

Abstract
Background:
Hemophagocytic lymphohistiocytosis (HLH) is an uncommon, potentially fatal, hyperinflammatory syndrome that may rarely complicate the clinical course of disseminated Mycobacterium tuberculosis (MTB). The clinical course of tuberculosis-associated HLH (TB-HLH) has been reported to be unpredictable.
Materials and Methods:
Here we describe the clinicopathological features, laboratory parameters, management, and outcome data of a patient who satisfied the 2004 diagnostic criteria for HLH secondary to disseminated MTB; we also do a systematic review of the international literature on TB-HLH. The literature review (January 1975–March 2014) found that HLH complicated the clinical course of 63 tuberculosis patients (41 males, 22 females, mean age = 45 ± 23.5 years) with a high mortality rate of 49% (31/63 died). The mean serum ferritin level (n = 44/63) was 5963 ng/mL (range 500–38,539 ng/mL); and a higher proportion (54.2%) of patients had pancytopenia at presentation. On univariate analysis (n = 53/63), age >30 years [hazard ratio (HR): 2.79, 95% confidence interval (CI):1.03–7.56, P = 0.03], presence of comorbidities (HR 4.59, CI: 1.08–19.52, P = 0.04), marked hemophagocytosis in bone marrow (HR: 2.65, CI: 1.16–6.05, P = 0.02), and nonusage/delayed usage of antitubercular therapy (ATT) (HR: 3.44, CI: 1.51–7.87, P = 0.003) were associated with decreased survival, though none of these parameters attained statistical significance (P > 0.05) in multivariate analysis. Usage of corticosteroids and/or immunomodulator drugs (HR 1.00, CI: 0.66–3.22, P = 0.35) did not alter the outcome in these patients.
Conclusion:
HLH should be considered as a differential diagnosis in patients with tuberculosis who present with cytopenias, organomegaly, and coagulopathy. Strong clinical suspicion and early usage of ATT might be useful in reducing the morbidity and mortality. The utility of immunosuppressive/immunomodulator therapy lacks general concensus among treating physicians, and warrants further studies.
KEY WORDS: Antitubercular therapy, hemophagocytic lymphohistiocytosis, survival, tuberculosis
INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is an often fatal syndrome of exacerbated but ineffective inflammatory responses, characterized by excessive macrophage and T-cell activation as well as impairment of the ability of natural killer (NK) cells and cytotoxic T lymphocytes to kill the target cells. This results in uncontrolled histiocytic phagocytosis of mature blood elements and their precursors throughout the reticuloendothelial organs, and associated cytokine-mediated multiorgan dysfunction.[1,2] Primary or familial HLH appears to have a genetic basis, whereas secondary or acquired HLH may be associated with infections (commonly the Epstein–Barr virus or EBV, bacteria, Rickettsia, etc.), hematological malignancies (mostly T/NK cell leukemias/lymphomas), rheumatological/autoimmune disorders (so-called macrophage activation syndrome), etc.[3,4] The diagnosis is established by fulfilling one of the following HLH 2004 criteria: i) Positive family history or molecular diagnosis consistent with HLH (mutations of PRF, SAP, or Munc13-4 genes), ii) any five out of the following eight criteria: Prolonged fever, unexplained progressive cytopenias involving at least two cell lines (hemoglobin ≤90 g/L, platelet count ≤100 × 109/L, absolute neutrophil count <1× 109/L); splenomegaly; hyperferritinemia (≥500 ng/mL); fasting hypertriglyceridemia (≥265 mg/dL) or hypofibrinogenemia (≤1.5 g/L); histiocytic hemophagocytosis in bone marrow, liver, spleen, or lymph nodes without evidence of malignancy; low or absent NK cell cytotoxicity; and elevated soluble CD25 levels (≥2400 IU/mL of interleukin-2Rα chain).[5]
Tuberculosis, being a chronic disease, remains a common health problem in Southeast Asian and other underdeveloped countries, with significant morbidity and mortality. The causative organism Mycobacterium tuberculosis (MTB) is known as a “great mimicker” and has a diverse range of clinical manifestations. Tuberculosis may rarely be complicated by HLH, which may be diagnostically challenging to the treating physicians, and in the absence of early and definitive therapy may lead to significant morbidity and mortality.[6]
In this manuscript, we describe a patient, with tuberculosis-associated HLH with a favorable outcome following early initiation of antitubercular therapy (ATT). We also present a brief, concise, systematic review of all relevant international literature regarding tuberculosis-associated HLH (TB-HLH) with regard to the clinicopathological characteristics, immunopathology, and therapeutic outcome.
