

Summary
Technological advances have paved the way for accelerated genomic discovery and are bringing precision medicine clearly into view. Epilepsy research in particular is well-suited to serve as a model for the development and deployment of targeted therapeutics in precision medicine because of the rapidly expanding genetic knowledge base in epilepsy, the availability of good in vitro and in vivo model systems to efficiently study the biological consequences of genetic mutations, the ability to turn these models into effective drug screening platforms, and the establishment of collaborative research groups. Moving forward, it is critical that we strengthen these collaborations, particularly through integrated research platforms to provide robust analyses both for accurate personal genome analysis and gene and drug discovery. Similarly, the implementation of clinical trial networks will allow the expansion of patient sample populations with genetically defined epilepsy so that drug discovery can be translated into clinical practice.
I. Introduction
In the decades after the initiation of the Human Genome Project, the idea that treatments could be targeted to genetically-defined subgroups of individuals has often been espoused but rarely realized. The advent of next-generation sequencing has promoted a new wave of enthusiasm, and a new name to go with it – precision medicine. In this Personal View, precision medicine refers to the scientific basis that underpins the personalization of medical care (cf1, “Moving toward precision medicine”, NRC committee report, 2011), particularly in the context of treatments targeted towards the precise molecular causes of disease. The realization of precision medicine is perhaps best illustrated in the specialty of cancer, in which mechanism-based treatments have successfully moved from bench to bedside.2 However, in most therapeutic areas, particularly in neurological disease, precision medicine remains aspirational.
Here we argue that, after cancer, epilepsy offers one of the most compelling opportunities to achieve precision medicine for the following fundamental and synergistic reasons: the rapid progress in epilepsy gene discovery; the existence of good animal and in-vitro models allowing the development of medications tailored to genetically defined subtypes of epilepsy, largely because neuronal excitability underlies epilepsy phenotypes and can often be accurately modelled in vitro; and the ability to assess efficacy of experimental targeted treatments in cost-effective, small, brief clinical trials. To realize the potential of precision medicine in epilepsy, however, many distinct areas of basic and translational research must coalesce into coherent and collaborative programmes. Here we outline a strategy for the development of an integrated programme for precision medicine in epilepsy, including the expansion of cohorts for research, the development of in vitro and in vivo animal models of disease, and strategies to perform genetically stratified clinical trials. We conclude that fostering integrated research teams to advance precision medicine in the epilepsies will improve health care that epilepsy could serve as a model for other therapeutic areas.
II. Precision Genetics for Precision Medicine
Molecular genetics research in epilepsy began more than 20 years ago and has entered a phase of rapid progress. This suggests that in the near future, at least some of the important genetic risk factors contributing to epilepsy will be identified in a substantial proportion of individuals with non-acquired epilepsy. Analyses of the genes implicated to date further indicate that genetically resolved epilepsies will eventually be grouped into larger sets that share common underlying biological causes or pathways.
Findings from traditional heritability studies3–7 and more recent genomic heritability analysis8 unequivocally show the important role of genetics in epilepsy risk. Before the development of next-generation sequencing, both linkage analyses and targeted candidate gene studies identified a number of epilepsy genes.9–21 Although these discoveries represented a substantial advance and illuminated novel aspects of disease pathophysiology, collectively these genes underlie epilepsy in only a small proportion of individuals with the disorder.
The role of common variation in epilepsy has also been assessed, both with candidate genes22 and comprehensive genome-wide association studies (GWAS) with generally limited findings.23–26 In parallel, chromosome microarrays have been used to identify copy-number variants that confer substantial risk of epilepsy.27–30 Although each copynumber variant confers significant risk, none is sufficient to cause epilepsy alone,27,31 and all variants are associated with several neuropsychiatric diseases.32,33 Collectively, these findings led researchers to focus on rare variants in epilepsy precisely when developments in next-generation sequencing facilitated the comprehensive interrogation of genomes.
The most common application of next-generation sequencing is to investigate the “exome,” or the set of nearly all protein-coding regions of the genome. Trio sequencing, in particular, in which the genomes of the individual with epilepsy and both parents are sequenced, is a successful method to identify new risk factors for the epileptic encephalopathies (panel),34,35 as well as for other neuropsychiatric diseases, including intellectual disability and autism spectrum disorders.36–40 In the EEs alone, trio-based analyses have led to the identification of ALG13,34 GABRB3,34 DNM1,35 HCN1,41 GRIN2A,42–44 GABRA145 GNAO1,46 KCNT1,47 SCN2A,48 SCN8A,49,50 and SLC35A251 as genes associated with epilepsy. Interestingly, and not surprisingly, many of the proteins encoded by these genes are involved in synaptic transmission.35 The characterization of the specific effects of mutations in these genes will help to resolve the precise biological pathways within synaptic transmission that are disrupted in epilepsy.52
Post-zygotic (somatic) de novo mutations (panel) that are present in only a subset of cells have also been identified as the cause of malformation syndromes associated with severe epilepsy. Recent examples include somatic mutations in AKT3, MTOR and PICK3CA as the cause of hemimegalencephaly and intractable seizures,53–55 and somatic mutations in DCX, LIS1, FLNA, and TUBB2B as the cause of double cortex syndrome, periventricular nodular heterotopia, and pachygyria.56
To date, progress has been modest in understanding less severe forms of epilepsy, almost certainly owing to a combination of very high locus and allelic heterogeneity and the possibility that combined effects of variants in multiple genes underlie susceptibility (Figure 1). We anticipate that larger sample sizes will soon be available to facilitate discovery in these more genetically complex epilepsies. These epilepsies might also depend on more subtle regulatory variants, requiring application of genomic and transcriptomic approaches, an emerging area of focus in common diseases including epilepsy. We note that epilepsies that occur in response to precipitating factors such as traumatic brain injury or brain tumours, although having some genetic component, will probably be less tractable targets for precision medicine than epilepsies whose aetiology is largely genetic.
Figure 1.
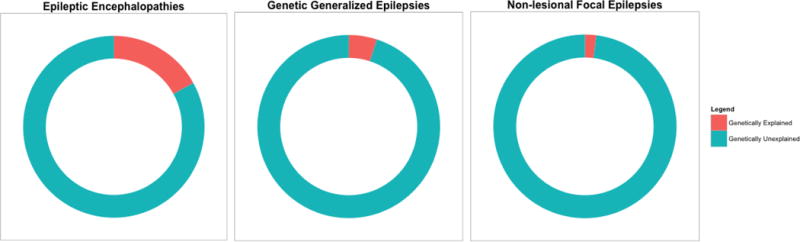
Estimated proportion of individuals with different types of epilepsy who carry a strong-acting, single mutation that either contributes substantially to or causes epilepsy.
