

Abstract
Background
Delayed afterdepolarizations (DADs) carried by Na+-Ca2+-exchange current (INCX) in response to sarcoplasmic reticulum (SR) Ca2+-leak can promote atrial fibrillation (AF). The mechanisms leading to DADs in AF-patients have not been defined.
Methods and Results
Protein levels (Western-blot), membrane-currents and action-potentials (patch-clamp), and [Ca2+]i (Fluo-3) were measured in right-atrial samples from 77 sinus-rhythm (Ctl) and 69 chronic-AF (cAF) patients. Diastolic [Ca2+]i and SR-Ca2+-content (integrated INCX during caffeine-induced-Ca2+-transient [cCaT]) were unchanged, whereas diastolic SR Ca2+-leak, estimated by blocking RyR2 with tetracaine, was ~50% higher in cAF vs. Ctl. Single-channel recordings from atrial RyR2 reconstituted into lipid-bilayers revealed enhanced open-probability in cAF-samples, providing a molecular basis for increased SR Ca2+-leak. Calmodulin-expression (+60%), CaMKII-autophosphorylation at Thr287 (+40%) and RyR2-phosphorylation at Ser2808 (PKA/CaMKII-site, +236%) and Ser2814 (CaMKII-site, +77%) were increased in cAF. The selective CaMKII-blocker KN-93 decreased SR Ca2+-leak, the frequency of spontaneous Ca2+-release events and RyR2 open-probability in cAF, whereas PKA-inhibition with H-89 was ineffective. Knock-in mice with constitutively-phosphorylated RyR2 at Ser2814 showed a higher incidence of Ca2+-sparks and increased susceptibility to pacing-induced AF vs. controls. The relationship between [Ca2+]i and INCX-density revealed INCX-upregulation in cAF. Spontaneous Ca2+-release events accompanied by inward INCX-currents and DADs/triggered-activity occurred more often and the sensitivity of resting membrane voltage to elevated [Ca2+]i (diastolic [Ca2+]i–voltage coupling gain) was higher in cAF vs. Ctl.
Conclusions
Enhanced SR Ca2+-leak through CaMKII-hyperphosphorylated RyR2, in combination with larger INCX for a given SR Ca2+-release and increased diastolic [Ca2+]i–voltage coupling gain, cause AF-promoting atrial DADs/triggered-activity in cAF patients.
Keywords: atrial fibrillation, ryanodine receptor, sarcoplasmic reticulum Ca2+-leak, Na+-Ca2+-exchanger, delayed afterdepolarizations
Atrial fibrillation (AF) induces self-promoting remodeling.1, 2 Altered intracellular Ca2+-signaling is a key contributor to the AF-maintaining substrate.3, 4 In normal hearts, Ca2+ enters cells through L-type Ca2+-channels (ICa,L) with each action potential (AP), triggering Ca2+-release from sarcoplasmic reticulum (SR) through type-2 ryanodine-receptor channels (RyR2). During diastole, cytosolic Ca2+ is removed via reuptake through the SR Ca2+-ATPase (Serca; cardiac-form=Serca2a) and transmembrane extrusion through the Na+-Ca2+-exchanger (NCX1).5
Reduced ICa,L contributes to AF-related AP duration (APD)-shortening, which promotes reentry.1, 6–8 Recent evidence points to RyR2 dysfunction and increased SR Ca2+-leak,9, 10 providing another arrhythmogenic mechanism.11, 12 Hyperphosphorylation increases RyR2-sensitivity to cytosolic Ca2+, making RyR2 more “leaky”.13 RyR2 is hyperphosphorylated at Ser2808/2809 (species-dependent; protein-kinase-A [PKA]-and Ca2+/calmodulin-dependent protein kinase-II [CaMKII]-site)11, 14 and Ser2814/2815 (species-dependent; exclusive CaMKII-site)15 in canine and human chronic-AF (cAF) paradigms.10, 13, 16 CaMKIIδ-activity is enhanced in clinical13, 16 and experimental AF.17, 18 CaMKIIδ-inhibition reduces Ca2+-spark frequency in AF.10 Although it is widely believed that altered Ca2+-signaling predisposes to delayed afterdepolarizations (DADs) and triggered activity in AF-patients, this has not been directly tested, nor have underlying mechanisms, been established in humans. The present study evaluated cardiomyocytes and tissues from right-atrial (RA) samples of sinus-rhythm versus cAF-patients, to establish the mechanisms underlying Ca2+-related DADs with the use of complementary biochemical and biophysical techniques, and to relate Ca2+-handling abnormalities directly to cellular arrhythmogenesis.
Methods (For details, see Online Data Supplement)
Human Tissue Samples
RA-appendages were dissected from 77 sinus-rhythm (Ctl) patients and 69 cAF-patients (Online Tables 1–2). Experimental protocols were approved by the ethics committees of Dresden University of Technology (EK790799) and Medical Faculty Mannheim, University of Heidelberg (#2011-216N-MA). Each patient gave written informed consent. After excision, atrial appendages were used for either myocyte isolation (41 Ctl, 31 cAF-patients) or snap-frozen in liquid-nitrogen for biochemical/biophysical studies (35 Ctl, 41 cAF-patients).
Human-myocyte Isolation
RA-myocytes isolated using a standard protocol19, 20 were suspended in EGTA-free storage solution.
Human-myocyte Intracellular-[Ca2+] Measurement
Intracellular [Ca2+] ([Ca2+]i) was quantified using Fluo-3-acetoxymethyl ester (Fluo-3 AM). In addition, Fluo-3 was included into the electrode solution. Fluorescence was excited at 488 nm and emitted light (<520 nm) converted to [Ca2+]i assuming
where kd=dissociation constant of Fluo-3 (864-nmol/L), F=Fluo-3 fluorescence; Fmax=Ca2+-saturated fluorescence obtained at the end of each experiment.21
Patch-clamp
Membrane currents and potentials were measured at 37°C in whole-cell ruptured-patch configuration using voltage-clamp and current-clamp techniques, with simultaneous [Ca2+]i measurement. Membrane capacitances did not differ between groups (cAF: 133.4±10.3 pF, n=40/20 [myocytes/patients] vs. Ctl: 114.8±5.9 pF, n=46/26). Currents are expressed as densities (pA/pF).
L-type Ca2+-current (ICa,L)-triggered [Ca2+]i-transients (CaTs) were recorded simultaneously (Figure 1A). Drugs were applied via a rapid-solution exchange system.
Figure 1. ICa,L-triggered Ca2+-transients (CaT) in sinus rhythm (Ctl) and cAF.
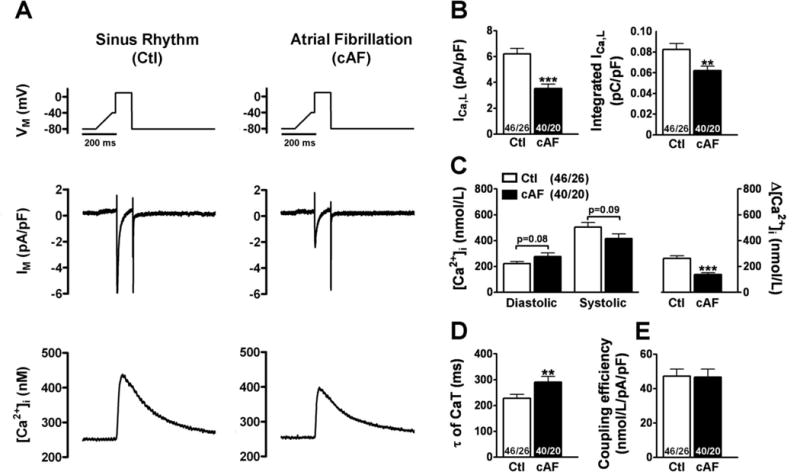
A, Top: Voltage-clam p protocol (0.5 Hz). Below: Simultaneous recording of ICa,L (middle) and triggered CaT (Fluo-3, bottom) in Ctl (left) and cAF (right) myocytes. B, Mean±SEM Peak-ICa,L (left) and integrated ICa,L (right). C, Mean±SEM diastolic and systolic [Ca2+]i (left) and resulting CaT-amplitude (right). D, Mean±SEM time-constant (τ) of decay of ICa,L-triggered CaT. E, Mean±SEM “coupling efficiency” of Ca2+-influx and SR Ca2+-release. **P<0.01 and ***P<0.001, respectively vs. corresponding means in Ctl. Numbers indicate myocytes/patients.
