google Scholar]
- 4.Solcia E, Kloppel G, Sobin LH. WHO International Classification of Tumours. 2. New York: Springer; 2000. Histological Typing of Endocrine Tumors. [Google Scholar]
- 5.Lamberts SW, van der Lely AJ, de Herder WW, Hofland LJ. Octreotide. N Engl J Med. 1996;334(4):246–254. doi: 10.1056/NEJM199601253340408. [DOI] [PubMed] [Google Scholar]
- 6.Krenning EP, Kwekkeboom DJ, Oei HY, et al. Somatostatin-receptor scintigraphy in gastroenteropancreatic tumors. An overview of European results. Ann N Y Acad Sci. 1994;733:416–424. doi: 10.1111/j.1749-6632.1994.tb17291.x. [DOI] [PubMed] [Google Scholar]
- 7.Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 8.Caplin ME, Pavel M, Cwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224–233. doi: 10.1056/NEJMoa1316158. [DOI] [PubMed] [Google Scholar]
- 9.Kwekkeboom DJ, de Herder WW, Kam BL, et al. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26(13):2124–2130. doi: 10.1200/JCO.2007.15.2553. [DOI] [PubMed] [Google Scholar]
- 10.Vinik AI, Woltering EA, O’Dorisio TM, Go VLW, Mamikunian G. Neuroendocrine Tumors: A Comprehenisve Guide to Diagnosis and Management. 5. California, USA: Inter Science Institute; 2012. [Google Scholar]
- 11.O’Dorisio TM, Redfern JS. Somatostatin and somatostatin-like peptides: Clinical research and clinical applications. In: Mazzaferri EL, Bar RS, Kreisberg RA, editors. Advances in Endocrinology and Metabolism. Vol. 1. Chicago: Mosby Year Book; 1990. pp. 174–230. [Google Scholar]
- 12.Oberndorfer S. Karzinoide Tumoren des dundarms. Frankf Z Pathol. 1907;1:426–432. [Google Scholar]
- 13.Modlin IM, Shapiro MD, Kidd M, Drozdav I, Gustafsson B. Siegfried Oberndorfer and the origins of carcinoid tumors. In: Modlin IM, Oberg K, editors. A Century of Advances in Neuroendocrine Tumor Biology and Treatment. Switzerland: Felsenstein; 2007. pp. 22–27. [Google Scholar]
- 14.Howe JR, Karnell LH, Menck HR, Scott-Conner C The American College of Surgeons Commission on Cancer and the American Cancer Society. Adenocarcinoma of the small bowel: review of the National Cancer Data Base, 1985–1995. Cancer. 1999;86(12):2693–2706. doi: 10.1002/(sici)1097-0142(19991215)86:12<2693::aid-cncr14>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 15.Bilimoria KY, Bentrem DJ, Wayne JD, Ko CY, Bennett CL, Talamonti MS. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg. 2009;249(1):63–71. doi: 10.1097/SLA.0b013e31818e4641. [DOI] [PubMed] [Google Scholar]
- 16.Drozdov I, Modlin IM, Kidd M, Goloubinov VV. From Leningrad to London: the saga of Kulchitsky and the legacy of the enterochromaffin cell. Neuroendocrinology. 2009;89(1):1–12. doi: 10.1159/000140663. [DOI] [PubMed] [Google Scholar]
- 17.Andrew A. Further evidence that enterochromaffin cells are not derived from the neural crest. J Embryol Exp Morphol. 1974;31(3):589–598. [PubMed] [Google Scholar]
- 18.Gershon MD. Review article: roles played by 5-hydroxytryptamine in the physiology of the bowel. Aliment Pharmacol Ther. 1999;13 (Suppl 2):15–30. [PubMed] [Google Scholar]
- 19.Vinik AI, Silva MP, Woltering EA, Go VLW, Warner R, Caplin M. Biochemical Testing for Neuroendocrine Tumors. Pancreas. 2009;38(8):876–889. doi: 10.1097/MPA.0b013e3181bc0e77. [DOI] [PubMed] [Google Scholar]