Source: Kalachikov et al (2002),14 EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4k Consortium (2014),35 Epilepsy Phenome/Genome Project Epi4K Consortium (2015),57 Mefford et al (2011),58 Olson et al (2014),59 Dibbens et al (2013),60 Ishida et al (2013),61 Picard et al (2014),62 and Thomas et al (2014).63
Despite the progress towards the identification of epilepsy risk genes, precision medicine depends on the identification of mutations that contribute to disease in individual patients. This represents a distinct and more difficult challenge than the simple determination that a gene is involved in risk of epilepsy at the population level. The development of methods to quantitatively assess the degree of confidence that specific mutations contribute to disease in particular individuals is therefore a priority in epilepsy precision medicine. One recently developed approach to help pinpoint pathogenic mutations involves comparison of the patterns of genetic variation, including both types of mutations and their frequencies, between the general population and the population of individuals with disease. These analyses indicate that disease genes, particularly genes associated with neuropsychiatric disorders, tend to have less common functional variation in the general population than expected given the overall predicted mutability of the genes.64,65 Indeed, bioinformatic signatures that integrate such “gene level” scores with established variant level scores have been shown to be predictive of causative mutations in the genomes of individuals with severe early-onset diseases.65,66 A key focus of emerging efforts in epilepsy precision medicine will be to develop new statistical genetic approaches to expand these research areas, including analyses in diverse ethnic groups, and analyses of mutation patterns in non-coding genomic regions. In view of the need for expanded sample sizes, both for gene discovery and to facilitate accurate interpretation of individual genomes, genetic data generated in different locations must be integrated as much as possible. Importantly, data are being collected in commercial genetic testing laboratories that do exome sequencing on a fee-for-service basis, but these data are largely unavailable for research at this time. To make more effective use of these data, the epilepsy community has come together to establish the Epilepsy Genetics Initiative (EGI) to integrate clinical data collected in medical centers and to allow the integration of clinical data with research data. EGI has created a database to house the clinically sequenced exomes (and, in due course, sequenced genomes) and phenotypic data of individuals with epilepsy, one unique purpose of which is to allow on-going iterative reassessment of unsolved cases. Thus, EGI is a resource that brings together people with epilepsy, clinicians, and researchers in a mutually beneficial effort to advance precision diagnostics and epilepsy research. Central to these discussions is the perspective of families living with epilepsy (TJD-S, unpublished). Taken together, developments in gene discovery, bioinformatic prioritization of putative pathogenic mutations, and large-scale genetic data integration suggest that the genetic basis for precision medicine in epilepsy is now within reach. As an illustration, the list of known epilepsy genes that can form the initial basis for epilepsy precision medicine is growing (Figure 2).
Figure 2.

Genes known to be associated with epilepsy, broken out by those with relevant in-vitro models and those with research support from family foundations (A) List of known epilepsy genes. (B) The estimated percentage of epilepsy genes for which an in-vitro assay is available to accurately assess the effects of mutations (in red). (C) The percentage of epilepsy genes for which research is actively being driven by family foundations (in red).
Precision medicine in the epilepsies also has an equally important role in facilitating avoidance of adverse reactions as in maximising efficacy, as illustrated by a number of recent examples. In some cases, improved diagnostics will enable avoidance of adverse reactions, as is the case for patients with epilepsy due to POLG1 mutations who might develop fatal hepatic failure when treated with valproate.67 In other cases the risk factors for a severe adverse reaction will be independent of factors responsible for the disease; for example, the HLA-B*15:02 allele is highly predictive of carbamazepine-induced Stevens-Johnson syndrome, a severe hypersensitivity reaction, in patients of Asian origin.68
III. Functional Modeling
Translation of genetic causes into new or more targeted treatments depends on effective model systems that illuminate the underlying biology and contribute to the development of new drug-screening protocols. Importantly, advances in the functional assessment of epilepsyassociated mutations in the past two decades have shown a remarkable ability to dissect disease mechanisms using both single cell and whole animal models.
There are multiple interrelated motivations for the Thorough characterization of the effects of identified mutations at the RNA, protein, cellular, tissue, and whole organism levels. First, a mechanistic understanding of how mutations confer risk provides new directions for drug development. Second, it is increasingly clear that in vitro functional readouts for individual mutations can provide important information about pathogenic mechanisms (e.g., gain vs. loss-of-function), prognosis, and in some cases, treatment choices.69 The enthusiasm to model genetic epilepsies, and the need to model them well, as illustrated by the rapidly growing number of family foundations focused on funding specific genetic epilepsies (Figure 2).
Features of an ideal pre-clinical model of genetic epilepsy include the following: 1) efficient expression of the genetic risk factors identified in human beings; 2) accurate Representation of fundamental disease mechanisms, and pharmacosensitivity in human beings; 3) sufficient “scalability” to enable high-throughput screening of compounds; and 4) the ability to represent the full pathological consequences of mutations, including both the effect of the primary molecular defect and the emergent disease pathways. No single model will satisfy all criteria, which is why we suggest an integrated approach across models. As an illustration, the fastest throughput in experimental screening will probably still be in-vitro models, whereas faithful reflection of the developmental consequences of epilepsy mutations will inevitably be lacking. Although many approaches to modelling epilepsy mutations exist, we discuss three broad classes of functional models that together meet the above criteria. This focus is not meant to imply that there is no value in other model systems, but rather that the application of the classes of models discussed will probably serve as a general approach for precision medicine in epilepsy.
Single-cell models
In the simplest models, the mutated gene is incorporated into a cell line that does not normally express that gene,70 allowing for functional assessment of the mutant gene product in isolation. By far the most commonly used cell-types have been the human–derived HEK293 cell line and Xenopus oocytes. These cell-based platforms have been successfully applied to study voltage-gated and ligand-gated ion channels. Additionally, these systems permit multiple assays of molecular trafficking and processing of gene products. Studies of GABA receptor mutations found in human epilepsies exemplify how these systems have been used.71–74
Although artificial when compared to an in situ neuron, these simple models provide efficient ways to reveal functional mechanisms. These systems are also amenable to optimization for high-throughput screening. For these reasons, heterologous expression systems will continue to be a mainstay for modeling ion channel and transporter mutations.
Network scale models
A major limitation of single cell systems is the absence of network and support cell environments. As we suspect that many genetic causes of epilepsy result in non-cell-autonomous defects, the pathogenic consequences of many epilepsy mutations will probably be apparent only in the context of a neuronal network. Moreover, treatments that control epilepsy through indirect compensation of mutation effects can only be discovered only if many neuronal processes are represented in the model. To address these needs, in vitro models of interacting neurons are necessary.
Brain slices from rodent models provide a ready source of neuronal networks to assess disease mechanism and drug action. Acute and organotypic brain slices have been used in epilepsy research,75 and although desirable because they retain many of the large-scale features of the brain regions from which they are derived, brain slices are not amenable to scaling sufficiently to allow effective screening.