Quantification of Diastolic SR Ca2+-leak
SR Ca2+-leak in intact myocytes was measured according to Shannon et al.22
RyR2 Single-channel Recordings
Single-channel recordings of Ctl and cAF RyR2 were obtained under voltage-clamp conditions as previously described.23
Intracardiac Electrophysiology and Ca2+-spark Measurements in S2814D Knock-in Mice
RyRS2814D knock-in mice (S2814D) were generated as described.12 In vivo electrophysiology was performed on S2814D and wildtype (WT) littermates 3–4 months-of-age in presence and absence of KN-93 (10-μmol/kg, i.p.).24 More than 1-second of AF or atrial flutter was considered an atrial arrhythmia episode.16, 25 Atrial arrhythmia inducibility was considered positive if at least 2 of 3 pacing trials induced AF or atrial flutter. Ca2+-sparks were recorded in Fluo-4-AM-loaded mouse atrial myocytes with LSM510 confocal microscope.
Biochemistry
Protein-expression of Ca2+-handling proteins was quantified by immunoblot.26 The mRNA levels of NCX1 and CaMKIIδ-isoforms were measured by real-time PCR.
Statistical Analysis
Differences between group means for continuous data were compared by unpaired Student’s t-test. Categorical data were analyzed with Fisher’s exact test. Data are mean±SEM. P<0.05 was considered statistically significant.
Results
ICa,L-triggered [Ca2+]i-transients in RA-myocytes from cAF-Patients
Figure 1A shows simultaneously depolarization-induced ICa,L and CaTs in Fluo-3-loaded myocytes. The peak-ICa,L amplitude was 42% lower in cAF, and the time integral of ICa,L was 22% smaller in cAF vs. Ctl (Figure 1B). Systolic CaT-amplitude was 50% lower and the CaT-decay was 28% slower in cAF vs Ctl, whereas diastolic [Ca2+]i tended to be increased (Figure 1C,D). No differences in the coupling-efficiency between Ca2+-influx and Ca2+-release were noted between Ctl and cAF (Figure 1E).
SR Ca2+-Content and NCX-Current
Atrial myocytes were preconditioned for 1-minute by the ICa,L-activation protocol in all subsequent voltage-clamp experiments. Caffeine (10-mmol/L) caused a rapid increase of [Ca2+]i as a result of SR Ca2+-release. The subsequent decay of [Ca2+]i results from sarcolemmal Ca2+-extrusion, mainly through NCX. This generates a transient-inward current (INCX): INCX amplitude and time course were monitored simultaneously with the CaT (Figure 2A).
Figure 2. Caffeine-induced Ca2+-transients (cCaT) to assess SR Ca2+ content and the corresponding transient inward currents (INCX) in atrial myocytes from Ctl and cAF patients.
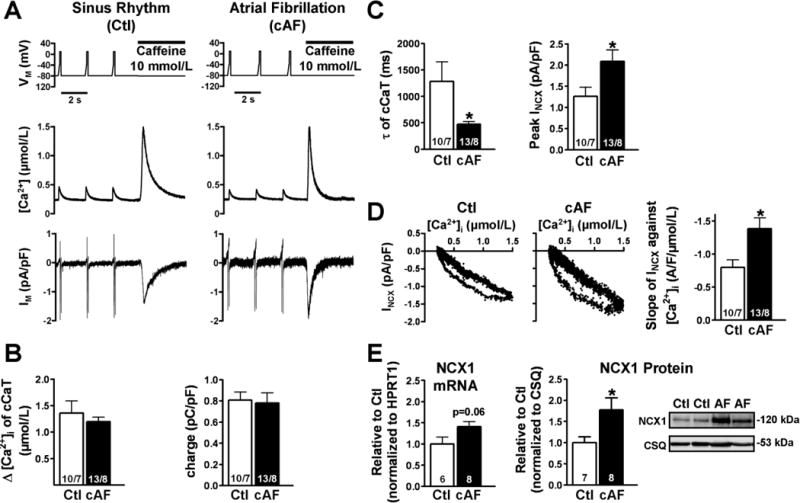
A, Voltage-clamp protocol to quantify SR Ca2+ content (Fluo-3) and corresponding INCX (top), cCaT (middle) and inward NCX current during caffeine (10-mmol/L) application (bottom) following steady-state stimulation for 1 -minute at 0.5 Hz in a Ctl (left) and in a cAF (right) myocyte. B, Mean±SEM cCaT-amplitude (left) and integrated inward NCX (right). C, Mean±SEM time constant (τ) of decay of cCaT (left) and Peak-INCX during caffeine (10-mmol/L) application (right). D, INCX as a function of [Ca2+]i obtained during CaT together with the mean±SEM slope-fit from linear regression obtained from decay phase. E, mRNA and protein levels of NCX1 in Ctl (sinus rhythm) and cAF atria. *P<0.05 vs. corresponding means in Ctl. Numbers indicate myocytes/patients (B, C, D) and tissue samples (E), respectively.
The amplitude of caffeine-induced CaT was comparable in Ctl and cAF, suggesting unchanged SR Ca2+-content (Figure 2B). As described previously,27 the time-integral of caffeine-induced INCX is a measure of Ca2+ extruded by NCX and an indicator of SR Ca2+-content. The mean integral (charge-transfer) of INCX was unchanged in cAF-myocytes (Figure 2B), confirming comparable SR Ca2+-contents between groups.
The caffeine-induced CaT decay-rate was accelerated in cAF, suggesting enhanced Ca2+-extrusion via NCX (Figure 2C). In addition, INCX-amplitude was larger in cAF (Figure 2C) and inward INCX for any given [Ca2+]i was greater (Figure 2D), indicating enhanced INCX. NCX1-expression was higher in cAF-atria vs. Ctl (Figure 2E), indicating that functional enhancement may be due to protein up-regulation.
Expression and Phosphorylation of RyR2
Since Ca2+-content was unchanged, we studied expression and phosphorylation of RyR2 and RyR2-regulatory proteins as a potential source of DAD-generation (Online-Figure 1). Total RyR2 was unchanged, whereas fractional phosphorylation of RyR2 at Ser2814 (CaMKII-site) and Ser2808 (PKA/CaMKII-site) was increased, by 77% and 236%, respectively, in cAF (Online-Figure 1A). Protein-expression of calsequestrin, junctin and triadin, major regulators of RyR2 function, was unchanged (Online-Figure 1B).
Consistent with RyR2-hyperphosphorylation, expression of cytosolic CaMKIIδC increased by 92% in cAF (Online-Figure 2A) and stimulatory autophosphorylation at Thr287 increased by 87%. Fractional autophosphorylation of CaMKIIδC at its inhibitory Thr306/307 site was reduced by 46%. The mRNA levels of the cytosolic (CaMKIIδC) and nuclear (CaMKIIδB) isoforms and the protein levels of CaMKIIδB were unchanged in cAF, suggesting posttranscriptional upregulation of CaMKIIδC (Online-Figure 2A,B) Calmodulin-expression was also increased, by 60% (Online-Figure 2C). In contrast, protein-levels of PKAc- and PKAIIα-subunits were similar (Online-Figure 2D). Overall, these data point to enhanced CaMKII-activity underlying RyR2-hyperphosphorylation and previously-reported increases in Ca2+-spark incidence.9, 10, 16
Diastolic SR Ca2+-leak
Increased Ca2+-spark frequency has been reported in cAF patients,9, 10 but SR Ca2+-leak estimation with Ca2+-spark recordings is semi-quantitative because of substantial non-spark SR Ca2+-release.28, 29 To directly quantify total SR Ca2+-leak we employed the tetracaine method of Shannon et al.,22 as illustrated in Figure 3A. Rapid application of tetracaine to voltage-clamped myocytes perfused with Na+- and Ca2+-free bath solution (to prevent transmembrane fluxes) reflects the magnitude of total SR Ca2+-leak.22
Figure 3. Quantification of diastolic SR Ca2+-leak with tetracaine in voltage-clamped atrial myocytes from sinus rhythm (Ctl) and cAF patients.