- 20.Norheim I, Oberg K, Theodorsson-Norheim E, et al. Malignant carcinoid tumors. An analysis of 103 patients with regard to tumor localization, hormone production, and survival. Ann Surg. 1987;206(2):115–125. doi: 10.1097/00000658-198708000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meijer WG, Kema IP, Volmer M, Willemse PH, de Vries EG. Discriminating capacity of indole markers in the diagnosis of carcinoid tumors. Clin Chem. 2000;46(10):1588–1596. [PubMed] [Google Scholar]
- 22.Bajetta E, Ferrari L, Martinetti A, et al. Chromogranin A, neuron specific enolase, carcinoembryonic antigen, and hydroxyindole acetic acid evaluation in patients with neuroendocrine tumors. Cancer. 1999;86(5):858–865. doi: 10.1002/(sici)1097-0142(19990901)86:5<858::aid-cncr23>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Allen KR, Degg TJ, Anthoney DA, Fitzroy-Smith D. Monitoring the treatment of carcinoid disease using blood serotonin and plasma 5-hydroxyindoleacetic acid: three case examples. Ann Clin Biochem. 2007;44(Pt 3):300–307. doi: 10.1258/000456307780480936. [DOI] [PubMed] [Google Scholar]
- 24.Kema IP, de Vries EG, Slooff MJ, Biesma B, Muskiet FA. Serotonin, catecholamines, histamine, and their metabolites in urine, platelets, and tumor tissue of patients with carcinoid tumors. Clin Chem. 1994;40(1):86–95. [PubMed] [Google Scholar]
- 25.Research MFfMEa. [Accessed April 30, 2015];Serotonin, Serum 2015. 2015 [Google Scholar]
- 26.Boudreaux JP, Klimstra DS, Hassan MM, et al. The NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the Jejunum, Ileum, Appendix, and Cecum. Pancreas. 2010;39(6):753–766. doi: 10.1097/MPA.0b013e3181ebb2a5. [DOI] [PubMed] [Google Scholar]
- 27.Stridsberg M, Eriksson B, Oberg K, Janson ET. A comparison between three commercial kits for chromogranin A measurements. J Endocrinol. 2003;177(2):337–341. doi: 10.1677/joe.0.1770337. [DOI] [PubMed] [Google Scholar]
- 28.Arnold R, Wilke A, Rinke A, et al. Plasma chromogranin A as marker for survival in patients with metastatic endocrine gastroenteropancreatic tumors. Clin Gastroenterol Hepatol. 2008;6(7):820–827. doi: 10.1016/j.cgh.2008.02.052. [DOI] [PubMed] [Google Scholar]
- 29.Jensen EH, Kvols L, McLoughlin JM, et al. Biomarkers predict outcomes following cytoreductive surgery for hepatic metastases from functional carcinoid tumors. Ann Surg Oncol. 2007;14(2):780–785. doi: 10.1245/s10434-006-9148-z. [DOI] [PubMed] [Google Scholar]
- 30.Welin S, Stridsberg M, Cunningham J, et al. Elevated plasma chromogranin A is the first indication of recurrence in radically operated midgut carcinoid tumors. Neuroendocrinology. 2009;89(3):302–307. doi: 10.1159/000179900. [DOI] [PubMed] [Google Scholar]
- 31.Research MFfMEa. [Accessed April 30, 2015];Chromogranin A. 2015 [Google Scholar]
- 32.O’Connor DT, Cadman PE, Smiley C, et al. Pancreastatin: multiple actions on human intermediary metabolism in vivo, variation in disease, and naturally occurring functional genetic polymorphism. J Clin Endocrinol Metab. 2005;90(9):5414–5425. doi: 10.1210/jc.2005-0408. [DOI] [PubMed] [Google Scholar]
- 33.O’Dorisio TM, Krutzik SR, Woltering EA, et al. Development of a Highly Sensitive and Specific Carboxy-Terminal Human Pancreastatin Assay to Monitor Neuroendocrine Tumor Behavior. Pancreas. 2010;39(5):611–616. doi: 10.1097/MPA.0b013e3181c68d7a. [DOI] [PubMed] [Google Scholar]