A promising new direction is the use of cultured neural networks (CNNs). The large-scale assessment of CNNs is facilitated by advances in non-invasive approaches to monitor activity in these networks. The development of multi-well, plate-based multi-electrode arrays (MEA) allows for screening of hundreds of networks per day. With the recent introduction of optophysiological and optogenetic approaches76 that can detect small and rapid changes in neuronal electrical state and can selectively stimulate neuronal populations, high-resolution interrogation of cultures has become possible. The use of CNNs, however, to study genetic epilepsy requires some form of manipulation of the cells in culture. Several approaches are possible. Most simply, CNNs can be developed by harvesting neuronal cells directly from mouse genetic models, allowing higher throughput screening than is possible in vivo. CNNs can also be developed directly using human cells from individuals with epilepsy, reprogramming those cells to become induced pluripotent stem cells (panel), and then differentiating them into appropriate cells, such as neurons and glia. An alternative approach is to introduce the mutation by genome editing using CRISPRCas (panel) or other editing approaches in a controlled isogenic line of induced pluripotent stem cells, followed by differentiation into relevant cell type(s). These approaches involving induced pluripotent stem cells provide an expandable source of cells to study humanspecific phenotypes and a human cellular model for replicating single-gene epilepsies. Both approaches have methodological challenges, particularly with respect to the development of sufficiently homogeneous populations of neuronal or glial cells. Neurons derived from induced pluripotent stem cells do not fully recapitulate mature neurons, but further elaborations on the horizon will allow more realistic in vitro models. These include cerebral organoids that develop more tissue-like cortical structure and lamination patterns77 and are amenable to so-called slice recording approaches. In principle, CNNs have all the advantages of a model that efficiently captures multiple aspects of the disease biology and pharmacology, and provide a direct pathway to the incorporation of human brain cells into the drug-discovery process.
Whole-animal models
Many model organisms have contributed to advances in epilepsy gene research, including rodents, flies, fish and invertebrates,78–83 with the mouse being the most successful and widely used. Experiments with mice have contributed substantial insights into epilepsy pathogenesis,84,85 and specific gene discoveries in the mouse have often anticipated later findings in human beings.83,86–88 Explicit gene targeting89–92 and editing84 now noticeably enhance the use of mice in precision medicine approaches. Whole animal models provide excellent behavioural and electroclinical correlates of seizure activity in the absence and presence of drugs. Because scalability in the whole animal model is a concern for in vivo screening, the use of lower organisms such as Caenorhabditis elegans (worm) or Danio rerio (zebrafish) afford improved throughput has also been undertaken. However, the advantage of scalability of lower organisms may be offset by the cost of losing the ability to evaluate some comorbid symptoms, including depression, anxiety, movement disorders, intellectual disability, and other cognitive impairments. Moreover, the identification of a drug that rescues a phenotype in a fish, worm, or fly will likely require validation in mice or in vitro human cell models before moving to clinical trials in human beings, although some promising therapeutic directions have emerged from studies in these organisms.93 Thus, mouse models of genetic epilepsy continue to be crucial as they fulfil several of the above criteria, including the provision of a ready source of neurons for ex-vivo single-cell and network analyses, most readily bridging clinically relevant spatiotemporal scales through sufficient behavioural complexity at the organism level. Importantly, use of mouse models allows screening for adverse effects on other organ systems.
Treatment modalities
Several factors must be considered in the search for candidate treatments for genetic subtypes of epilepsy, including potential toxicities of the treatment and the severity and prevalence of the disorder. Development strategies should be informed, to the extent possible, by the genetic and biological causes of the disease. The EEs illustrate all of these considerations most clearly since they often result from highly penetrant mutations in genes with at least partially elucidated biological roles in epileptogenesis.
To facilitate the development of new treatments for genetic epilepsies, a clear functional readout of the mutations that is both related to how the mutation causes disease and whether it is amenable to screening is essential. Once such information is available, assessment of drugs approved for other indications that revert the phenotype of interest (so-called “repurposing”) is an obvious priority. However, the fortuitous coincidence that an effective compound for a particular genetic epilepsy is already clinically available cannot be relied upon, and systems amenable to high-throughput screens for a range of new compounds should be a major priority.
A key first consideration in any screening effort is the “druggability” of the relevant gene product. Some researchers suggest that only 5% of human genes are both druggable and disease relevant.94 Although new small molecule strategies might increase the number of druggable targets, the development of alternative drug discovery approaches that rely on targeting alternative proteins in the same disease networks and pathways will be essential. For example, treatment of epilepsy caused by a loss-of-function mutation might be limited to treatment with compounds that activate compensatory mechanisms. Although a mutation can be appropriately expressed, some in vitro models may not have enough complexity to enable the assessment of compounds that compensate for deficiencies created by mutant proteins. However, we expect that the assessment of drug effects in CNNs, human cells, and mouse models in concert will be able to overcome these limitations.
In cases for which no active drugs are known and the screening of approved drug libraries fails to identify new treatments, consideration must be given to screening new chemical entities. Bioactive peptides from venom libraries95 are a rich source of molecules biologically selected to act on nervous system targets. Anti-sense oligonucleotides, currently in clinical trials for various disorders,96 can cause long-term knockdown of specific genes and are therefore compatible with gain-offunction mutations, and could potentially be applied in an allele-specific manner. Small molecule drug screening can provide a longer-term strategy for delivery of novel drugs using single cell and network assays as primary screens. Finally, viral delivery of genes or knockdown oligonucleotides has shown promise in mouse models,97,98 and efforts to target, “de-target,” and ensure the safe delivery of these products in human beings are on the horizon. These methods could prove to be particularly useful for loss-of-function mutations for which Replacement strategies may be the only approach. We note that drug discovery efforts within this framework will not be limited to academic research. In fact, pharmaceutical companies are showing interest in pursuing treatments for genetic conditions, as emphasized by the development of genetically targeted treatments for cystic fibrosis (CFTR), and by many drug development efforts inspired by human genetics (eg, PCSK9). Importantly, pharmaceutical companies are also much better equipped than academic laboratories to rapidly screen for potential adverse reactions to candidate therapeutic compounds not yet tested in human beings.
The availability of multiple avenues of treatment development, in combination with the emerging high-throughput efficacy screening methods in cellular models, makes tractable treatments for genetic epilepsies an increasingly realistic goal for the near future.
Frameworks for testing genetically targeted Therapies
Although precision diagnostics in the clinical management of epilepsy is not new,99–101 advances in genetics suggest that the possibility of considerably improved targeting of treatments to precise underlying causes will not only control seizures, but also improve neurodevelopmental outcomes. However, there are practical barriers to clinical implementation of precision medicine that need to be overcome. Access to clinical genetic testing needs to be increased and should be widely available for epilepsies such as the epileptic encephalopathies, for which abundant evidence suggests that the current diagnostic yield could be 15–20%,35,58,59,102–105 and the rate of gene discovery suggests that this proportion will continue to increase. Making such diagnostic evaluation available to all individuals with epilepsy, even in resource-poor environments, is crucial. Dedicated multidisciplinary epilepsy clinics with expertise in genetic epilepsy, not currently available to all patients, will be required to facilitate early expert clinician involvement in diagnosis and optimization of treatment; such clinics would be ideally placed to act as clinical trial centers. Furthermore, application of precision diagnostics and treatment has ethical, legal, and social implications of genetics for people with epilepsy and their family members; consideration of individual preferences, psychosocial impacts, and equity of access should be a priority.