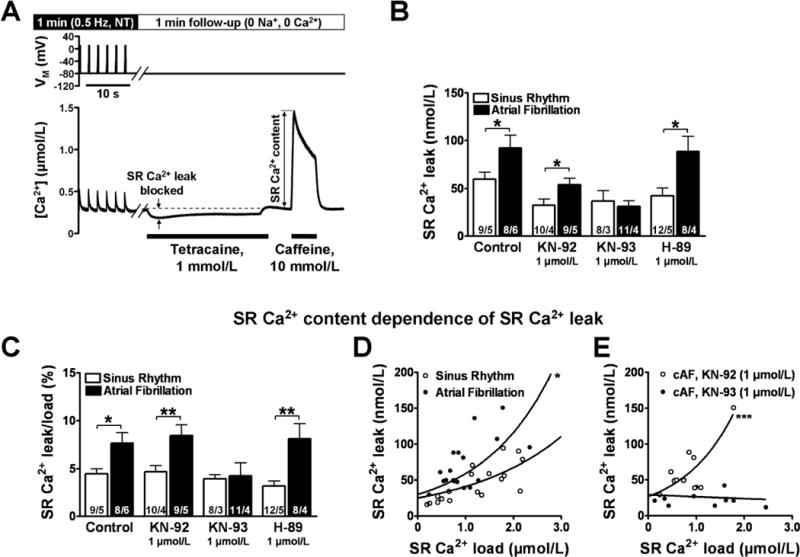
A, Experimental protocol for determination of SR Ca2+-leak (Fluo-3). After steady-state stimulation for 1-minute at 0.5 Hz, the bath solution is rapidly switched to sodium- and calcium-free (0Na+, 0Ca2+) solution. Tetracaine (1-mmol/L) blocks RyR2, and the shift downward in resting [Ca2+]i is proportional to SR Ca2+-leak. After at least 30-seconds and tetracaine washout, SR Ca2+-content is measured with 10-mmol/L caffeine. B,C, Mean±SEM tetracaine-dependent decrease in resting [Ca2+]i (SR Ca2+-leak; B) and for SR Ca2+-leak normalized to SR Ca2+-content (C) in control myocytes and myocytes pretreated (30-minutes) with the CaMKII-inhibitor KN-93 (1-μmol/L), its inactive analogue KN-92 (1-μmol/L) and the PKA-inhibitor H-89 (1-μmol/L), respectively. *P<0.05 and **P<0.01 vs. corresponding means in Ctl. Numbers within columns indicate myocytes/patients. D,E, SR Ca2+-leak plotted vs. SR Ca2+-load. Curves are from exponential regression. *P<0.05 and ***P<0.001, respectively vs. corresponding rate constant in Ctl (F-test).
The tetracaine-induced Ca2+-shift was significantly enhanced in cAF-myocytes (Figure 3B). The contributions of CaMKII and PKA to enhanced SR Ca2+-leak in cAF were assessed in myocytes pretreated for 30 minutes with KN-93 to inhibit CaMKII or H-89 to inhibit PKA. Pretreatment with KN-93, its inactive analogue KN-92 or H-89 did not significantly affect diastolic [Ca2+]i, whereas the cAF-related increase in SR Ca2+-leak was abolished by KN-93, but not by KN-92 or H-89 (Figure 3B). Similar results were obtained when diastolic SR Ca2+-leak was related to SR Ca2+ content, estimated by caffeine-induced CaT in each myocyte (Figure 3A,C). Moreover, the “leak-load” relationship was leftward-shifted in cAF; this was reversed by KN-93 but not KN-92 (Figure 3D,E), indicating increased CaMKII-mediated SR Ca2+-leak at any given SR Ca2+-content.
During decay of ICa,L-induced CaT, cytosolic Ca2+ is extruded by Ca2+-reuptake into the SR via Serca and into extracellular space via NCX and plasmalemmal Ca2+-ATPase (PMCA). We estimated the Ca2+ transport-rate of Serca, NCX and PMCA by the rate-constants of exponential curves fitted to ICa,L- and caffeine-evoked CaT decays, as previously described (Online-Figure 3).30 Ca2+-removal mechanisms switched from a predominance of Serca over NCX in Ctl to an equal contribution of Serca and NCX in cAF (Online-Figure 3D).
Enhanced Open-Probability (Po) of RyR2 in cAF
To compare directly the Ca2+-sensitivities of RyR2-channels in Ctl vs. cAF, single-RyR2 currents were recorded from RyR2-channels obtained from cAF (42 channel-recordings/10 patients) and Ctl patients (38 channel-recordings/11 patients; Figure 4A,B). Under conditions that mimic diastole (i.e., 150-nmol/L cytosolic [Ca2+]), cAF-patients exhibited enhanced opening-frequencies and open-probabilities (Figure 4A,B), and shorter closed-times (Tc: cAF, 827±266 ms vs. Ctl, 3687±930 ms; P<0.05). Open-times were unchanged (To: cAF, 5.82±1.07 ms vs. Ctl, 6.32±0.83 ms). cAF RyR2-channels had left-shifted Ca2+-dependence such that they were activated at resting cytosolic-Ca2+ levels (Figure 4C). The enhanced RyR2 Po in cAF provides a molecular correlate for increased SR Ca2+-leak in intact myocytes (Figure 3).
Figure 4. RyR2 single-channel recordings of sinus rhythm (Ctl) and cAF patients.
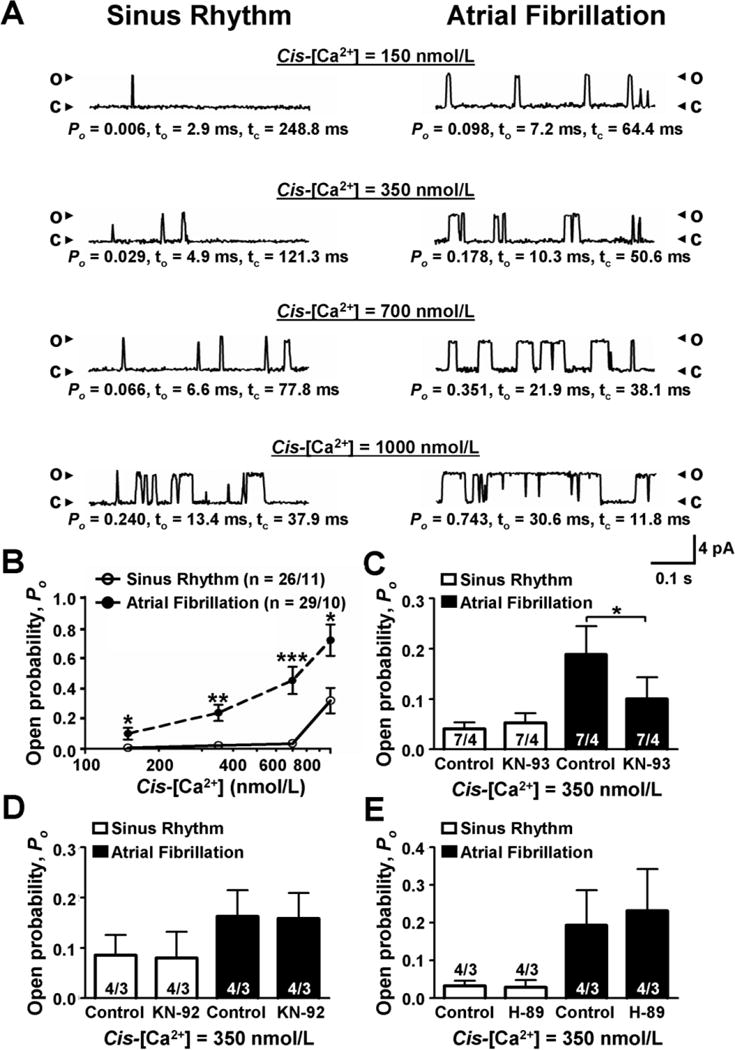
A, Single-channel tracings were obtained using planar lipid-bilayer recordings of RyR2 channels from sinus rhythm (left) and cAF (right) patients, respectively. Channel openings (o) are shown as upward deflections from the closed (c) level. The measurements were performed at indicated cytosolic (cis) [Ca2+] levels. The open probability (Po), mean open time (To), and mean closed time (Tc) are shown below each respective tracing. B, Mean+SEM RyR2 Po in Ctl and cAF at increasing cis [Ca2+]. C–E, Mean±SEM RyR2 Po before and after the CaMKII-inhibitor KN-93 (10-μmol/L, C), its inactive analog KN-92 (10-μmol/L, D) and the PKA-inhibitor H-89 (10-μmol/L, E) in Ctl and cAF, respectively. *P<0.05, **P<0.01 and ***P<0.001, respectively vs. corresponding control values in cAF. Numbers indicate channels/patients.