MATERIALS AND METHODS
Presentation of a case
A 17-year-old Indian male, student by occupation, required admission into the intensive care unit of our Institute with persistent high-grade fever, cough with expectoration, worsening breathlessness, tachypnea, bilateral coarse crepitation, and hypoxemia suggestive of acute respiratory distress syndrome (ARDS), thus requiring ventilator support. Prior to the present admission he had been evaluated at an outside hospital for a 2-month history of persistent low-grade fever, night sweats, and weight loss (5 kg in the last month), possibly of tubercular origin. On examination he was found to be thin of build, febrile (oral temperature: 103°F) with significant conjunctival pallor, scleral icterus, bilateral subcentimeter cervical and axillary lymphadenopathy, hepatosplenomegaly (liver 4 cm, spleen 6 cm below the costal margin), bilateral coarse crepitation (anterior and midaxillary lines), and mild ascites. Radiological evaluation showed miliary shadows in bilateral lung fields, hepatosplenomegaly without focal lesions, and ascites. Routine laboratory evaluation on day 2 revealed microcytic hypochromic anemia [hemoglobin: 68 g/L (120–140 g/L), mean corpuscular volume (MCV): 70 fL (80–98 fL)], leukopenia [leukocyte count: 1.3 × 109/L (ref.; 4–11 × 109/L), absolute neutrophil count (ANC): 790/cmm], total platelet count: 160 × 109/L (150–450 × 109/L), total bilirubin: 3.4 mg/dL (direct 1.6 mg/dL), raised liver transaminases (>1.5 times the upper limit of normal), prolonged prothrombin time [27 s, control 12 s, international normalized ratio (INR) 1.2] and activated partial thromboplastin time (45 s, control 26 sec), and negative D-dimer. Plasma fibrinogen and serum triglyceride levels were within normal reference range. Microbiological and serological work-up were negative for human immunodeficiency virus (HIV), hepatitis B and C viruses, dengue, malaria, Leptospira, Brucella, and scrub typhus; and microbial cultures of blood, urine, sputum, and brochoalveolar lavage fluid were sterile. In view of persistent fever, worsening cytopenias, and organomegaly, bone marrow aspiration and trephine biopsy were performed on day 4 post admission, which showed multiple well-formed, caseating epithelioid granulomas, suggestive of tuberculosis. Besides these, there was evidence of a marked degree of histiocytic hemophagocytosis in the bone marrow aspirate smears. The assessment of histiocytic hemophagocytosis on bone marrow aspirate smears was done as per the method devised by Ho et al. (low/mild: 1–5 hemophagocytic cells per entire slide, moderate: 6–10 hemophagocytic cells per entire slide; and >10 hemophagocytic cells per entire slide).[7] Biochemical evaluation revealed hyperferritinemia (>2000 ng/mL), raised lactate dehydrogenase (903 IU/L), hypoalbuminemia (2.5 g/dL), and hyponatremia (125 meq/L). Thus, the clinicolaboaratory parameters satisfied five of six diagnostic criteria for HLH,[5] as NK cell activity and soluble CD25 levels were not tested due to lack of facilities. The patient was managed with broad-spectrum intravenous antibiotics and supportive measures. Furthermore, he was started on four-drug ATT with isoniazid, rifampicin, pyrazinamide, and ethambutol. He responded dramatically to ATT and was finally discharged in a stable condition on day 14 post admission with hemoglobin of 80 g/L, ANC of 1300/cmm, and platelet count of 170 × 109/L. On follow-up he was found to be responding well to ATT, and is presently on a two-drug continuation phase of ATT with isoniazid and rifampicin.
Collection of data on TB-HLH
A systematic search of HLH that complicated the clinical course of tuberculosis, over the last 39 years (January 1975–March 2014), was done by searching the PubMed, PubMed Central, Medline, and Directory of Open Access Journal databases. The following keywords were used for the literature search: Tuberculosis-associated HLH, tuberculosis-associated hemophagocytosis/hemophagocytic syndrome, macrophage activation and tuberculosis, and tuberculosis complicated with HLH. The references of all articles were cross-checked for relevant articles. In 2006, Brastianos et al. reported a case of TB-HLH and reviewed 36 similar cases reported around the world till that time.[7] A systematic search found 27 more articles from 2006 till date. A total of 55 articles describing nearly 70 cases of TB-HLH (including 37 reviewed by Brastianos et al.) were found in the world literature till March 2014.[7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34] The following data were collected for TB-HLH cases and placed in Microsoft office Excel 2007 for statistical analysis: Age, gender, duration of symptoms, associated comorbidities, organomegaly, complete blood count, serum biochemical parameters (ferritin, triglyceride, fibrinogen, liver transaminases, alkaline phosphatase, total bilirubin, albumin, and lactate dehydrogenase), coagulation abnormalities, evidence of hemophagocytosis on bone marrow evaluation, source and method of isolation of the organism, usage of ATT and/or immunosuppressive/immunomodulators drugs, and final outcome (alive or dead). Finally, complete information was available in 63 out of 70 cases, and the remaining 7 were excluded due to inaccessibility of the complete articles and/or lack of adequate information.