Examples of effective precision therapy in genetic epilepsies are already emerging. The first is in glucose transporter deficiency syndrome, generally caused by mutations in the SLC2A1 gene and treated with a ketogenic diet.99 The second is for pyridoxine-dependent epilepsy, typically caused by mutations in the ALDH7A1 gene and treated with pyridoxine (vitamin B6).99,101 These treatments became established through decades of anecdotal treatments, case reports, and case series rather than randomized controlled trials. Currently, diagnosis of these and other potentially treatable disorders100 is still often delayed because of wide phenotypic variability leading to delay in testing. This delay will be rectified only by large-scale comprehensive genomic testing across the epilepsies.
Several genetic epilepsies now amenable to prompt genetic diagnosis106 are candidates for controlled trials of potentially effective, disease-modifying precision therapies. In some instances, suggestions of targeted therapies have been based on the results of elegant invitro studies showing reversibility of underlying functional defects and suggesting application in human cases.102 These cases include individuals with KCNT1 mutations treated with quinidine,107,108 children with KCNQ2 mutations treated with ezogabine (retigabine),109 individuals with GRIN2A mutations treated with memantine,110,111 and patients with so-called mTORopathies treated with everolimus.112–114 Epilepsies with these mutations can present in infancy with encephalopathy and seizures, can be diagnosed with targeted gene panels or exome sequencing, often do not respond to currently available treatments, and are obvious candidates for organized, multi-center clinical and translational research initiatives. Although it is far from clear that these initial candidate therapies will prove to be safe and effective, they illustrate a precedent for the use of appropriate functional models to identify new, targeted treatments for assessment in the clinic.
An initial step to improve the evidence base for epilepsy precision treatment is a curated registry of therapeutic trials for rare genetic epilepsies. A registry is a key requirement and will facilitate the development of appropriately powered clinical trials to assess treatment effects. It is inevitable that some targeted treatments of genetic epilepsies will take place outside of multi-center, controlled trials as these are often disorders with devastating consequences, and families might be unwilling to wait for the results of properly designed trials. Such registries, when combined with careful phenotyping data about specific genes and functional profiling of mutations, will provide information on dosing and side effects and generate hypotheses for more rigorous trials. A platform such as EGI, for example, will provide a phenotype and genotype data repository and could be expanded to include data on successful and unsuccessful precision treatments, side effects, and other consequences. A registry such as this would provide a portal for analysis of the evidence for a personalized treatment choice for each disorder.
In addition to registries, multi-center, randomized, controlled trials are needed – and are feasible. The high seizure burden in genetic EEs, combined with the potential for a rapid therapeutic effect of some precision treatments,108 makes the cross-over trial design, lasting 2–6 weeks and comparing precision therapy with placebo or conventional therapy, a valid and robust study design to assess short-term efficacy. Long-term efficacy, safety, and neurodevelopmental outcomes can be assessed with open-label extensions of the initial controlled trials. On the basis of early case reports,108 the therapeutic effect could be rapid and pronounced, and as few as 10–20 study participants might be sufficient to assess some precision therapeutics in a controlled trial. For epilepsies or investigational compounds needing a trial of longer duration, controlled trials are still possible with careful attention to inclusion and exclusion criteria as well as outcome measures. Multi-center collaborations using EGI, the Pediatric Epilepsy Research Consortium (PERC) (http://www.pediatricerc.com/), and the US National Institutes of Health NeuroNEXT trial network present an opportunity for future research initiatives. Moreover, extending these networks to allow input from global networks of clinicians will enable more rapid accrual of outcomes and inform improvements in clinical care. Non-profit epilepsy advocacy groups and social media will be vital in raising awareness of trials and in directing potential participants to study sites. In some cases, drugs being investigated might have broader therapeutic implications and pharmaceutical companies could be sponsors or valuable partners. In other cases, just as with quinidine to treat individuals with KCNT1 mutations, the drug might be widely available as a generic drug with little profit potential, so support for trials from government and non-profit organizations will be needed.
Since specific mutations in epilepsy genes (i.e., mutations causing gain- or loss-of-function) can have profound implications for precision therapeutics, a clinical trial network will ideally be directly linked to a collaborative group of geneticists and experts in in vitro functional models. All newly identified mutations in known epilepsy genes must be analyzed expeditiously to determine their in vitro pathological effects and potential for therapeutic correction. As knowledge of epilepsy gene pathways, such as the mTOR pathway, evolves, the prospect of treatments that target the pathways could be assessed in a similarly collaborative way. Since the underlying molecular mechanisms of the genetic epilepsies and potential therapeutic targets converge on specific homeostatic pathways, such as disruption in cell signaling, cell growth, vesicle fusion and release, and ion channel function, the molecular mechanisms behind genetic epilepsies become tangible and targetable. Therefore, an organized effort to characterize mutations and assess possible treatments is of great interest to clinicians, academic basic scientists, the pharmaceutical industry, and most of all individuals with epilepsy and their families.
IV. Conclusions
The considerations and examples outlined above make it clear that precision medicine could transform clinical care in epilepsy, and in so doing will establish a new paradigm in the treatment of the epilepsies (Figure 3). Precision medicine in epilepsy will probably emerge first in epilepsies that are most strongly determined by single mutations of major effect that can be accurately modeled in vitro, in particular the EEs (Figure 2). Although this prediction could seem to imply that only the strongly genetic epilepsies will benefit from precision medicine, it is worth appreciating that some targeted treatments for genetic epilepsies might find wider applications in other epilepsies. Although this nascent specialty already has a number of successful examples of genetics guiding therapy, the development of precision medicine in epilepsy needs a broad range of tools, techniques, and approaches that are never present in single laboratories and are often difficult to assemble without the appropriate mechanisms and structures.
Figure 3.
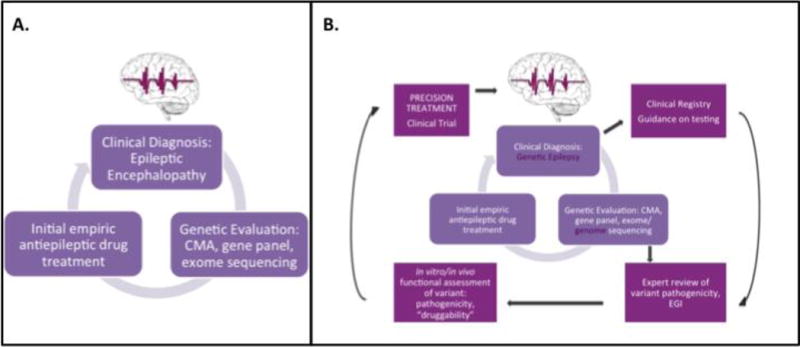
Current practice for genetic diagnosis in epilepsy and the envisioned future of precision medicine in epilepsy (A) Current practice for genetic diagnosis in epilepsy. (B) New additions to the approach of precision medicine are highlighted in purple boxes. In the envisioned model of precision medicine in epilepsy, all types of non-acquired epilepsy will be assessed, and basic, clinical, and translational science will be closely integrated to drive the development of precision therapies.