Next, we tested whether CaMKII- and PKA-phosphorylation inhibition reduces RyR2 Po in cAF. KN-93 (at 350-nmol/L cytosolic [Ca2+]) did not alter RyR2 Po for Ctl-myocytes, but significantly decreased RyR2 Po in cAF, whereas KN-92 and the PKA-inhibitors H-89 and PKI were ineffective (Figure 4C–E, Online-Figure 4B). These results suggest that the increased SR Ca2+-leak in cAF results from a CaMKII-mediated increase in Po of RyR2.
Atrial Myocytes from cAF Patients Display Enhanced Frequency of Spontaneous Ca2+-release Events (SCaEs)
To establish the relationship between NCX and arrhythmogenic SCaEs, we quantified SCaEs and corresponding INCX in the same cells, in which we measured the susceptibility to SCaEs by increasing bath [Ca2+] to 5-mmol/L (Figure 5A). SCaEs were defined as unstimulated [Ca2+]i-increases following a 1-minute period of ICa,L-triggered CaTs at 0.5-Hz. cAF-myocytes were more susceptible to SCaEs than Ctls (Figure 5B). In myocytes showing SCaEs, SCaE amplitude was ~3-fold higher and the coupling interval between the first SCaE and the last regular ICa,L-triggered CaT was significantly shorter in cAF (Figure 5C). The SCaEs in cAF-myocytes were completely suppressed by 10-μmol/L ryanodine (n=3; not shown), supporting the critical role of dysfunctional RyR2. SCaEs were accompanied by INCX currents, the amplitude of which was ~2.5-fold greater in cAF vs. Ctl (Figure 5C). The [Ca2+]i-INCX relationship was comparable in both groups (not shown).
Figure 5. Spontaneous SR Ca2+-release events (SCaEs) with corresponding inward INCX in myocytes from Ctl and cAF patients.
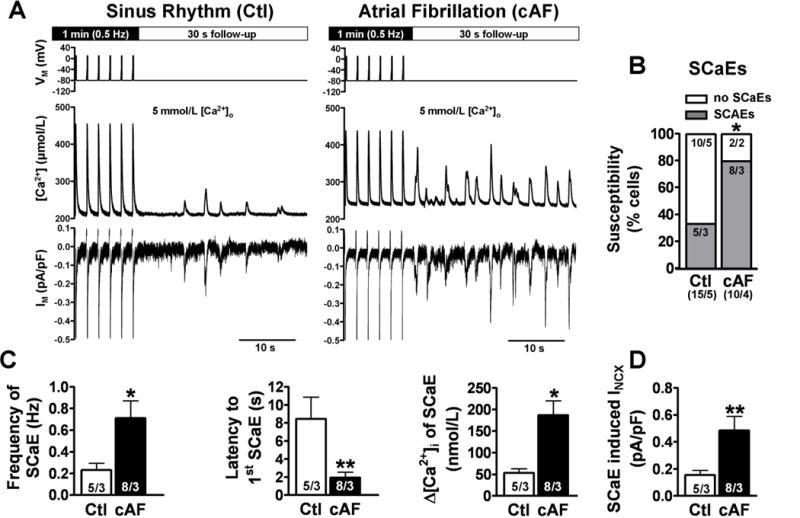
A, Voltage-clamp protocol (top) and representative recordings of SCaEs (Fluo-3, middle) and corresponding INCX (bottom) from a Ctl (left) and cAF (right) myocyte, respectively following steady-state stimulation for 1-minute at 0.5 Hz. B, Susceptibility to SCaEs in Ctl and cAF. *P<0.05 vs. Ctl (Fisher’s exact test). C, Mean±SEM frequency (left), latency (middle), and amplitude (right) of SCaEs in Ctl and cAF. D, Mean±SEM amplitude of SCaE-generated INCX. C,D, *P<0.05 and **P<0.01 vs. corresponding means in Ctl. Numbers indicate myocytes/patients.
Membrane-voltage Response to SCaEs and DAD-susceptibility
In normal cardiomyocytes, safety factors limit the ability of SCaEs to cause DADs.31, 32 In cAF, however, besides increased incidence of SCaEs, the same Δ[Ca2+]i generates a larger depolarizing INCX (Figure 2D), pointing to an increase in intracellular [Ca2+]i–membrane voltage (Vm) coupling gain, enhancing the risk of DADs/triggered activity.
Figure 6A shows representative AP-triggered CaTs in a Ctl- and a cAF-myocyte, respectively. Resting-membrane potential and AP-amplitude did not differ between Ctl and cAF, whereas AP-duration was significantly shorter in cAF (Figure 6B). Diastolic [Ca2+]i was non-significantly increased, whereas amplitude of the systolic CaT was 48% smaller in cAF vs. Ctl, consistent with the results of ICa,L-triggered CaTs. Figure 7A shows simultaneous Vm and CaT recordings in current-clamped myocytes immediately after 1-minute period of AP-triggered CaTs at 0.5-Hz. DADs were defined as a SCaE-induced Vm-change greater than 20-mV, because only DADs of such magnitude cause arrhythmogenic triggered activity in whole hearts.33 Susceptibility to SCaEs and SCaE-induced DADs were significantly increased in cAF (Figure 7B). Triggered activity in cAF was accompanied by prominent diastolic Vm-oscillations that appeared as DADs (6/13 myocytes) or triggered APs (2/13 myocytes). In the one Ctl-myocyte showing DADs, triggered APs were also observed. The Vm-oscillations were likely due to INCX-upregulation, because the NCX-blocker Ni2+ (10-mmol/L) prevented DADs/triggered activity without changing SCaE incidence (n=3, data not shown).
Figure 6. Simultaneous recordings of membrane voltage (Vm) and [Ca2+]i in atrial myocytes from Ctl and cAF patients.