- 34.Calhoun K, Toth-Fejel S, Cheek J, Pommier R. Serum peptide profiles in patients with carcinoid tumors. Am J Surg. 2003;186(1):28–31. doi: 10.1016/s0002-9610(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 35.Raines D, Chester M, Diebold AE, et al. A Prospective Evaluation of the Effect of Chronic Proton Pump Inhibitor Use on Plasma Biomarker Levels in Humans. Pancreas. 2012;41(4):508–511. doi: 10.1097/MPA.0b013e318243a0b6. [DOI] [PubMed] [Google Scholar]
- 36.Rustagi S, Warner RR, Divino CM. Serum pancreastatin: the next predictive neuroendocrine tumor marker. J Surg Oncol. 2013;108(2):126–128. doi: 10.1002/jso.23359. [DOI] [PubMed] [Google Scholar]
- 37.Sherman SK, Maxwell JE, O’Dorisio MS, O’Dorisio TM, Howe JR. Pancreastatin predicts survival in neuroendocrine tumors. Ann Surg Oncol. 2014;21(9):2971–2980. doi: 10.1245/s10434-014-3728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner GB, Johnston BT, McCance DR, et al. Circulating markers of prognosis and response to treatment in patients with midgut carcinoid tumours. Gut. 2006;55(11):1586–1591. doi: 10.1136/gut.2006.092320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diebold AE, Boudreaux JP, Wang YZ, et al. Neurokinin A levels predict survival in patients with stage IV well differentiated small bowel neuroendocrine neoplasms. Surgery. 2012;152(6):1172–1176. doi: 10.1016/j.surg.2012.08.057. [DOI] [PubMed] [Google Scholar]
- 40.Kulke MH, Shah MH, Benson AB, 3rd, et al. Neuroendocrine tumors, version 1.2015. J Natl Compr Canc Netw. 2015;13(1):78–108. doi: 10.6004/jnccn.2015.0011. [DOI] [PubMed] [Google Scholar]
- 41.Pape UF, Perren A, Niederle B, et al. ENETS Consensus Guidelines for the management of patients with neuroendocrine neoplasms from the jejuno-ileum and the appendix including goblet cell carcinomas. Neuroendocrinology. 2012;95(2):135–156. doi: 10.1159/000335629. [DOI] [PubMed] [Google Scholar]
- 42.Kunz PL, Reidy-Lagunes D, Anthony LB, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013;42(4):557–577. doi: 10.1097/MPA.0b013e31828e34a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuo JH, Lee JA, Chabot JA. Nonfunctional pancreatic neuroendocrine tumors. Surg Clin North Am. 2014;94(3):689–708. doi: 10.1016/j.suc.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 44.Franko J, Feng W, Yip L, Genovese E, Moser AJ. Non-functional neuroendocrine carcinoma of the pancreas: incidence, tumor biology, and outcomes in 2,158 patients. J Gastrointest Surg. 2010;14(3):541–548. doi: 10.1007/s11605-009-1115-0. [DOI] [PubMed] [Google Scholar]
- 45.Bilimoria KY, Tomlinson JS, Merkow RP, et al. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: analysis of 9,821 patients. J Gastrointest Surg. 2007;11(11):1460–1467. doi: 10.1007/s11605-007-0263-3. discussion 1467–1469. [DOI] [PubMed] [Google Scholar]
- 46.Ehehalt F, Saeger HD, Schmidt CM, Grutzmann R. Neuroendocrine tumors of the pancreas. Oncologist. 2009;14(5):456–467. doi: 10.1634/theoncologist.2008-0259. [DOI] [PubMed] [Google Scholar]
- 47.Schimmack S, Svejda B, Lawrence B, Kidd M, Modlin IM. The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch Surg. 2011;396(3):273–298. doi: 10.1007/s00423-011-0739-1. [DOI] [PubMed] [Google Scholar]