A systematic approach to precision medicine in epilepsy will require the following: 1) large cohorts of individuals with epilepsy who have been carefully characterized phenotypically and genomically; 2) standardized functional characterization of mutations in each of the epilepsy genes and careful co-interpretation of functional readouts for studied mutations incorporating information about the frequency of mutations and mutation types in cases and controls; and 3) the initiation of well-designed clinical trials when functional work identifies new targeted therapeutics. Meeting these goals depends on the development of collaborative and integrated research groups that bring together researchers with clinical, genetic, and biological expertise.
Acknowledgments
We thank all participants of the Epilepsy Genetics in the Era of Precision Medicine Meeting in Half Moon Bay, CA, USA, for their insight and contributions to the meeting.
In September 2014, 100 scientists, clinicians, and representatives from lay organizations, industry, and government assembled for the Epilepsy Genetics in the Era of Precision Medicine Meeting in Half Moon Bay, California to map out to develop a strategy for the advancement of precision medicine in epilepsy. This perspective reflects the discussions from the two day conference. The meeting was organized by Dan Lowenstein and was funded by John and Barbara Vogelstein, Ron and Sanne Higgins, The Joseph and Vera Long Foundation, Richard Thalheimer, Citizens United for Research in Epilepsy (CURE), Epilepsy Foundation, Ambry Genetics, GeneDx, Biogen, and Lundbeck. All of the authors participated in the meeting. We would like to thank all participants for their insight and contributions to the meeting.
Authors of this manuscript were supported by grants from the NINDS [U01NS077303 (DBG, ELH, HCM), U01NS077274 (DBG, SFB, DHL), U01NS077276 (SFB, DHL, DJD, IES), R37NS036654 (SFT), K23NS069784 (AP), R01NS069605 (HM)], NHMRC (IES, SFB, SP), CURE (DBG), Janssen (SFT), Burroughs Wellcome Fund (HCM), UCB Pharma (SFB), the German Research Foundation [HE 5415 3-1, 5-1, 6-1 (IH)] including the EuroEPINOMICS framework of the Eurocore program by the European Science Foundation, and intramural funds of the University of Kiel, Germany (IH).
Glossary
Glossary of Terms
- CRISPR-Cas system
A novel gene-editing system derived from a prokaryotic immune system that confers resistance to foreign genetic elements. The system delivers the Cas9 protein and appropriate guide RNAs into a cell, allowing the organism"s genome to be cut at any desired location and edited to include specific sequences of interest.
- Cultured neural networks (CNNs)
Cultures of neurons and support cells used to create an in-vitro model of communicating neurons. Cultured neural networks provide a controlled environment in which to investigate neuronal activity and the effect of mutations on that activity. The behaviour of cultured neural networks can be monitored with non-invasive approaches, including multielectrode array technology and optogenetic approaches.
- De-novo mutation
Agenetic mutation present in a child that is not present in either parent (ie, not detectable by conventional means of assaying DNA from leucocytes) and usually arises in an individual as a result of a germ-cell (egg or sperm) mutation in one of the parents.
- Druggability
The extent to which a particular protein can be modulated by a drug acting on that target (ie, largely on the basis of how drugs work).
- Epileptic encephalopathies
Severe forms of epilepsy, generally beginning in childhood, typically associated with intellectual disability, where the epileptic progress may contribute to severe cognitive and behavioural impairments.
- HEK293 cell line
A cell line originally derived from human embryonic kidney (HEK); HEK293 cells are easy to grow in culture and have been used widely for gene expression studies and heterologous gene studies.
- Induced pluripotent stem cells (iPSCs)
Cells that are reprogrammed into a state that is capable, in theory, of differentiating into any type of human cell with the right types of stimulation.
- Locus heterogeneity and allelic heterogeneity
Locus heterogeneity relates to the number of genes that can affect a trait. Allelic heterogeneity relates to the number of different alleles at a locus that can affect a trait.
- Multi-electrode array (MEA)
A system to monitor non-invasively the electrical activity of cultured neural networks. This system is typically used in single-well or multiwell (12–96 wells per plate) formats with embedded electrodes, which monitor the action potentials of a small subset of the neurons in each of the cultured neural networks.
- Optogenetics
In the context of this Personal View, optogenetics is a method that uses rhodopsin proteins to allow optical control and monitoring of neuronal membrane potentials.
- Precision diagnostics
The use of genomic and related technologies to determine the precise cause of epilepsy in individual patients.
- Precision medicine
While precision medicine generally refers to the scientific basis that underpins the personalization of medical care the term here is primarily used in the context of targeting treatments to the precise molecular and physiological causes of disease
EpiPM Consortium Members
Prof S F Berkovic MD
Epilepsy Research Centre, Department of Medicine, University of Melbourne, Austin Health, VIC, Australia
Norman Delanty, M.D.
Molecular and Cellular Therapeutics Department, Royal College of Surgeons in Ireland, Dublin, Ireland Division of Neurology, Beaumont Hospital, Dublin, Ireland
Tracy J. Dixon-Salazar, Ph.D.
CURE Epilepsy, Chicago, IL 60654, USA
Prof D J Dlugos MD
Division of Neurology, The Children’s Philadelphia, PA, USA
Department of Neurology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
Wayne N. Frankel, Ph.D.
The Jackson Laboratory, Bar Harbor, Maine 04609
Prof D B Goldstein PhD
Department of Genetics and Development, Columbia University, New York, NY 10032, USA.
Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA
Erin L. Heinzen, Pharm.D., Ph.D.
Department of Pathology and Cell Biology, Columbia University, New York, NY 10032, USA.
Institute for Genomic Medicine, Columbia University, New York, NY 10032, USA
Ingo Helbig, M.D.
Division of Neurology, The Children’s Hospital of Philadelphia, Philadelphia, PA
Department of Neuropediatrics, University Medical Center Schleswig-Holstein, Christian-Albrechts-University, Kiel, Germany
Prof D H Lowenstein MD
Department of Neurology, University of California, San Francisco, USA
Heather C. Mefford, M.D., Ph.D.
Department of Pediatrics, Division of Genetic Medicine, University of Washington, Seattle, Washington, U.S.A
Jack M. Parent, Ph.D.
Department of Neurology, University of Michigan Medical Center, Ann Arbor, MI
Prof S Petrou PhD
The Florey Institute for Neuroscience and Mental Health, The University of Melbourne, Australia The Centre for Neural Engineering, The University of Melbourne, Australia
Annapurna Poduri, M.D., M.P.H.