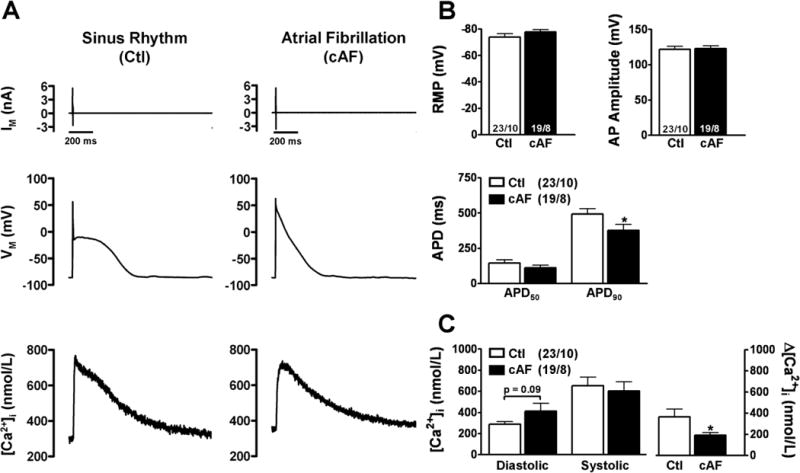
A, Current-clamp protocol (0.5 Hz, top) together with simultaneous recordings of triggered AP (middle) and CaT (Fluo-3, bottom) in a Ctl (left) and in a cAF (right) myocyte. B, Mean±SEM resting membrane potential, AP amplitude, APD50 and APD90, respectively. C, Mean±SEM diastolic and systolic [Ca2+]i levels (left) and resulting CaT-amplitude (right). *P<0.05 vs. corresponding means in Ctl. Numbers indicate myocytes/patients.
Figure 7. Incidence of SCaEs and corresponding DADs in current-clamped atrial myocytes from Ctl and cAF patients.
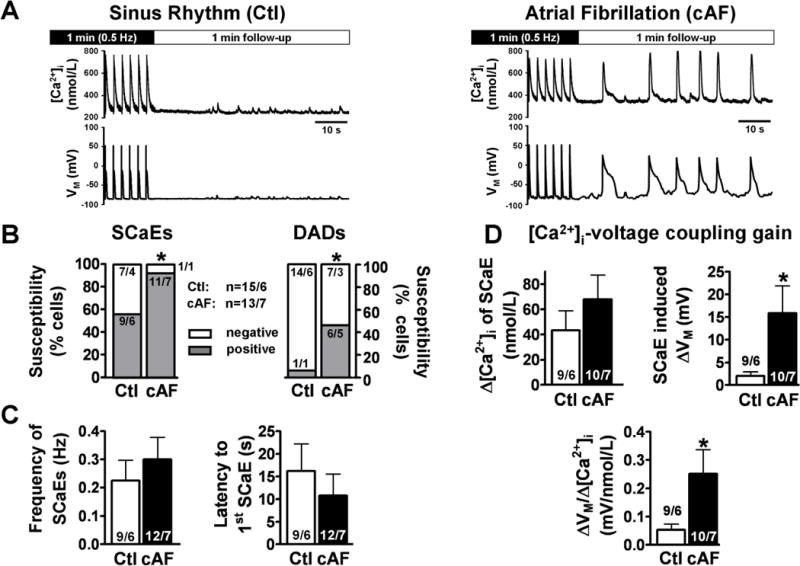
A, Representative recording of [Ca2+]i (Fluo-3) and corresponding membrane-voltage (Vm) oscillations (DADs/triggered APs) in a Ctl and a cAF-myocyte, respectively, following steady-state stimulation for 1-minute at 0.5 Hz. B, Enhanced susceptibility to spontaneous Ca2+-release events (SCaEs) and SCaE-induced DADs in cAF vs. Ctl. *P<0.05 vs. Ctl (Fisher’s exact test). C, Mean±SEM for frequency (left) and latency (right) of SCaEs. D, Mean±SEM amplitude of SCaEs (top left), magnitude of corresponding Vm-change (top right), and the calculated [Ca2+]i-membrane voltage coupling gain (bottom). *P<0.05 vs. corresponding means in Ctl. Numbers indicate myocytes/patients.
Consistent with Figure 5C, the intrinsic frequency of SCaEs tended to be higher, whereas the coupling interval to the first SCaE tended to be shorter in cAF (Figure 7C). The amplitude of SCaEs was non-significantly larger, whereas the SCaE-induced Vm change was significantly higher in cAF vs. Ctl (Figure 7D). Diastolic [Ca2+]i–Vm coupling gain, defined as the ratio of SCaE-induced Vm-change magnitude to SCaE-amplitude, was 5-fold greater in cAF than in Ctl, indicating that changes in diastolic [Ca2+]i produce stronger Vm-depolarizations in cAF vs. Ctl. Because of possible changes in cytosolic-constituents with tight-seal patch-clamp, we repeated selected experiments with perforated-patch methods. Qualitatively-similar results were obtained for basic AP-properties and CaTs in perforated-patch clamped myocytes (Online-Figure 5) compared to tight-seal results (Figure 6). Perforated-patch clamped myocytes also showed similar cAF-related promotion of SCaEs/DADs (Online Figures 6A–C) to tight-seal patched cells (Figure 7A–C), and their Ctl-cAF SCaE/DAD frequency-differential persisted despite PKA-inhibition with H-89, whereas subsequent CaMKII-inhibition with KN-93 normalized the frequency of SCaEs/DADs in cAF to Ctl-levels (Online-Figure 6C). Similar trends were observed for SCaE-associated Vm-change (Online-Figure 6D).
S2814D Knock-in Mice Display Enhanced SR Ca2+-leak and Susceptibility to AF
To determine whether CaMKII-phosphorylation of RyR2 alone can cause SCaEs and AF, we studied a knock-in mouse model in which a constitutive phosphorylation of RyR2 at S2814 is mimicked (S2814D-mice).12 The Ca2+-spark frequency (CaSF) was significantly higher in myocytes from homozygous S2814D vs. wildtype (WT) mice (Figure 8A,B). Full-width-half-maximum (FWHM) and full-duration-half-maximum (FDHM) of Ca2+ sparks were increased in S2814D vs. WT, and there was a trend towards increased Ca2+-spark amplitude (Online-Figure 7). SR Ca2+-content was ~25% lower in S2814D vs. WT (Online-Figure 7). KN-93 decreased Ca2+-spark frequency, but Ca2+-spark frequency remained significantly higher than in WT, unmasking the specific contribution of S2814 (Figure 8A,B).
Figure 8. Increased AF susceptibility and SR Ca2+-leak in S2814D knock-in mice.
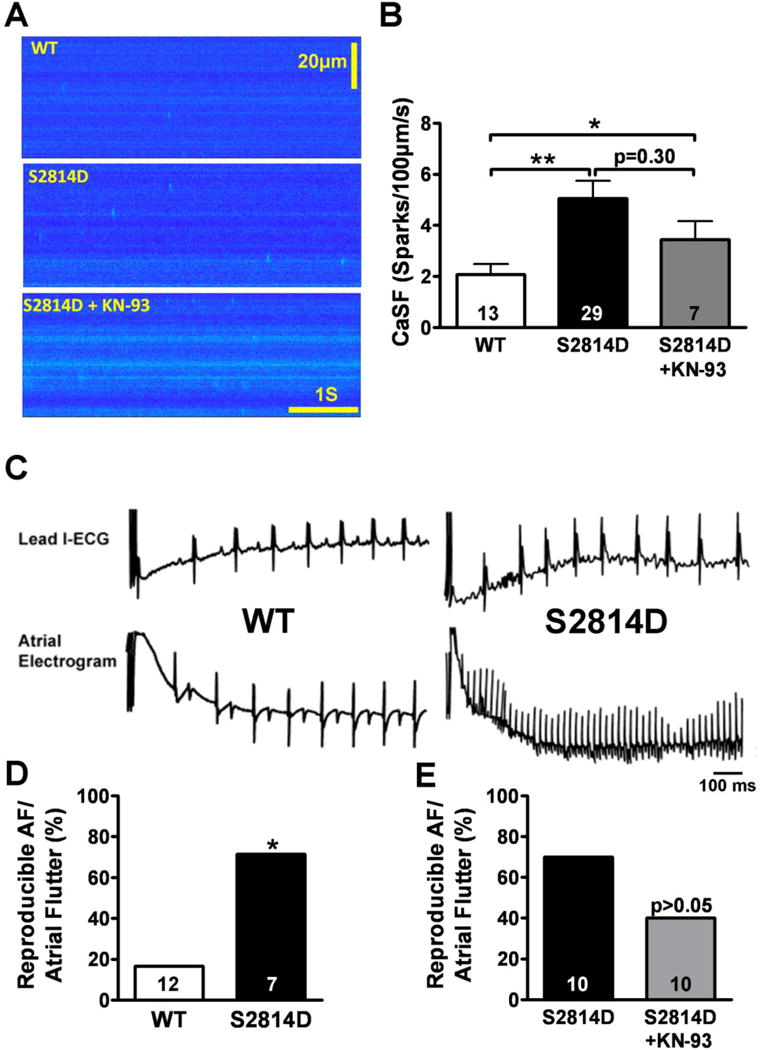
A, Representative traces of Ca2+-spark (Fluo-4) recordings in WT and S2814D myocytes in absence and presence of KN-93 (10-μmol/L). B, Mean±SEM Ca2+-spark frequency (CaSF). Numbers indicate myocytes from at least 3 mice in each group. *P<0.05 and **P<0.01 vs. WT. C, Surface L1-ECG and intracardiac atrial electrogram showing P-wave absence and irregular RR-intervals in S2814D mice following atrial-burst pacing (right), suggestive of AF. WT-mice typically reverted to sinus rhythm immediately following atrial pacing (left). D,E Bar graphs summarizing the incidence of reproducible AF in WT, S2814D-control mice and S2814D-mice treated with KN-93 (10-μmol/kg, i.p.). Numbers indicate mice.