- 48.Stabile BE, Morrow DJ, Passaro E., Jr The gastrinoma triangle: operative implications. Am J Surg. 1984;147(1):25–31. doi: 10.1016/0002-9610(84)90029-1. [DOI] [PubMed] [Google Scholar]
- 49.Jensen RT, Niederle B, Mitry E, et al. Gastrinoma (duodenal and pancreatic) Neuroendocrinology. 2006;84(3):173–182. doi: 10.1159/000098009. [DOI] [PubMed] [Google Scholar]
- 50.Wang SC, Parekh JR, Zuraek MB, et al. Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg. 2010;145(3):276–280. doi: 10.1001/archsurg.2010.10. [DOI] [PubMed] [Google Scholar]
- 51.McGuigan JE, Wolfe MM. Secretin injection test in the diagnosis of gastrinoma. Gastroenterology. 1980;79(6):1324–1331. [PubMed] [Google Scholar]
- 52.Kulke MH, Anthony LB, Bushnell DL, et al. NANETS treatment guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas. 2010;39(6):735–752. doi: 10.1097/MPA.0b013e3181ebb168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti MP, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic system. World J Gastroenterol. 2008;14(35):5377–5384. doi: 10.3748/wjg.14.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Piovesan A, Pia A, Visconti G, et al. Proinsulin-secreting neuroendocrine tumor of the pancreas. J Endocrinol Invest. 2003;26(8):758–761. doi: 10.1007/BF03347360. [DOI] [PubMed] [Google Scholar]
- 55.Priest WM, Alexander MK. Isletcell tumour of the pancreas with peptic ulceration, diarrhoea, and hypokalaemia. Lancet. 1957;273(7006):1145–1147. doi: 10.1016/s0140-6736(57)92051-2. [DOI] [PubMed] [Google Scholar]
- 56.Verner JV, Morrison AB. Islet Cell Tumor and a Syndrome of Refractory Watery Diarrhea and Hypokalemia. American Journal of Medicine. 1958:370–380. doi: 10.1016/0002-9343(58)90075-5. [DOI] [PubMed] [Google Scholar]
- 57.O’Dorisio TM, O’Dorisio MS. Endocrine tumors of the gastroenteropancreatic (GEP) axis. In: Mazzaferri EL, editor. Endocrinology. New York: Medical Examination Publishing; 1985. pp. 76–81. [Google Scholar]
- 58.Bloom SR, Polak JM. VIPomas. In: Said SI, editor. Vasoactive Intestinal Peptide. New York: Raven Press; 1982. pp. 457–463. [Google Scholar]
- 59.O’Dorisio TM, Mekljian HS., VI . Poma Syndrome. In: Cohen S, Soloway RD, editors. Contemporary Issues in Gastroenterology. Edinburgh: Churchill Livingston; 1984. pp. 101–116. [Google Scholar]
- 60.O’Dorisio TM, Vinik AI. Pancreatic polypeptides and mixed hormone-producing tumors of the gastrointestinal tract. In: Cohen S, Soloway RD, editors. Contemporary Issues in Gastroenterology. Endinburgh: Churchill-Livingston; 1984. pp. 117–128. [Google Scholar]
- 61.Vinik AI, Strodel WE, O’Dorisio TM. Endocrine tumors of the gastroenteropancreatic axis. In: Santern A, editor. Endocrine Related Tumors. Amsterdam: Martino Nijolof; 1984. pp. 305–345. [Google Scholar]
- 62.Maxwell JE, O’Dorisio TM, Bellizzi AM, Howe JR. Elevated pancreatic polypeptide levels in pancreatic neuroendocrine tumors and diabetes mellitus: causation or association? Pancreas. 2014;43(4):651–656. doi: 10.1097/MPA.0000000000000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Falconi M, Bartsch DK, Eriksson B, et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology. 2012;95(2):120–134. doi: 10.1159/000335587. [DOI] [PubMed] [Google Scholar]