Statistical analysis
Baseline characteristics were described using mean (±standard deviation) for continuous variables, and count (percentage) for nominal variables. Among 53 of 63 cases, the follow-up duration from the diagnosis was not reported; hence the symptom duration till diagnosis for each case was taken as the observation period for analysis and taken as overall survival for the patients. Cox proportional hazard models were used to estimate the hazard ratio (HR) and the 95% confidence interval (CI) for death due to TB. Factors that had P values less than 0.10 in univariate analysis were included in the multivariate analysis. In addition, the Kaplan–Meier method was used to determine survival patterns for variables identified as having a significant effect on survival. Differences between survival curves were evaluated using log-rank tests. All statistical tests were two-sided; P values of less than 0.05 were considered to be statistically significant. Analyses were performed using IBM SPSS statistics (version 20.0).
DISCUSSION
The hallmark pathophysiologic mechanism of HLH is exacerbated but deregulated Th1 cell-mediated immune response against an intracellular pathogen, macrophage hyperactivity, widespread hemophagocytosis, and hypercytokinemia leading to multiorgan dysfunction. This results due to impaired or suppressed function of cytotoxic T cells and NK cells to effectively clear the antigenic stimulus and thus turn off the inflammatory response.[1] Splenomegaly is the result of activation and proliferation of splenic macrophages, as evidenced by the increased expression of major histocompatibility complex (MHC)-I and MHC-II molecules, as well as macrophage colony-stimulating factor (M-CSF) receptors. The cytokine milieu in HLH is characterized by increased levels of interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), granulocyte monocytes colony-stimulating factor (GM-CSF), and interleukin-18 (IL-18). The bi/pancytopenia in HLH is likely the consequence of high TNF-α and IFN-γ leading to suppressed hematopoiesis and increased apoptosis. While TNF-α– and IFN-γ–mediated inhibition of lipoprotein lipase leads to hypertriglyceridemia, macrophage overactivity might explain excess release of ferritin (an acute-phase reactant) in response to inflammatory cytokines, tissue infiltration leading to organomegaly, and fever (secondary to IL-1, IL-6).[1] Therefore MTB, being an obligate intracellular pathogen, may exacerbate Th1 cell-mediated cytotoxicity and macrophage overactivity, which may lead to HLH in susceptible individuals, as is evidenced by increased serum levels of IFN-γ, M-CSF, and TNF-α in patients with tuberculosis.[31]
Associated comorbidities in TB-HLH
Concise, systematic details of the clinicopathological characteristics of all the 63 cases of TB-HLH are presented in Tables 1 and 2. There were 41 males and 22 females, and the mean age was 45 years (±23.5 years) (range 14 days–83 years). A high proportion (41/63, 65%) of patients had underlying comorbidities; 11 of 41 (26.8%) patients had end-stage renal disease and were receiving either hemodialysis (n = 8)or had undergone renal transplant (n = 3); 4 had type 2 diabetes mellitus (2 with nephropathy), 6 had a past history of malignancy (2 Hodgkin lymphoma, 2 non-Hodgkin lymphoma, 1 myelodysplastic syndrome, 1 small-cell lung carcinoma), 5 had underlying autoimmune diseases (2 systemic lupus erythematosus, 1 ankylosing spondylitis, 1 Wegener's granulomatosis, 1 Crohn's disease), 1 had sarcoidosis, and 3 had HIV/acquired immune deficiency syndrome (AIDS) (1 with coexistent EBV infection). Prior history of tuberculosis was present in 3 adults, and 2 neonates contracted the infection from their mothers (perinatal tuberculosis). Chamsi-Pasha et al. reported the only case of Mycobacterium avium complex (MAC)-associated HLH in a sickle cell patient (homozygous) with a fatal outcome.[10] Naha et al., reported a case of disseminated tuberculosis with secondary HLH and the presence of tuberculosis-associated reactive arthritis (Poncet's disease), in an immunocompetent male.[12] Mancebo et al. reported the first case of familial HLH in an adult patient with PRF1 gene mutation and coexistent tuberculosis.[33] The mean duration of symptoms before diagnosis of TB-HLH was 51 days (±47.3 days) (median 45 days), and in 8 patients (12.7%) the diagnosis of tuberculosis was made at autopsy; this was an indirect reflection of disease chronicity, delay in diagnosis, and thus a delay in initiation of ATT. In 3 patients, HLH was reported to be exacerbated after initiation of ATT, which required stoppage of ATT or a change of drug regimen.[8,25]
Table 1.