S2814D-mice did not develop spontaneous AF. However, following RA-pacing using a catheter inserted via the jugular vein,24 S2814D-mice were more susceptible to atrial-arrhythmia induction, which occurred in 71% of S2814D vs. 17% of WT-mice (P<0.05) (Figure 8C,D), indicating that SR Ca2+-leak via constitutively CaMKII-phosphorylated RyR2 predisposes to AF-induction. To define the specific contribution of the S2814-phophorylation to AF-inducibility in a separate cohort we injected S2814D-mice with KN-93 (Figure 8E). The susceptibility to pacing-induced AF decreased from 70% before to 40% after KN-93, but remained significantly higher than in WT (17%), suggesting the presence of an arrhythmogenic substrate specifically related to the phosphomimetic (S2814D) RyR2-channel (Figure 8E).
Discussion
Here, we detailed the Ca2+-signaling mechanisms leading to cellular ectopic activity in cAF. cAF-myocytes showed greater SR Ca2+-leak for any given SR Ca2+-content as a consequence of CaMKII-mediated increase in Po of RyR2. Inhibition of CaMKII normalized RyR2 Po and SR Ca2+-leak, suggesting a direct link between defective RyR2 function and enhanced SR Ca2+-leak. SCaEs and potentially-arrhythmogenic DADs occurrence was increased in cAF. A given [Ca2+]i generated greater depolarizing INCX and a stronger Vm-depolarization in cAF, suggesting increased diastolic [Ca2+]i-Vm coupling gain as a mechanism of increased ectopic activity in cAF. CaMKII-inhibition reduced the AF-related increase in SCaE-occurrence. Phosphomimetic S2814D-mice showed enhanced SR Ca2+-leak and increased susceptibility to pacing-induced AF, validating the direct mechanistic link between CaMKII-phophorylated RyR2 and atrial arrhythmogenesis in clinical AF.
Comparison to Previous Studies
In previous work atrial tissue and myocytes from cAF patients showed abnormal SR Ca2+-handling9, 10, 13, 16, 26 and increased incidence of Ca2+-sparks,9, 10 pointing to enhanced SR Ca2+-leak, a well-established contributor to cardiac arrhythmogenesis.11, 12 Since SR Ca2+-content was not increased9, 10 and RyR2 was hyperphosphorylated at Ser2808 (PKA- and CaMKII-site) and Ser2814 (CaMKII-site),10, 13, 16 it was assumed that the higher Ca2+-spark frequency results from altered RyR2 function. Consistent with this hypothesis, inhibition of CaMKII, which is enhanced in cAF (see below),16 reduced Ca2+-spark frequency in cAF-myocytes,10 suggesting RyR2-hyperphosphorylation as a major mechanism of increased Ca2+-sparks frequency in cAF. However, the direct contribution of RyR2 to SR Ca2+-leak and the mechanistic link between CaMKII, RyR2 dysfunction and SR Ca2+-leak underlying AF pathogenesis were not addressed.
RyR2-hyperphosphorylation sensitizes RyR2-channels to Ca2+, and here we found increased Ca2+-sensitivity along with enhanced Po of RyR2 in cAF. Increased Po was due to increased frequency of openings, with no change in open time. These data, the first direct demonstration of RyR2 dysfunction in human atrium, suggest RyR2-mediated SR Ca2+-leak in cAF. Indeed, using tetracaine,22 we found a higher total SR Ca2+-leak for any given SR Ca2+-content in cAF. Interestingly, CaMKII-inhibition normalized both the higher Po of RyR2 and the larger SR Ca2+-leak, whereas PKA-inhibition was ineffective. These results further validate the culprit role of CaMKII in RyR2-mediated SR Ca2+-leak.
Continuous SR Ca2+-leak and discontinuous SCaEs are arrhythmogenic because cytosolic Ca2+ activates forward-mode INCX that may produce DADs.5 Expression of NCX1 is increased in sheep and patients with cAF10, 26, 34, 35 and [Ca2+]i-INCX coupling gain is increased in cAF sheep,32 although SR Ca2+-leak was not studied in this model. Here we show an increase in [Ca2+]i-INCX coupling gain in cAF-myocytes, which along with the higher SR Ca2+-leak should increase the propensity for arrhythmogenic DADs/triggered activity.
Although the increased incidence of Ca2+-sparks in cAF-patients was postulated to predispose to DADs/triggered activity by producing larger depolarizing INCX,9, 10 we are not aware of any study demonstrating higher susceptibility to DADs in clinical AF. In this study, we conducted for the first time simultaneous recordings of Ca2+ and membrane current or potentials, respectively, in human atrial myocytes. We observed 42% lower ICa,L and 23% shorter APD90, two hallmarks of atrial remodeling in cAF patients.6, 7, 36 In addition, we detected 50% and 48% lower amplitudes of ICa,L- and AP-triggered CaT, respectively, and a tendency of diastolic [Ca2+]i to be higher in cAF than in Ctl, which was consistent with previous results.10 Most important, we found that myocytes from cAF patients have increased SCaE frequency and amplitude, accompanied by greater INCX currents than Ctls. The magnitude of membrane depolarization due to SCaE-induced INCX depends not only on the amplitude of SCaEs but also on the latency of the SCaE from the last regular CaT.31 Consistent with the greater leakiness of RyR2 in cAF, the coupling interval to the first SCaE was significantly shorter in cAF vs. Ctl myocytes, suggesting that the intrinsic ability of the Ca2+-release mechanism to become refractory after release is impaired in cAF. These findings shed light on the factors that govern the rate of cytosolic SR Ca2+-release during SCaEs.
In normal hearts, plasmalemmal safety factors constrain the ability of SCaEs to promote DADs,5 protecting the heart against arrhythmias.31 We demonstrated more frequent burst emergence of SCaEs in cAF, accompanied by Vm-oscillations in the form of DADs and triggered APs. The Vm-oscillations disappeared after NCX-inhibition with Ni2+, whereas SCaEs remained unaffected, indicating a cause-effect relationships among SCaEs, NCX and DADs. The size of a DAD depends on at least 2-factors: the latency and amplitude of SCaE and the sensitivity of resting Vm to [Ca2+]i and here we detected a 5-fold increase in [Ca2+]i–Vm coupling gain in cAF compared to Ctl. The [Ca2+]i–Vm coupling gain is determined by the amplitude of depolarizing INCX and membrane resistance, set by background conductances like the inward-rectifier potassium current IK1,37 with enhanced INCX and/or reduced IK1 both promoting DADs. Expression of NCX1, and [Ca2+]i-corrected INCX amplitude are greater in cAF than Ctl; however, IK1 is upregulated in cAF-patients,36, 38–40 suggesting that augmented SCaEs and increased INCX, rather than reduced IK1, accounts for the stronger [Ca2+]i–Vm coupling gain in cAF. In addition, the relative contribution of Serca to Ca2+ removal from the cytosol was decreased, whereas the relative contribution of NCX was increased in cAF, suggesting that a larger part of the SR Ca2+-leak and SCaEs is removed by NCX, which might contribute to the increased NCX function in cAF. Further extensive experimentation is needed to dissect the molecular determinants of increased diastolic [Ca2+]i–Vm coupling gain in cAF.