- 64.Modlin IM, Drozdov I, Kidd M. The identification of gut neuroendocrine tumor disease by multiple synchronous transcript analysis in blood. PLoS One. 2013;8(5):e63364. doi: 10.1371/journal.pone.0063364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kerr SE, Schnabel CA, Sullivan PS, et al. Multisite validation study to determine performance characteristics of a 92-gene molecular cancer classifier. Clin Cancer Res. 2012;18(14):3952–3960. doi: 10.1158/1078-0432.CCR-12-0920. [DOI] [PubMed] [Google Scholar]
- 66.Kerr SE, Schnabel CA, Sullivan PS, et al. A 92-gene cancer classifier predicts the site of origin for neuroendocrine tumors. Mod Pathol. 2014;27(1):44–54. doi: 10.1038/modpathol.2013.105. [DOI] [PubMed] [Google Scholar]
- 67.Sherman SK, Maxwell JE, Carr JC, et al. Gene expression accurately distinguishes liver metastases of small bowel and pancreas neuroendocrine tumors. Clin Exp Metastasis. 2014;31(8):935–944. doi: 10.1007/s10585-014-9681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maxwell JE, Sherman SK, Stashek KM, O’Dorisio TM, Bellizzi AM, Howe JR. A practical method to determine the site of unknown primary in metastatic neuroendocrine tumors. Surgery. 2014;156(6):1359–1365. doi: 10.1016/j.surg.2014.08.008. discussion 1365–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Givi B, Pommier SJ, Thompson AK, Diggs BS, Pommier RF. Operative resection of primary carcinoid neoplasms in patients with liver metastases yields significantly better survival. Surgery. 2006;140(6):891–897. doi: 10.1016/j.surg.2006.07.033. discussion 897–898. [DOI] [PubMed] [Google Scholar]
- 70.Hill JS, McPhee JT, McDade TP, et al. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer. 2009;115(4):741–751. doi: 10.1002/cncr.24065. [DOI] [PubMed] [Google Scholar]
- 71.Mayo SC, de Jong MC, Pulitano C, et al. Surgical management of hepatic neuroendocrine tumor metastasis: results from an international multi-institutional analysis. Ann Surg Oncol. 2010;17(12):3129–3136. doi: 10.1245/s10434-010-1154-5. [DOI] [PubMed] [Google Scholar]
- 72.Graff-Baker AN, Sauer DA, Pommier SJ, Pommier RF. Expanded criteria for carcinoid liver debulking: Maintaining survival and increasing the number of eligible patients. Surgery. 2014;156:1369–1377. doi: 10.1016/j.surg.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 73.Chambers AJ, Pasieka JL, Dixon E, Rorstad O. Role of imaging in the preoperative staging of small bowel neuroendocrine tumors. J Am Coll Surg. 2010;211(5):620–627. doi: 10.1016/j.jamcollsurg.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 74.Bushnell DL, Baum RP. Standard Imaging Techniques for Neuroendocrine Tumors. Endocrinol Metab Clin N Am. 2011;40:153–162. doi: 10.1016/j.ecl.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 75.Sahani DV, Bonaffini PA, Fernandez-Del Castillo C, Blake MA. Gastroenteropancreatic Neuroendocrine Tumors: Role of Imaging in Diagnosis and Management. Radiology. 2013;266(1):38–61. doi: 10.1148/radiol.12112512. [DOI] [PubMed] [Google Scholar]
- 76.Woodbridge LR, Murtagh BM, Yu DFQC, Planche KL. Midgut Neuroendocrine Tumors: Imaging Assessment for Surgical Resection. Radiographics. 2014;34:413–426. doi: 10.1148/rg.342135504. [DOI] [PubMed] [Google Scholar]