Clinicopathological characteristics of tuberculosis associated hemophagocytic lymphohistiocytosis published in the world literature (January 1975- March 2014) (n=63)
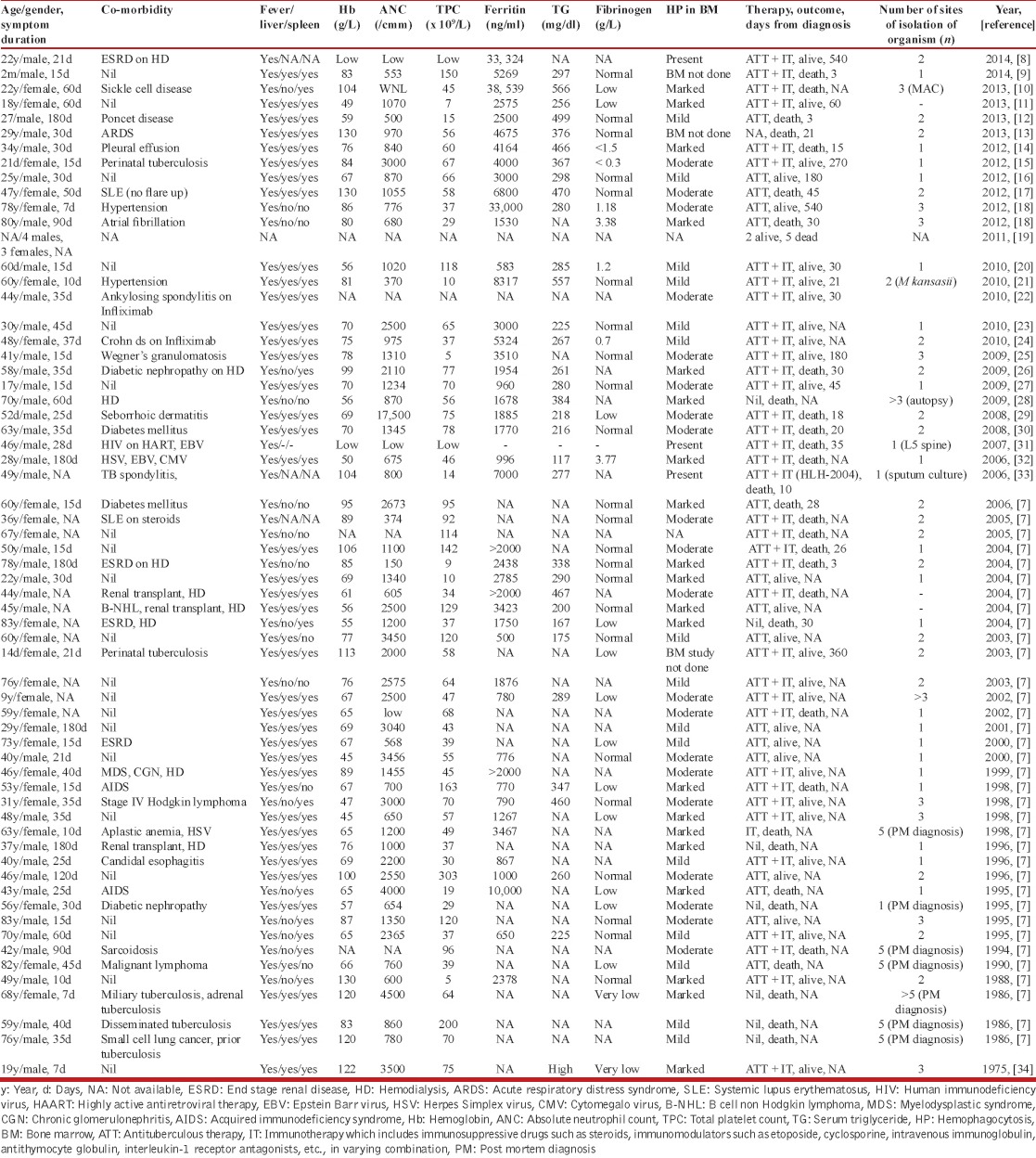
Table 2.
Clinicopathological characteristics of patients with tuberculosis associated hemophagocytic lymphohistiocytosis (January 1975 till March 2014)

HLH diagnostic features
HLH diagnosis was made in all cases by a constellation of fever, organomegaly, cytopenia (s), elevated serum ferritin and triglyceride levels with or without lower plasma fibrinogen, and demonstration of histiocytic hemophagocytosis on bone marrow examination. Fever was the most common clinical presentation and was present in all cases. Hepatosplenomegaly and lymphadenopathy were observed in 43 of 61 cases (70.5%), and 6 (9.8%) cases had isolated splenomegaly. A higher proportion [32/59 (54.2%)] of patients had pancytopenia, and 22/59 (37.2%) had bicytopenia, which was present at the time of initial presentation or developed during the course of disease evolution. The mean hemoglobin (Hb) (n = 56), absolute neutrophil count (ANC) (n = 55), and total platelet count (TPC) (n = 58) of cases (where data were available) were 78.45 ± 22.36 g/L, 1548 ± 1052/cmm, and 67 ± 51.9 × 109/L respectively. Similarly, the median and interquartile range of Hb, ANC, and TPC among cases were 72.5 g/L (65.0–88.75), 1100/cmm (760–2500), and 57.5 × 109/L (37.0–77.25), respectively. The mean and median serum ferritin values (n = 44) among cases were 5963 ng/mL and 1955 ng/mL (interquartile range 960–3510) respectively. Serum triglyceride levels were measured in 31 cases; the mean and median values were found to be 307.5 ± 108.6 mg/dL, and 285 mg/dL (interquartile range: 225–376), respectively. Furthermore, among 45 cases where data were available, plasma fibrinogen was reported as low (<1.5 g/L) among 22 (49%) cases; and it was within the normal reference range among the remaining 23 cases (51%). Bone marrow examination performed in all 63 cases revealed the presence of histiocytic hemophagocytosis in 58 (92%), which was reported as moderate to marked in intensity among 40 (69%) cases. This was possibly an indirect reflection of the persistent disease activity reflected by cytopenia (s), hyperferritinemia (mean: 5963 ng/mL), hepatic dysfunction, and consequent coagulation abnormality.