Novel Findings and Potential Significance
Atrial remodeling is a key element of the AF-maintaining substrate1, 2 and emerging evidence suggests a critical involvement of abnormal subcellular Ca2+ signaling.3, 4, 9, 10, 13, 16, 26 Our study shows existence of RyR2-mediated SR Ca2+-leak and up-regulated NCX in cAF, which predispose to diastolic SCaEs, enhancing the susceptibility to cellular DADs/triggered activity in cAF. Defective RyR2 and up-regulated INCX likely contribute to atrial arrhythmogenesis in vivo by creating an arrhythmogenic substrate and acting as triggers for AF. We suggest that the CaMKII-mediated increase in SR Ca2+-leak in cAF creates an arrhythmogenic substrate by increasing the susceptibility to SCaEs, whereas due to the increase in diastolic [Ca2+]i–Vm coupling gain SCaEs generate a larger INCX and a given INCX greater DADs in cAF, providing a trigger for AF.
Here we noted reduced inhibitory Thr306-phosphorylation of CaMKIIδC and increased calmodulin expression in cAF vs. Ctl. The mRNA levels of CaMKIIδC were unchanged, suggesting a posttranscriptional mechanism. The increased atrial expression and Thr287-phosphorylation of CaMKIIδC are typical for dogs with atrial tachycardia remodeling18 and goats with sustained AF,17 suggesting that the greater CaMKII activity might be a consequence of the high-atrial rate during AF. Atrial dilatation in goats17 and ventricular tachypacing-induced heart failure in dogs,41 two frequent causes of cAF, are also associated with increased expression and Thr287 phosphorylation of CaMKIIδC. Thus, it is possible, but not proven, that an increase in CaMKII activity may also be a risk factor for AF, suggesting that CaMKII-activation may be both a cause and consequence of AF. This finding adds to our appreciation of the phenomenon of “AF begets AF”, indicating that in addition to the well-established tendency of AF to promote its own reentrant substrate, it can also enhance spontaneous ectopic activity that induces reentry.
Abnormal Ca2+ handling can contribute to atrial arrhythmogenesis through multiple mechanisms. For instance, changes in Ca2+-homeostasis modify the function of ion channels and the shape and the dynamics of the AP, creating tissue properties (vulnerable substrate) that may initiate and maintain reentry. SCaEs may cause subcellular Ca2 alternans and abrupt repolarization changes that may increase the dispersion of atrial refractoriness, facilitating reentry.42 Thus, the Ca2+-handling abnormalities we observed may promote AF by mechanisms additional to triggered activity, a notion that merits assessment in future studies.
Potential Limitations
We focused on the role of abnormal subcellular Ca2+-signaling for atrial arrhythmogenesis, but these alterations may critically contribute to AF-related atrial hypocontractility and myofilament dysfunction.43–45 SCaEs can impair contractile function by causing dyssynchrony of myocyte contraction. The potential link of altered subcellular Ca2+-signaling to impaired myocyte-contraction should be a subject of future investigations.
For myocytes isolation, we used only right-atrial tissue collected from only one region (right-atrial appendages). Thus, our findings may not apply fully to other atrial regions. In statistical comparisons, patients may contribute more than one observation to each sub-analysis suggesting that observations are not necessarily independent. Since due to the low sample size within-patient correlations were not taken into account in statistical comparisons, our results should be interpreted with caution.
We used 5-mmol/L extracellular Ca2+ to provoke SCaEs and these conditions test SCaE vulnerability rather than occurrence under normal physiological conditions. However, many AF-patients do not immediately show atrial ectopy or AF-recurrence immediately post-cardioversion, indicating that they likely have a latent predisposition to develop arrhythmia, perhaps related to changes in autonomic tone or other factors. Our results provide, for the first time, insights into the magnitude and dynamics of SCaEs and underlying factors in man, essential information to understand why an increase in SR Ca2+-leak and SCaE occurrence are potentially arrhythmogenic in cAF-patients.
In the sub-analysis including SCaEs-positive myocytes only, Ca2+-sensitivity of INCX appeared comparable between Ctl and cAF. However, since Ca2+ is a strong modulator of NCX-function46 and these experiments were primarily carried out at 5-mmol/L extracellular Ca2+ to test SCaE vulnerability, the complex patterns of Ca2+ modulation of NCX under these conditions might have masked the differences in Ca2+-sensitivity of NCX between Ctl and cAF that we detected using caffeine in unselected atrial myocytes and at physiological extracellular Ca2+. Since our results with caffeine showing increased Ca2+-sensitivity of INCX were consistent with recent work in sheep with persistent AF,34 increased Ca2+-sensitivity of INCX might be a typical finding in myocytes during persistent AF.
Previous results from perforated-patch clamped human atrial myocytes showed that Ca2+-waves disappear 10 minutes after patch-break47 and that H-89 reduces the frequency of Iti-currents48, suggesting a role for baseline cAMP-formation and PKA-activation.49, 50 Here, we did not detect a contribution of PKA to the increased SR Ca2+-leak in cAF. One reason could be that cAMP was washed out of the myocyte in the ruptured-patch whole-cell configuration. However, we detected a 2-fold increase in cAMP levels in cAF (Online-Figure 8E) and found that application of isoprenaline and inhibition of phosphodiesterases with IBMX both increased ICa,L and CaT amplitudes in Ctl and cAF-myocytes, without significant differences in magnitude between the rhythm groups (Online-Figures 8–9). Since cAMP/PKA are essential for both signals, washout of cAMP is an unlikely explanation for the lack of any effect of PKA-inhibition in Figures 3–4. In addition, we tested directly the specific contributions of CaMKII and PKA to the enhanced susceptibility to SCaEs and the generation of DADs in perforated-patch clamped myocytes with preserved cAMP levels. We found that enhanced SCaEs/DADs persisted in cAF-myocytes in the presence of PKA-inhibition with H-89, and that subsequent CaMKII-inhibition with KN-93 reduced the frequency of SCaEs/DADs. Perhaps because of the limited number of cells available for these experiments, while these studies all provided results consistent with the more extensive experiments shown elsewhere in the paper, some differences between groups did not achieve formal statistical significance (Online-Figures 5–6), which needs to be considered in their interpretation.
Our approach of preincubating the myocytes with H-89 did not allow us to establish baseline SCaE-incidence, preventing an evaluation of the acute effects of PKA inhibition on SCaEs. However, since SCaEs were seen in about 83% of cAF-myocytes incubated in H-89 (Online-Figure 6), PKA-activity is clearly not essential to SCaE expression. A more systematic approach and much larger number of cells would be needed to determine precisely the role of PKA in SCaE-occurrence under physiological conditions in human atrial myocytes. While interesting and relevant, the required work is beyond the scope of the present study. Furthermore, notwithstanding our finding of a primary role for CaMKII in the RyR2 abnormalities and in the specific AF-related DAD-substrate, cAMP and PKA contribute importantly to determining atrial myocyte Ca2+-entry and SR Ca2+-load. Thus, although PKA-phosphorylation may not importantly control intrinsic RyR2 abnormalities, it could certainly contribute to DAD-generation and arrhythmias in AF-patients by enhancing SR Ca2+-loading and unmasking their vulnerability to DADs/triggered activity.
Finally, transgenic mouse models are useful research tools but they do not phenocopy all important aspects of clinical AF. Nevertheless, they are quite useful because it is difficult to assess directly the specific mechanisms controlling CaMKII and PKA phosphorylation of RyR2 and their consequences in AF patients. Further extensive work in large-animal AF models is required to define the specific contributions of CaMKII and PKA to AF-pathophysiology under conditions similar to those causing AF in man.