- 77.Sailer J, Zacherl J, Schima W. MDCT of small bowel tumours. Cancer Imaging. 2007;7:224–233. doi: 10.1102/1470-7330.2007.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sundin A, Vullierme MP, Kaltsas G, Plockinger U Mallorca Consensus Conference p, European Neuroendocrine Tumor S. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: radiological examinations. Neuroendocrinology. 2009;90(2):167–183. doi: 10.1159/000184855. [DOI] [PubMed] [Google Scholar]
- 79.Dahdaleh FS, Lorenzen A, Rajput M, et al. The value of preoperative imaging in small bowel neuroendocrine tumors. Ann Surg Oncol. 2013;20(6):1912–1917. doi: 10.1245/s10434-012-2836-y. [DOI] [PubMed] [Google Scholar]
- 80.Khashab MA, Yong E, Lennon AM, et al. EUS is still superior to multidetector computerized tomography for detection of pancreatic neuroendocrine tumors. Gastrointest Endosc. 2011;73(4):691–696. doi: 10.1016/j.gie.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 81.Versari A, Camellini L, Carlinfante G, et al. Ga-68 DOTATOC PET, Endoscopic Ultrasonography, and Multidetector CT in the Diagnosis of Duodenopancreatic Neuroendocrine Tumors. Clin Nucl Med. 2010;35:321–328. doi: 10.1097/RLU.0b013e3181d6677c. [DOI] [PubMed] [Google Scholar]
- 82.Foti G, Boninsegna L, Falconi M, Mucelli RP. Preoperative assessment of nonfunctioning pancreatic endocrine tumours: role of MDCT and MRI. Radiol Med. 2013;118(7):1082–1101. doi: 10.1007/s11547-013-0956-5. [DOI] [PubMed] [Google Scholar]
- 83.Bartlett EK, Roses RE, Gupta M, et al. Surgery for metastatic neuroendocrine tumors with occult primaries. J Surg Res. 2013;184(1):221–227. doi: 10.1016/j.jss.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 84.Chiti A, Fanti S, Savelli G, et al. Comparison of somatostatin receptor imaging, computed tomography and ultrasound in the clinical management of neuroendocrine gastro-entero-pancreatic tumours. European Journal of Nuclear Medicine. 1998;25:1396–1403. doi: 10.1007/s002590050314. [DOI] [PubMed] [Google Scholar]
- 85.Manfredi R, Bonatti M, Mantovani W, et al. Non-hyperfunctioning neuroendocrine tumours of the pancreas: MR imaging appearance and correlation with their biological behaviour. Eur Radiol. 2013;23(11):3029–3039. doi: 10.1007/s00330-013-2929-4. [DOI] [PubMed] [Google Scholar]
- 86.Dromain C, de Baere T, Lumbroso J, et al. Detection of liver metastases from endocrine tumors: a prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. J Clin Oncol. 2005;23(1):70–78. doi: 10.1200/JCO.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 87.Sundin A. Radiological and nuclear medicine imaging of gastroenteropancreatic neuroendocrine tumours. Best Pract Res Clin Gastroenterol. 2012;26(6):803–818. doi: 10.1016/j.bpg.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 88.Ferrara K, Pollard R, Borden M. Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annu Rev Biomed Eng. 2007;9:415–447. doi: 10.1146/annurev.bioeng.8.061505.095852. [DOI] [PubMed] [Google Scholar]
- 89.Hoeffel C, Job L, Ladam-Marcus V, Vitry F, Cadiot G, Marcus C. Detection of hepatic metastases from carcinoid tumor: prospective evaluation of contrast-enhanced ultrasonography. Dig Dis Sci. 2009;54(9):2040–2046. doi: 10.1007/s10620-008-0570-x. [DOI] [PubMed] [Google Scholar]
- 90.Delle Fave G, Kwekkeboom DJ, Van Cutsem E, et al. ENETS Consensus Guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology. 2012;95(2):74–87. doi: 10.1159/000335595. [DOI] [PubMed] [Google Scholar]