Diagnosis of tuberculosis
A schematic representation of diagnostic algorithm in all cases with tuberculosis is presented in Flow chart 1. Overall, among 40 of 60 cases where complete data were available (66.6%), the diagnosis of tuberculosis was made at ≥2 different anatomic sites by using either histopathological evaluation (n = 40, 66.6%) or by microbiological methods [demonstration of acid-fast bacilli by the Ziehl–Neelsen stain (n = 36, 60%), isolation by microbial culture (n = 33, 55%), or polymerase chain reaction (PCR) (n = 8, 13.3%)]. On the contrary, clinicoradiological features consistent with tuberculosis were present in only 10 cases (16.7%). On histopathological examination, granulomatous inflammations were demonstrated most commonly in the bone marrow (n = 23), lungs and/or pleura (n = 16), spleen (n = 10), liver (n = 8), and lymph nodes (n = 8). Similarly, among 33 cases where the culture results were positive, the organisms were isolated from 52 different anatomic samples such as the respiratory tract (n = 16), extrapulmonary sites (n = 23) [bone marrow (n = 15), liver (n = 2), spleen (n = 2), and lymph node (n = 4)], and body fluids (n = 13). The isolate was reported as MAC in a sickle cell patient,[10] Mycobacterium kansasii in another case;[21] in the remaining cases it was reported as MTB. Therefore, it can be postulated that cases with disseminated tuberculosis were complicated by the process of HLH.
Flow chart 1.
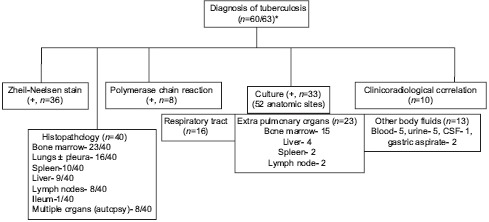
Diagnostic algorithm of all cases of tuberculosis who presented with hemophagocytic lymphohistiocytosis. +; positive, *; in 3 cases, no information was available. Note: in 33 of 60 cases, the disease was evident in ≥ 2 anatomic sites. Mycobacterium kansasii[21] and Mycobacterium avium intracellulare complex (MAC)[10] were isolated in one case each; and rest cases were attributable to Mycobacterium tuberculosis
Management and outcome of TB-HLH
The biological behavior of cases with TB-HLH was unpredictable, with a mortality rate of 49% (31/63 died); the overall survival at 3 months was 45% [Figure 1a]. Fifty-four of the 62 cases where data were available received treatment: either ATT alone (n = 16; 10 survived, 62.5%) or in combination with immunosuppressive (steroids and/or intravenous immunoglobulin) and/or immunomodulators (n = 37; 20 survived, 54%). The following immunomodulator drugs were used: Cyclosporine in 3, etoposide in 2, cyclosporine and etoposide in 1, vincristine in 1, chlorambucil and fludarabine in 1, and IL-1 receptor antagonist in 2 cases. None of the 8 cases who did not receive any therapy survived, and 1 who received only immunotherapy also did not survive. In most cases, the failure of therapy was attributed to delayed diagnosis and/or initiation of therapy late in the course of the illness. On univariate analysis, age >30 years (HR: 2.79, 95% CI: 1.03–7.56, P = 0.03), presence of comorbidities (HR: 4.59, CI: 1.08–19.52, P = 0.04), presence of marked hemophagocytosis in bone marrow (HR: 2.65, CI: 1.16–6.05, P = 0.02), and delayed/nonusage of ATT (HR: 3.44, CI: 1.51–7.87, P = 0.003) were significantly associated with decreased survival [Table 3, Figure 1b-g].
Figure 1.

(Survival patterns in patients with tuberculosis associated hemophagocytic lymphohistiocytosis (TB-HLH) in relation to different parameters by Kaplan-Meier analysis using log-rank test. (a) Overall survival in patients with TB-HLH was approximately 45% after 3 months. On univariate analysis, (b) age > 30 years (P = 0.03); (c) presence of co-morbidity (P = 0.02); (d) evidence of moderate to marked degree of bone marrow hemophagocytosis (P = 0.01); and (e) non usage/delayed usage of antitubercular therapy (P = 0.001) were significantly associated with decreased survival. Usage of immunomodulators and/or immunosuppressive drugs (f) did not contribute significantly (P = 0.33) to the improved survival. High ferritin (>1000 ng/ml) was associated with poor survival; though it was not statistically significant (P = 0.25) (g)d
Table 3.