Epilepsy Genetics Program, Division of Epilepsy and Clinical Neurophysiology, Department of Neurology, Boston Children’s Hospital, Boston, MA
Prof I E Scheffer PhD
Epilepsy Research Centre, Department of Medicine, University of Melbourne, Austin Health, Australia
Department of Pediatrics, University of Melbourne, Royal Children’s Hospital, Australia
Florey Institute, Melbourne, Australia
Stephen F. Traynelis, Ph.D.
Department of Pharmacology, Emory University School of Medicine, Atlanta, GA, USA.
Conflicts of Interest
Dr. Berkovic reports personal fees from Bionomics and Athena in the past. Dr. Berkovic also has a patent for DEPDC5 testing applied for by University of South Australia and University of Melbourne pending, and a patent for SCN1A testing held by Bionomics Inc. and licensed to various diagnostic companies.
Dr. Goldstein reports personal fees from LabCorp, Clarus Ventures, Astra Zeneca, Johnson & Johnson, Takeda, Teva, and Eli Lilly.
Dr. Mefford reports personal fees from SFARI Gene Advisory Board and Sera Prognostics.
Dr. Petrou reports personal fees from Bionomics Limited in the past and patents for SCN1A testing held by Bionomics Limited and licensed to various diagnostic companies.
Dr. Scheffer reports personal fees from UCB, Athena Diagnostics, Transgenomics, GlaxoSmithKline, Sanofi, Eisai. In addition, Dr. Scheffer has a patent Diagnostic and Therapeutic Methods for EFMR (Epilepsy and Mental Retardation Limited to Females) with royalties paid.
Dr. Traynelis reports personal fees from NeurOp Inc. and Janssen. In addition, Dr. Traynelis is a co-inventor on US Patents 7,375,136 and 8,420,680 licensed to NeurOp Inc, and is a co-founder of NeurOp Inc., a pharmaceutical company developing NMDA receptor modulators for the treatment of neurological diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author’s Contributions
All authors provided sections of text covering their area of expertise and participated in the development of the perspectives presented. DBG and ELH compiled the manuscript, and all authors reviewed, edited, and approved the final version.
References
- 1.Moving toward precision medicine. Lancet. 2011;378(9804):1678. doi: 10.1016/S0140-6736(11)61725-X. [DOI] [PubMed] [Google Scholar]
- 2.Collisson EA, Cho RJ, Gray JW. What are we learning from the cancer genome? Nature reviews Clinical oncology. 2012;9(11):621–30. doi: 10.1038/nrclinonc.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Annegers JF, Hauser WA, Anderson VE, Kurland LT. The risks of seizure disorders among relatives of patients with childhood onset epilepsy. Neurology. 1982;32(2):174–9. doi: 10.1212/wnl.32.2.174. [DOI] [PubMed] [Google Scholar]
- 4.Corey LA, Berg K, Pellock JM, Solaas MH, Nance WE, DeLorenzo RJ. The occurrence of epilepsy and febrile seizures in Virginian and Norwegian twins. Neurology. 1991;41(9):1433–6. doi: 10.1212/wnl.41.9.1433. [DOI] [PubMed] [Google Scholar]
- 5.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43(4):435–45. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- 6.Lennox WG, Lennox MA. The genetics of epilepsy. In: Brown L, editor. Epilepsy and related disorders. Vol. 1. Boston: 1960. pp. 532–74. [Google Scholar]
- 7.Peljto AL, Barker-Cummings C, Vasoli VM, et al. Familial risk of epilepsy: a population-based study. Brain. 2014;137(Pt 3):795–805. doi: 10.1093/brain/awt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speed D, O’Brien TJ, Palotie A, et al. Describing the genetic architecture of epilepsy through heritability analysis. Brain. 2014;137(Pt 10):2680–9. doi: 10.1093/brain/awu206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beck C, Moulard B, Steinlein O, et al. A nonsense mutation in the alpha4 subunit of the nicotinic acetylcholine receptor (CHRNA4) cosegregates with 20q-linked benign neonatal familial convulsions (EBNI) Neurobiol Dis. 1994;1(1–2):95–9. doi: 10.1006/nbdi.1994.0012. [DOI] [PubMed] [Google Scholar]
- 10.Baulac S, Picard F, Herman A, et al. Evidence for digenic inheritance in a family with both febrile convulsions and temporal lobe epilepsy implicating chromosomes 18qter and 1q25-q31. Ann Neurol. 2001;49(6):786–92. doi: 10.1002/ana.1014. [DOI] [PubMed] [Google Scholar]
- 11.Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19(4):366–70. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 12.Dibbens LM, Feng HJ, Richards MC, et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004;13(13):1315–9. doi: 10.1093/hmg/ddh146. [DOI] [PubMed] [Google Scholar]
- 13.Moulard B, Guipponi M, Chaigne D, Mouthon D, Buresi C, Malafosse A. Identification of a new locus for generalized epilepsy with febrile seizures plus (GEFS+) on chromosome 2q24-q33. Am J Hum Genet. 1999;65(5):1396–400. doi: 10.1086/302621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30(3):335–41. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24(4):343–5. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 16.Baulac S, Gourfinkel-An I, Picard F, et al. A second locus for familial generalized epilepsy with febrile seizures plus maps to chromosome 2q21-q33. Am J Hum Genet. 1999;65(4):1078–85. doi: 10.1086/302593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klepper J, Willemsen M, Verrips A, et al. Autosomal dominant transmission of GLUT1 deficiency. Hum Mol Genet. 2001;10(1):63–8. doi: 10.1093/hmg/10.1.63. [DOI] [PubMed] [Google Scholar]
- 18.Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40(6):782–8. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 19.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68(6):1327–32. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalscheuer VM, Tao J, Donnelly A, et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet. 2003;72(6):1401–11. doi: 10.1086/375538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40(6):776–81. doi: 10.1038/ng.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavalleri GL, Weale ME, Shianna KV, et al. Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol. 2007;6(11):970–80. doi: 10.1016/S1474-4422(07)70247-8. [DOI] [PubMed] [Google Scholar]
- 23.Kasperaviciute D, Catarino CB, Heinzen EL, et al. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain. 2010;133(Pt 7):2136–47. doi: 10.1093/brain/awq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International League Against Epilepsy Consortium on Complex Epilepsies. Electronic address e-auea. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol. 2014;13(9):893–903. doi: 10.1016/S1474-4422(14)70171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium E, Leu C, de Kovel CG, et al. Genome-wide linkage meta-analysis identifies susceptibility loci at 2q34 and 13q31.3 for genetic generalized epilepsies. Epilepsia. 2012;53(2):308–18. doi: 10.1111/j.1528-1167.2011.03379.x. [DOI] [PubMed] [Google Scholar]
- 26.Epicure Consortium. Consortium EM, Steffens M, et al. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3 and 17q21.32. Hum Mol Genet. 2012;21(24):5359–72. doi: 10.1093/hmg/dds373. [DOI] [PubMed] [Google Scholar]