- 91.Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic Ultrasound is Highly Accurate and Directs Management in Patients with Neuroendocrine Tumors of the Pancreas. Am J Gastroenterol. 2000;95:2271–2277. doi: 10.1111/j.1572-0241.2000.02480.x. [DOI] [PubMed] [Google Scholar]
- 92.Bellutti M, Fry LC, Schmitt J, et al. Detection of neuroendocrine tumors of the small bowel by double balloon enteroscopy. Dig Dis Sci. 2009;54(5):1050–1058. doi: 10.1007/s10620-008-0456-y. [DOI] [PubMed] [Google Scholar]
- 93.van Tuyl SA, van Noorden JT, Timmer R, Stolk MF, Kuipers EJ, Taal BG. Detection of small-bowel neuroendocrine tumors by video capsule endoscopy. Gastrointest Endosc. 2006;64(1):66–72. doi: 10.1016/j.gie.2006.01.054. [DOI] [PubMed] [Google Scholar]
- 94.Alsohaibani F, Bigam D, Kneteman N, Shapiro AMJ, Sandha GS. The impact of preoperative endoscopic ultrasound on the surgical management of pancreatic neuroendocrine tumors. Can J Gastroenterol. 2008;22(10):817–820. doi: 10.1155/2008/868369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sundin A, Eriksson B, BergstrÖM M, LÅNgstrÖM B, ÖBerg K, ÖRlefors H. PET in the Diagnosis of Neuroendocrine Tumors. Annals of the New York Academy of Sciences. 2004;1014(1):246–257. doi: 10.1196/annals.1294.027. [DOI] [PubMed] [Google Scholar]
- 96.Bhate K, Mok WY, Tran K, Khan S, Al-Nahhas A. Functional assessment in the multimodality imaging of pancreatic neuroendocrine tumours. Minerva Endocrinologica. 2010;35(1):17–25. [PubMed] [Google Scholar]
- 97.Pasquali C, Rubello D, Sperti C, et al. Neuroendocrine Tumor Imaging: Can 18F-Fluorodeoxyglucose Positron Emission Tomography Detect Tumors with Poor Prognosis and Aggressive Behavior? World J of Surgery. 1998;22:588–592. doi: 10.1007/s002689900439. [DOI] [PubMed] [Google Scholar]
- 98.Bahri H, Laurence L, Edeline J, et al. High prognostic value of 18F-FDG PET for metastatic gastroenteropancreatic neuroendocrine tumors: a long-term evaluation. J Nucl Med. 2014;55(11):1786–1790. doi: 10.2967/jnumed.114.144386. [DOI] [PubMed] [Google Scholar]
- 99.Reubi JC, Krenning E, Lamberts SWJ. Distribution of Somatostatin Receptors in Normal and Tumor Tissue. Metabolism: Clinical and Experimental. 1990;39(9):78–81. doi: 10.1016/0026-0495(90)90217-z. [DOI] [PubMed] [Google Scholar]
- 100.Patel YC. Somatostatin and Its Receptor Family. Front Neuroendocrinol. 1999;20:157–198. doi: 10.1006/frne.1999.0183. [DOI] [PubMed] [Google Scholar]
- 101.Kwekkeboom DJ, Krenning EP, Scheidhauer K, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: somatostatin receptor imaging with (111)In-pentetreotide. Neuroendocrinology. 2009;90(2):184–189. doi: 10.1159/000225946. [DOI] [PubMed] [Google Scholar]
- 102.Reubi JC. Somatostatin and Other Peptide Receptors as Tools for Tumor Diagnosis and Treatment. Neuroendocrinology. 2004;80 (Suppl 1):51–56. doi: 10.1159/000080742. [DOI] [PubMed] [Google Scholar]
- 103.Lu SJ, Gnanasegaran G, Buscombe J, Navalkissoor S. Single photon emission computed tomography/computed tomography in the evaluation of neuroendocrine tumours: a review of the literature. Nucl Med Commun. 2013;34(2):98–107. doi: 10.1097/MNM.0b013e32835bd59d. [DOI] [PubMed] [Google Scholar]
- 104.Ambrosini V, Campana D, Tomassetti P, Fanti S. 68Ga-labelled peptides for diagnosis of gastroenteropancreatic NET. Eur J Nucl Med Mol Imaging. 2012;39:s52–s60. doi: 10.1007/s00259-011-1989-4. [DOI] [PubMed] [Google Scholar]