Cox proportional Hazard model result in tuberculosis associated hemophagocytic lymphohistiocytosis (n=53/63)
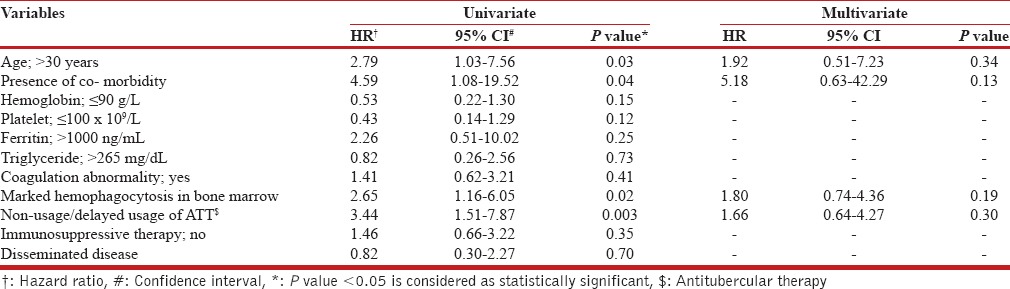
Highly elevated ferritin is strongly associated with HLH and its levels may provide a prognostic marker. Lin et al. suggested that a rapid rate of fall in ferritin levels following therapy initiation was associated with decreased mortality.[35] However, Park et al. in their cohort of 23 patients with secondary HLH found that the rate of decline in ferritin was not associated with survival, and that high fibrinogen at the time of diagnosis was significantly associated with survival.[36] We found that high serum ferritin (>1000 ng/mL) was an important indicator of disease severity and was associated with decreased survival, though the impact was statistically insignificant on univariate analysis (HR: 2.26, CI: 0.51–10.02, P = 0.25) [Figure 1g]. Paradoxically, the literature review also showed that patients with low hemoglobin (≤90 g/L) (P = 0.15) and low platelet count (≤100 × 109/L) (P = 0.12) did not affect the outcome in a Cox regression model [Table 3]. Similarly, the presence of disseminated disease, coagulation abnormalities, and usage of immunosuppressive and/or immunomodulators in therapy [Figure 1f] were not significantly associated with survival. However, the interpretation of outcomes from univariate analysis must be interpreted with great caution, as the cohort of patients reported in the literature is small and heterogenous, and the therapeutic strategies differed from case to case.
Comparative analysis of intensive chemotherapy and conventional combination therapy, in the management of HLH, has not shown any difference in outcomes. However, there exists general agreement that early treatment may improve outcomes. Imashuku et al.[37] have suggested that young adult patients receiving early etoposide treatment have a better prognosis than those not treated with etoposide or treated late. However, a recent Korean study by Park et al.[36] did not find any differences in patient outcomes between the two groups based on therapy (HLH protocol vs immunosuppressive therapy). Furthermore, as rightly pointed out by Park et al.,[36] in the patients with severe disease and/or associated sepsis or multiple organ failure at the time of diagnosis, it may be difficult to use cytotoxic agents such as etoposide. In such circumstances, immunosuppression with corticosteroids and/or cyclosporine remains the foundation of early management as it can control systemic inflammation. At present, there are no guidelines as to when and which patients with secondary HLH require the complete HLH 2004 regimen, although targeted therapy for the underlying disease is crucial in determining outcome.
CONCLUSION AND FUTURE PERSPECTIVE
HLH should be considered as a differential diagnosis in patients with tuberculosis who present with cytopenia(s), organomegaly, and coagulopathy. The existing literature points to the fact that TB-HLH may have an unpredictable and/or unfavorable outcome with or without ATT. Early diagnosis and initiation of ATT, even in the presence of disseminated disease, might alter the final outcome in these cases. First-line antituberculous drugs such as rifampicin have enzyme-inducing activity, which can lower the efficacy of drugs such as cyclosporine and etoposide used in the HLH 2004 protocol. Besides, HLH per se leads to significant derangement of liver functions, making the administration of ATT as well as etoposide difficult. Moreover, as was evident in 3 cases, HLH may even be exacerbated after initiation of ATT, which may be challenging to treat.[8,25] Therefore, it is open to speculation whether HLH per se or the delay in initiation of ATT is the predictor of unfavorable outcome in these cases. The best approach would be determination of treatment priorities based on the clinical condition of the patient and individualization of the treatment plan according to clinicolaboratory parameters, as was evident in our case. Finally, the utility of the HLH 2004 protocol in patients with TB-HLH seems to be controversial at present, and warrants larger future prospective studies.