- 27.Helbig I, Mefford HC, Sharp AJ, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41(2):160–2. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharp AJ, Mefford HC, Li K, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40(3):322–8. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Kovel CG, Trucks H, Helbig I, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(1):23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heinzen EL, Radtke RA, Urban TJ, et al. Rare Deletions at 16p13.11 Predispose to a Diverse Spectrum of Sporadic Epilepsy Syndromes. Am J Hum Genet. 2010;86(5):707–18. doi: 10.1016/j.ajhg.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mefford HC, Muhle H, Ostertag P, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6(5):e1000962. doi: 10.1371/journal.pgen.1000962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ben-Shachar S, Lanpher B, German JR, et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009;46(6):382–8. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grayton HM, Fernandes C, Rujescu D, Collier DA. Copy number variations in neurodevelopmental disorders. Prog Neurobiol. 2012;99(1):81–91. doi: 10.1016/j.pneurobio.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Epi4K Consortium and Epilepsy Phenome/Genome Project. Allen AS, Berkovic SF, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217–21. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Euro Epinomics- R.E. S. Consortium, Epilepsy Phenome/Genome Project, Epi4k Consortium. De Novo Mutations in Synaptic Transmission Genes Including DNM1 Cause Epileptic Encephalopathies. Am J Hum Genet. 2014;95(4):360–70. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43(6):585–9. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanders SJ, Ercan-Sencicek AG, Hus V, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70(5):863–85. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–5. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380(9854):1674–82. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 40.de Ligt J, Willemsen MH, van Bon BW, et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N Engl J Med. 2012;367(20):1921–9. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 41.Nava C, Dalle C, Rastetter A, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet. 2014;46(6):640–5. doi: 10.1038/ng.2952. [DOI] [PubMed] [Google Scholar]
- 42.Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45(9):1073–6. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Endele S, Rosenberger G, Geider K, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42(11):1021–6. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 44.Lesca G, Rudolf G, Bruneau N, et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013;45(9):1061–6. doi: 10.1038/ng.2726. [DOI] [PubMed] [Google Scholar]
- 45.Carvill GL, Weckhuysen S, McMahon JM, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 2014;82(14):1245–53. doi: 10.1212/WNL.0000000000000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakamura K, Kodera H, Akita T, et al. De Novo Mutations in GNAO1, Encoding a Galphao Subunit of Heterotrimeric G Proteins, Cause Epileptic Encephalopathy. Am J Hum Genet. 2013;93(3):496–505. doi: 10.1016/j.ajhg.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44(11):1255–9. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ogiwara I, Ito K, Sawaishi Y, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73(13):1046–53. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohba C, Kato M, Takahashi S, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55(7):994–1000. doi: 10.1111/epi.12668. [DOI] [PubMed] [Google Scholar]
- 50.Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90(3):502–10. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kodera H, Nakamura K, Osaka H, et al. De Novo Mutations in SLC35A2 Encoding a UDP-Galactose Transporter Cause Early-Onset Epileptic Encephalopathy. Hum Mutat. 2013;34(12):1708–14. doi: 10.1002/humu.22446. [DOI] [PubMed] [Google Scholar]
- 52.Dhindsa RS, Bradrick SS, Yao X, et al. Epileptic encephalopathy-causing mutations in DNM1 impair synaptic vesicle endocytosis. Neurology Genetics. 2015;1:1–9. doi: 10.1212/01.NXG.0000464295.65736.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gleeson JG, Minnerath S, Kuzniecky RI, et al. Somatic and germline mosaic mutations in the doublecortin gene are associated with variable phenotypes. Am J Hum Genet. 2000;67(3):574–81. doi: 10.1086/303043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poduri A, Evrony GD, Cai X, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74(1):41–8. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.D’Gama AM, Geng Y, Couto JA, et al. mTOR pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77(4):720–5. doi: 10.1002/ana.24357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jamuar SS, Lam AT, Kircher M, et al. Somatic Mutations in Cerebral Cortical Malformations. N Engl J Med. 2014;371(8):733–43. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Epilepsy Phenome/Genome Project Epi4K Consortium. Copy number variant analysis from exome data in 349 patients with epileptic encephalopathy. Ann Neurol. 2015;78:323–28. doi: 10.1002/ana.24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mefford HC, Yendle SC, Hsu C, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Ann Neurol. 2011;70:974–85. doi: 10.1002/ana.22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olson H, Shen Y, Avallone J, et al. Copy number variation plays an important role in clinical epilepsy. Ann Neurol. 2014;75:943–58. doi: 10.1002/ana.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dibbens LM, de Vries B, Donatello S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45:546–51. doi: 10.1038/ng.2599. [DOI] [PubMed] [Google Scholar]
- 61.Ishida S, Picard F, Rudolf G, et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013;45:552–55. doi: 10.1038/ng.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Picard F, Makrythanasis P, Navarro V, et al. DEPDC5 mutations in families presenting as autosomal dominant nocturnal frontal lobe epilepsy. Neurology. 2014;82:2101–06. doi: 10.1212/WNL.0000000000000488. [DOI] [PubMed] [Google Scholar]
- 63.Thomas RH, Berkovic SF, et al. The hidden genetics of epilepsy—a clinically important new paradigm. Nat Rev Neurol. 2014;10:283–92. doi: 10.1038/nrneurol.2014.62. [DOI] [PubMed] [Google Scholar]
- 64.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB, et al. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Samocha KE, Robinson EB, Sanders SJ, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–50. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015 doi: 10.1038/gim.2014.191. published online Jan 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hynynen J, Komulainen T, Tukiainen E, et al. Acute liver failure after valproate exposure in patients with POLG1 mutations and the prognosis after liver transplantation. Liver Transpl. 2014;20:1402–12. doi: 10.1002/lt.23965. [DOI] [PubMed] [Google Scholar]
- 68.Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428:486. doi: 10.1038/428486a. [DOI] [PubMed] [Google Scholar]
- 69.Mikati MA, Jiang Y, Carboni M, et al. Quinidine in the treatment of KCNT1 positive epilepsies. Ann Neurol. 2015 doi: 10.1002/ana.24520. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015;47:39–46. doi: 10.1038/ng.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Todd E, Gurba KN, Botzolakis EJ, Stanic AK, Macdonald RL, et al. GABAA receptor biogenesis is impaired by the gamma2 subunit febrile seizure-associated mutation, GABRG2(R177G) Neurobiol Dis. 2014;69:215–24. doi: 10.1016/j.nbd.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 72.Kang JQ, Shen W, Macdonald RL, et al. Trafficking-deficient mutant GABRG2 subunit amount may modify epilepsy phenotype. Ann Neurol. 2013;74:547–59. doi: 10.1002/ana.23947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tian M, Macdonald RL, et al. The intronic GABRG2 mutation, IVS6+2T->G, associated with childhood absence epilepsy altered subunit mRNA intron splicing, activated nonsense-mediated decay, and produced a stable truncated gamma2 subunit. J Neurosci. 2012;32:5937–52. doi: 10.1523/JNEUROSCI.5332-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF, et al. Ionotropic GABA and glutamate receptor mutations and human neurologic diseases. Mol Pharmacol. 2015;88:203–17. doi: 10.1124/mol.115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dyhrfjeld-Johnsen J, Berdichevsky Y, Swiercz W, Sabolek H, Staley KJ. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clin Neurophysiol. 2010;27:418–24. doi: 10.1097/WNP.0b013e3181fe0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hochbaum DR, Zhao Y, Farhi SL, et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat Methods. 2014;11:825–33. doi: 10.1038/nmeth.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lancaster MA, Knoblich JA, et al. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9:2329–40. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hortopan GA, Dinday MT, Baraban SC, et al. Zebrafish as a model for studying genetic aspects of epilepsy. Dis Model Mech. 2010;3:144–48. doi: 10.1242/dmm.002139. [DOI] [PubMed] [Google Scholar]