- 105.Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228–252. doi: 10.1016/j.yfrne.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 106.Brandon D, Alazraki A, Halkar RK, Alazraki NP. The role of single-photon emission computed tomography and SPECT/computed tomography in oncologic imaging. Semin Oncol. 2011;38(1):87–108. doi: 10.1053/j.seminoncol.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 107.Bural GG, Muthukrishnan A, Oborski MJ, Mountz JM. Improved Benefit of SPECT/CT Compared to SPECT Alone for the Accurate Localization of Endocrine and Neuroendocrine Tumors. Mol Imaging Radionucl Ther. 2012;21(3):91–96. doi: 10.4274/Mirt.80299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zanzonico P. Principles of nuclear medicine imaging: planar, SPECT, PET, multimodality, and autoradiography systems. Radiat Res. 2012;177(4):349–364. doi: 10.1667/rr2577.1. [DOI] [PubMed] [Google Scholar]
- 109.Krausz Y, Keidar Z, Kogan I, et al. SPECT/CT hybrid imaging with 111In-pentetreotide in assessment of neuroendocrine tumours. Clinical Endocrinology. 2003;59:565–573. doi: 10.1046/j.1365-2265.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 110.Kumbasar B, Kamel IR, Tekes A, Eng J, Fishman EK, Wahl RL. Imaging of neuroendocrine tumors: accuracy of helical CT versus SRS. Abdom Imaging. 2004;29(6):696–702. doi: 10.1007/s00261-003-0162-3. [DOI] [PubMed] [Google Scholar]
- 111.Shi W, Johnston CF, Buchanan KD, et al. Localization of neuroendocrine tumours with 111In-DTPA-octreotide scintigraphy (Octreoscan): a comparitive study with CT and MR imaging. Q J Med. 1998;91:295–301. doi: 10.1093/qjmed/91.4.295. [DOI] [PubMed] [Google Scholar]
- 112.Gabriel M, Decristoforo C, Kendler D, et al. 68Ga-DOTA-Tyr3-Octreotide PET in Neuroendocrine Tumors: Comparison with Somatostatin Receptor Scintigraphy and CT. Journal of Nuclear Medicine. 2007;48(4):508–518. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]
- 113.Maxwell JE, Sherman SK, Menda Y, Wang D, O’Dorisio TM, Howe JR. Limitations of somatostatin scintigraphy in primary small bowel neuroendocrine tumors. J Surg Res. 2014;190(2):548–553. doi: 10.1016/j.jss.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Savelli G, Lucignani G, Seregni E, et al. Feasibility of somatostatin receptor scintigraphy in the detection of occult primary gastro-entero-pancreatic (GEP) neuroendocrine tumors. Nuclear Medicine Communications. 2004;25(5):445–449. doi: 10.1097/00006231-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 115.Naswa N, Sharma P, Kumar A, et al. Gallium-68-DOTA-NOC PET/CT of Patients with Gastroenteropancreatic Neuroendocrine Tumors: A Prospective Single-Center Study. AJR. 2011;197:1221–1228. doi: 10.2214/AJR.11.7298. [DOI] [PubMed] [Google Scholar]
- 116.Prasad V, Ambrosini V, Hommann M, Hoersch D, Fanti S, Baum RP. Detection of unknown primary neuroendocrine tumours (CUP-NET) using (68)Ga-DOTA-NOC receptor PET/CT. Eur J Nucl Med Mol Imaging. 2010;37(1):67–77. doi: 10.1007/s00259-009-1205-y. [DOI] [PubMed] [Google Scholar]
- 117.Buchmann I, Henze M, Engelbrecht S, et al. Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2007;34(10):1617–1626. doi: 10.1007/s00259-007-0450-1. [DOI] [PubMed] [Google Scholar]