Author contribution
SP did the conceptual design, collection, acquisition, and interpretation of the data, writing, and editing of the final manuscript. KR did the statistical analysis. JPS and AB did the collection and acquisition of data, and reviewed the literature. RGV reviewed and analyzed the manuscript for the intellectual content. All authors agreed as to the final content of the manuscript.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Rosado FG, Kim AS. Hemophagocytic lymphocytosis: An update on diagnosis and pathogenesis. Am J Clin Pathol. 2013;139:713–27. doi: 10.1309/AJCP4ZDKJ4ICOUAT. [DOI] [PubMed] [Google Scholar]
- 2.Bode SF, Lehmberg K, Maul-Pavicic A, Vraetz T, Janka G, Stadt UZ, et al. Recent advances in the diagnosis and treatment of hemophagocytic lymphohistiocytosis. Arthritis Res Ther. 2012;14:213. doi: 10.1186/ar3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: Clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6:137–54. doi: 10.1586/eci.09.58. [DOI] [PubMed] [Google Scholar]
- 4.Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with hemophagocytic syndrome. Lancet Infect Dis. 2007;7:814–22. doi: 10.1016/S1473-3099(07)70290-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 6.Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated hemophagocytic syndrome. Lancet Infect Dis. 2006;6:447–54. doi: 10.1016/S1473-3099(06)70524-2. [DOI] [PubMed] [Google Scholar]
- 7.Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment of hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014;141:62–71. doi: 10.1309/AJCPMD5TJEFOOVBW. [DOI] [PubMed] [Google Scholar]
- 8.Seminari E, Contardi G, Rubert L, Fronti E, Comoli P, Minoli L, et al. Tuberculosis-induced haemophagocytic syndrome in a patient on haemodialysis treated with anti-thymocyte globulin. Int J Tuberc Lung Dis. 2014;18:248–9. doi: 10.5588/ijtld.13.0533. [DOI] [PubMed] [Google Scholar]
- 9.Dey A, Shah I, Paikrao P, Iyenger V. Tuberculosis with hemophagocytic lymphohistiocytosis in an infant. Indian J Pediatr. 2014;81:214–5. doi: 10.1007/s12098-013-1022-y. [DOI] [PubMed] [Google Scholar]
- 10.Chamsi-Pasha MA, Alraies MC, Alraiyes AH, Hsi ED. Mycobacterium avium complex-associated hemophagocytic lymphohistiocytosis in a sickle cell patient: An Unusual Fatal Association. Case Rep Hematol 2013. 2013 doi: 10.1155/2013/291518. 291518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherif E, Feki NB, Hassine LB, Khalfallah N. Hemophagocytic syndrome with disseminated intravascular coagulation associated with tuberculosis. BMJ Case Rep. 2013;2013:bcr2013008743. doi: 10.1136/bcr-2013-008743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naha K, Dasari S, Vivek G, Prabhu M. Disseminated tuberculosis presenting with secondary haemophagocytic lymphohistiocytosis and Poncet's disease in an immunocompetent individual. BMJ Case Rep 2013. 2013 doi: 10.1136/bcr-2012-008265. pii: bcr2012008265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talluri MR, Uppin S, Paritala V, Kumar N, Challa S, Rao N, et al. Miliary tuberculosis: A rare cause of hemophagocytic lymphohistiocytosis. Chest. 2013;144:211A. [Google Scholar]
- 14.Aggarwal P, Kumar G, Dev N, Kumari P. Haemophagocytic lymphohistiocytosis: A cause for rare but fatal outcome in tuberculosis. BMJ Case Rep 2012. 2012 doi: 10.1136/bcr-2012-006982. pii: bcr2012006982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maheshwari P, Chhabra R, Yadav P. Perinatal tuberculosis associated hemophagocytic lymphohistiocytosis. Indian J Pediatr. 2012;79:1228–9. doi: 10.1007/s12098-011-0675-7. [DOI] [PubMed] [Google Scholar]
- 16.Rakotoson JL, Rakotonirainy OH, Moroyandsa F, Rakotoharivelo H, Rakotomizao JR, Andrianarisoa AC. Hemophagocytic syndrome secondary to cavitary pulmonary tuberculosis. Med Sante Trop. 2012;22:99–101. doi: 10.1684/mst.2012.0020. [DOI] [PubMed] [Google Scholar]
- 17.Halabi H, Hafiz W, Bawayan M, Maulawi A, Almoallim H. Mycobacterium tuberculosis-associated hemophagocytic syndrome in systemic lupus erythematosus: A case report. Turk J Rheumatol. 2012;27:267–70. [Google Scholar]
- 18.Shea YF, Chan JF, Kwok WC, Hwang YY, Chan TC, Ni MY, et al. Haemophagocytic lymphohistiocytosis: An uncommon clinical presentation of tuberculosis. Hong Kong Med J. 2012;18:517–25. [PubMed] [Google Scholar]
- 19.Tseng YT, Sheng WH, Lin BH, Lin CW, Wang JT, Chen YC, et al. Causes, clinical symptoms, and outcomes of infectious diseases associated with hemophagocytic lymphohistiocytosis in Taiwanese adults. J Microbiol Immunol Infect. 2011;44:191–7. doi: 10.1016/j.jmii.2011.01.027. [DOI] [PubMed] [Google Scholar]