- 79.Williams SN, Locke CJ, Braden AL, Caldwell KA, Caldwell GA, et al. Epileptic-like convulsions associated with LIS-1 in the cytoskeletal control of neurotransmitter signaling in Caenorhabditis elegans. Hum Mol Genet. 2004;13:2043–59. doi: 10.1093/hmg/ddh209. [DOI] [PubMed] [Google Scholar]
- 80.Parker L, Padilla M, Du Y, Dong K, Tanouye MA, et al. Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the para sodium channel gene that leads to seizures. Genetics. 2011;187:523–34. doi: 10.1534/genetics.110.123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jones NC, O’Brien TJ, Powell KL. Morphometric changes and molecular mechanisms in rat models of idiopathic generalized epilepsy with absence seizures. Neurosci Lett. 2011;497:185–93. doi: 10.1016/j.neulet.2011.02.039. [DOI] [PubMed] [Google Scholar]
- 82.Paemka L, Mahajan VB, Ehaideb SN, et al. Seizures are regulated by ubiquitin-specific peptidase 9 X-linked (USP9X), a de-ubiquitinase. PLoS Genet. 2015;11:e1005022. doi: 10.1371/journal.pgen.1005022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Noebels JL. Single-gene models of epilepsy. Adv Neurol. 1999;79:227–38. [PubMed] [Google Scholar]
- 84.Smith M, Wilcox KS, White HS, et al. Discovery of antiepileptic drugs. Neurotherapeutics. 2007;4:12–17. doi: 10.1016/j.nurt.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leite JP, Garcia-Cairasco N, Cavalheiro EA. New insights from the use of pilocarpine and kainate models. Epilepsy Res. 2002;50:93–103. doi: 10.1016/s0920-1211(02)00072-4. [DOI] [PubMed] [Google Scholar]
- 86.Burgess DL, Jones JM, Meisler MH, Noebels JL, et al. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–92. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 87.Fletcher CF, Lutz CM, O’’Sullivan TN, et al. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–17. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 88.Boumil RM, Letts VA, Roberts MC, et al. A missense mutation in a highly conserved alternate exon of dynamin-1 causes epilepsy in fitful mice. PLoS Genet. 2010;6:e1001046. doi: 10.1371/journal.pgen.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J, et al. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci USA. 2006;103:19152–57. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–49. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 91.Tan HO, Reid CA, Single FN, et al. Reduced cortical inhibition in a mouse model of familial childhood absence epilepsy. Proc Natl Acad Sci USA. 2007;104:17536–41. doi: 10.1073/pnas.0708440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reid CA, Leaw B, Richards KL, et al. Reduced dendritic arborization and hyperexcitability of pyramidal neurons in a Scn1b-based model of Dravet syndrome. Brain. 2014;137:1701–15. doi: 10.1093/brain/awu077. [DOI] [PubMed] [Google Scholar]
- 93.Baraban SC, Dinday MT, Hortopan GA, et al. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun. 2013;4:2410. doi: 10.1038/ncomms3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cheng AC, Coleman RG, Smyth KT, et al. Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotech. 2007;25:71–75. doi: 10.1038/nbt1273. [DOI] [PubMed] [Google Scholar]
- 95.King GF. Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin Biol Ther. 2011;11:1469–84. doi: 10.1517/14712598.2011.621940. [DOI] [PubMed] [Google Scholar]
- 96.Lee JJ, Yokota T, et al. Antisense therapy in neurology. J Pers Med. 2013;3:144–76. doi: 10.3390/jpm3030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meyer K, Ferraiuolo L, Schmelzer L, et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther. 2015;23:477–87. doi: 10.1038/mt.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.de Lucas Cerrillo AM, Bond WS, Rex TS. Safety and angiogenic effects of systemic gene delivery of a modified erythropoietin. Gene Ther. 2015;22:365–73. doi: 10.1038/gt.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klepper J, Scheffer H, Leiendecker B, et al. Seizure control and acceptance of the ketogenic diet in GLUT1 deficiency syndrome: a 2- to 5-year follow-up of 15 children enrolled prospectively. Neuropediatrics. 2005;36:302–08. doi: 10.1055/s-2005-872843. [DOI] [PubMed] [Google Scholar]
- 100.van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S. The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Gen Metab. 2014;111:428–38. doi: 10.1016/j.ymgme.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 101.Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307–09. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- 102.Boutry-Kryza N, Labalme A, Ville D, et al. Molecular characterization of a cohort of 73 patients with infantile spasms syndrome. Eur J Med Genet. 2015;58:51–58. doi: 10.1016/j.ejmg.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 103.Helbig I, Swinkels ME, Aten E, et al. Structural genomic variation in childhood epilepsies with complex phenotypes. Eur J Hum Genet. 2014;22:896–901. doi: 10.1038/ejhg.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kodera H, Kato M, Nord AS, et al. Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia. 2013;54:1262–69. doi: 10.1111/epi.12203. [DOI] [PubMed] [Google Scholar]
- 105.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–30. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Poduri A. A channel for precision diagnosis and treatment in genetic epilepsy. Ann Neurol. 2014;76:323–24. doi: 10.1002/ana.24243. [DOI] [PubMed] [Google Scholar]
- 107.Milligan CJ, Li M, Gazina EV, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. 2014;75:581–90. doi: 10.1002/ana.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM, et al. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. 2014;76:457–61. doi: 10.1002/ana.24229. [DOI] [PubMed] [Google Scholar]
- 109.Orhan G, Bock M, Schepers D, et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann Neurol. 2014;75:382–94. doi: 10.1002/ana.24080. [DOI] [PubMed] [Google Scholar]
- 110.Pierson TM, Yuan H, Marsh ED, et al. Mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol. 2014;1:190–98. doi: 10.1002/acn3.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yuan H, Hansen KB, Zhang J, et al. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy. Nat Commun. 2014;5:3251. doi: 10.1038/ncomms4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 113.Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–87. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- 114.Parker WE, Orlova KA, Parker WH, et al. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Sci Transl Med. 2013;5:182ra53. doi: 10.1126/scitranslmed.3005271. [DOI] [PMC free article] [PubMed] [Google Scholar]