- 20.Deshpande A, Nayar PS, Pradhan AM, Manchanda RV. Miliary tuberculosis and hemophagocytosis in a two months old infant. Indian J Hematol Blood Transfus. 2010;26:115–7. doi: 10.1007/s12288-010-0038-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou YH, Hsu MS, Sheng WH, Chang SC. Disseminated Mycobacterium kansasii infection associated with hemophagocytic syndrome. Int J Infect Dis. 2010;14:e262–4. doi: 10.1016/j.ijid.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 22.Troncoso Mariño A, Campelo Sánchez E, Martínez López de Castro N, Inaraja Bobo MT. Haemophagocytic syndrome and paradoxical reaction to tuberculosis after treatment with Infliximab. Pharm World Sci. 2010;32:117–9. doi: 10.1007/s11096-010-9369-x. [DOI] [PubMed] [Google Scholar]
- 23.Sandrini I, Beucher AB, Rousselet MC, Gardembas M, Lavigne C. Tuberculosis with a hemophagocytic syndrome. Med Mal Infect. 2010;40:476–9. doi: 10.1016/j.medmal.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Namakura T, Matsuura Y, Takiguchi H, Hara Y, Ameku K, Takashi H. Tuberculosis associated hemophagocytic syndrome complicated by treatment with Infliximab. Nihon Kokyuki Gakkai Zasshi. 2010;48:449–53. [PubMed] [Google Scholar]
- 25.Balkis MM, Bazzi L, Taher A, Salem Z, Uthman I, Kanj N, et al. Severe hemophagocytic syndrome developing after treatment initiation for disseminated Mycobacterium tuberculosis: Case report and literature review. Scan J Infect Dis. 2009;41:535–7. doi: 10.1080/00365540902978075. [DOI] [PubMed] [Google Scholar]
- 26.Su NW, Chen CK, Chen GS, Hsieh RK, Chang MC. A case of tuberculosis-induced hemophagocytic lymphohistiocytosis in a patient under hemodialysis. Int J Hematol. 2009;89:298–301. doi: 10.1007/s12185-009-0265-x. [DOI] [PubMed] [Google Scholar]
- 27.Gupta AP, Parate SN, Bobhate SK; Anupriya. Hemophagocytic syndrome: A cause for fatal outcome in tuberculosis. Indian J Pathol Microbiol. 2009;52:260–2. doi: 10.4103/0377-4929.48939. [DOI] [PubMed] [Google Scholar]
- 28.Hori M, Yoshida R, Aoyama I, Ichida S. Case of tuberculosis-associated hemophagocytic syndrome in a hemodialysis patient under steroid therapy. Nippon Jinzo Gakkai Shi. 2009;51:1091–5. [PubMed] [Google Scholar]
- 29.Balsubramanian S, Kaarthigeyan K, Aparna V, Srinivas S. Tuberculosis associated hemophagocytic syndrome in infancy. Indian Pediatr. 2008;45:593–5. [PubMed] [Google Scholar]
- 30.Lee SW, Wang CY, Lee BJ, Kuo CY, Kuo CL. Hemophagocytic syndrome in miliary tuberculosis presenting with noncaseating granulomas in bone marrow and liver. J Formos Med Assoc. 2008;107:495–9. doi: 10.1016/S0929-6646(08)60158-8. [DOI] [PubMed] [Google Scholar]
- 31.Wong CK, Wong BC, Chan KC, Joynt GM, Yap FY, Lam CW, et al. Cytokine profile in fatal human immunodeficiency virus tuberculosis Epstein-Barr virus associated hemophagocytic syndrome. Arch Intern Med. 2007;167:1901–3. doi: 10.1001/archinte.167.17.1901. [DOI] [PubMed] [Google Scholar]
- 32.Khan FY, Fawzy Z, Siddiqui I, Yassin MA. Hemophagocytosis and miliary tuberculosis in a patient in the intensive care unit. Indian J Crit Care Med. 2006;10:112–4. [Google Scholar]
- 33.Mancebo E, Allende LM, Guzmán M, Paz-Artal E, Gil J, Urrea-Moreno R, et al. Familial hemophagocytic lymphohistiocytosis in an adult patient homozygous for A91V in the perforin gene, with tuberculosis infection. Haematologica. 2006;91:1257–60. [PubMed] [Google Scholar]
- 34.Chandra P, Chaudhery SA, Rosner F, Kagen M. Transient histiocytosis with striking phagocytosis of platelets, leukocytes, and erythrocytes. Arch Int Med. 1975;135:989–91. [PubMed] [Google Scholar]
- 35.Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56:154–5. doi: 10.1002/pbc.22774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park HS, Kim DY, Lee JH, Lee JH, Kim SD, Park YH, et al. Clinical features of adult patients with secondary hemophagocytic lymphohistiocytosis from causes other than lymphoma: An analysis of treatment outcome and prognostic factors. Ann Hematol. 2012;91:897–904. doi: 10.1007/s00277-011-1380-3. [DOI] [PubMed] [Google Scholar]
- 37.Imashuku S, Kuriyama K, Sakai R, Nakao Y, Masuda S, Yasuda N, et al. Treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: A report from the HLH study center. Med Pediatr Oncol. 2003;41:103–9. doi: 10.1002/mpo.10314. [DOI] [PubMed] [Google Scholar]