Conclusions
Increased diastolic RyR2-mediated SR Ca2+-leak, together with up-regulated NCX and enhanced diastolic [Ca2+]i–Vm coupling gain, predispose to cellular DADs/triggered activity, contributing to pathogenesis of human AF. Recent16, 25 and present work validated the mechanistic link between dysfunctional RyR2, along with underlying CaMKII-hyperphosphorylation, and susceptibility to AF, highlighting the importance of molecular RyR2 defects for AF pathobiology. Although the contribution of these cellular Ca2+-mediated proarrhythmic events to atrial foci in AF patients in vivo has to be established, development of new drugs specifically targeting diastolic SR Ca2+-leak might open novel therapeutic avenues to prevent atrial arrhythmogenesis by normalizing SR Ca2+-cycling.
Supplementary Material
Acknowledgments
The authors thank Trautlinde Thurm, Annett Opitz and Claudia Liebetrau for excellent technical assistance and the cardiosurgeons of Dresden and Heidelberg Heart Centers for providing human atrial tissue.
Sources of Funding
These studies were supported by the European-North American Atrial Fibrillation Research Alliance (ENAFRA, 07CVD03) and the Alliance for Calmodulin Kinase Signaling in Heart Disease (08CVD01) grants of Fondation Leducq, the European Network for Translational Research in Atrial Fibrillation (EUTRAF, 261057), the German Federal Ministry of Education and Research through the Atrial Fibrillation Competence Network (01Gi0204) and the German Center for Cardiovascular Research, the Canadian Institutes of Health Research (MOP 44365), and NIH grants R01-HL089598 and R01-HL091947. Dr. Wehrens is a W.M. Keck Foundation Distinguished Young Scholar in Medical Research. Dr. Li is the recipient of the Michel Mirowski International Fellowship in Cardiac Pacing and Electrophysiology from the Heart Rhythm Society (2009-2010) and the American Heart Association South Central Fellowship (2010–2012).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures
None.
References
- 1.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1:62–73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- 2.Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent Advances in the Molecular Pathophysiology of Atrial Fibrillation. J Clin Invest. 2011;121:2955–2968. doi: 10.1172/JCI46315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobrev D, Nattel S. Calcium handling abnormalities in atrial fibrillation as a target for innovative therapeutics. J Cardiovasc Pharmacol. 2008;52:293–299. doi: 10.1097/FJC.0b013e318171924d. [DOI] [PubMed] [Google Scholar]
- 4.Dobrev D, Voigt N, Wehrens XH. The ryanodine receptor channel as a molecular motif in atrial fibrillation: pathophysiological and therapeutic implications. Cardiovasc Res. 2011;89:734–743. doi: 10.1093/cvr/cvq324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 6.Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM, Nerbonne JM. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–436. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- 7.Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D. L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation. 2004;110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A. [DOI] [PubMed] [Google Scholar]
- 8.Grandi E, Pandit SV, Voigt N, Workman AJ, Dobrev D, Jalife J, Bers DM. Human Atrial Action Potential and Ca2+ Model: Sinus Rhythm and Chronic Atrial Fibrillation. Circ Res. 2011;109:1055–1066. doi: 10.1161/CIRCRESAHA.111.253955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 10.Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106:1134–1144. doi: 10.1161/CIRCRESAHA.109.203836. [DOI] [PubMed] [Google Scholar]
- 11.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 12.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XH. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez P, Bhogal MS, Colyer J. Stoichiometric phosphorylation of cardiac ryanodine receptor on serine 2809 by calmodulin-dependent kinase II and protein kinase A. J Biol Chem. 2003;278:38593–38600. doi: 10.1074/jbc.C301180200. [DOI] [PubMed] [Google Scholar]
- 15.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 16.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–1951. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greiser M, Neuberger HR, Harks E, El-Armouche A, Boknik P, de Haan S, Verheyen F, Verheule S, Schmitz W, Ravens U, Nattel S, Allessie MA, Dobrev D, Schotten U. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol. 2009;46:385–394. doi: 10.1016/j.yjmcc.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 18.Wakili R, Yeh YH, Yan Qi X, Greiser M, Chartier D, Nishida K, Maguy A, Villeneuve LR, Boknik P, Voigt N, Krysiak J, Kaab S, Ravens U, Linke WA, Stienen GJ, Shi Y, Tardif JC, Schotten U, Dobrev D, Nattel S. Multiple potential molecular contributors to atrial hypocontractility caused by atrial tachycardia remodeling in dogs. Circ Arrhythm Electrophysiol. 2010;3:530–541. doi: 10.1161/CIRCEP.109.933036. [DOI] [PubMed] [Google Scholar]
- 19.Voigt N, Makary S, Nattel S, Dobrev D. Voltage-clamp-based methods for the detection of constitutively active acetylcholine-gated IK,ACh channels in the diseased hea rt. Methods Enzymol. 2010;484:653–675. doi: 10.1016/B978-0-12-381298-8.00032-0. [DOI] [PubMed] [Google Scholar]
- 20.Dobrev D, Wettwer E, Himmel HM, Kortner A, Kuhlisch E, Schuler S, Siffert W, Ravens U. G-Proteinβ3-subunit 825T allele is associated with enhanced human atrial inward rectifier potassium currents. Circulation. 2000;102:692–697. doi: 10.1161/01.cir.102.6.692. [DOI] [PubMed] [Google Scholar]
- 21.Trafford AW, Diaz ME, Eisner DA. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Arch. 1999;437:501–503. doi: 10.1007/s004240050808. [DOI] [PubMed] [Google Scholar]
- 22.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 23.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 24.Li N, Wehrens XH. Programmed electrical stimulation in mice. J Vis Exp. 2010:e1730. doi: 10.3791/1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm. 2008;5:1047–1054. doi: 10.1016/j.hrthm.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–680. doi: 10.1161/CIRCULATIONAHA.106.636845. [DOI] [PubMed] [Google Scholar]
- 27.Diaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, Rios E, Shannon TR. Ca sparks do not explain all ryanodine receptor-mediated SR Ca leak in mouse ventricular myocytes. Biophys J. 2010;98:2111–2120. doi: 10.1016/j.bpj.2010.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi HS, Eisner DA. The role of sarcolemmal Ca2+-ATPase in the regulation of resting calcium concentration in rat ventricular myocytes. J Physiol. 1999;515:109–118. doi: 10.1111/j.1469-7793.1999.109ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res. 2008;103:509–518. doi: 10.1161/CIRCRESAHA.108.176677. [DOI] [PubMed] [Google Scholar]
- 32.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 33.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D’Hooge J, Heidbuchel H, Sipido KR, Willems R. Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circ Res. 2009;105:876–885. doi: 10.1161/CIRCRESAHA.109.206276. [DOI] [PubMed] [Google Scholar]
- 35.Schotten U, Greiser M, Benke D, Buerkel K, Ehrenteidt B, Stellbrink C, Vazquez-Jimenez JF, Schoendube F, Hanrath P, Allessie M. Atrial fibrillation-induced atrial contractile dysfunction: a tachycardiomyopathy of a different sort. Cardiovasc Res. 2002;53:192–201. doi: 10.1016/s0008-6363(01)00453-9. [DOI] [PubMed] [Google Scholar]
- 36.Dobrev D, Graf E, Wettwer E, Himmel HM, Hala O, Doerfel C, Christ T, Schuler S, Ravens U. Molecular basis of downregulation of G-protein-coupled inward rectifying K+ current IK,ACh in chronic human atrial fibrillation: decrease in GIRK4 mRNA correlates with reduced IK,ACh and muscarinic receptor-mediated shortening of action potentials. Circulation. 2001;104:2551–2557. doi: 10.1161/hc4601.099466. [DOI] [PubMed] [Google Scholar]
- 37.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 38.Voigt N, Trausch A, Knaut M, Matschke K, Varro A, Van Wagoner DR, Nattel S, Ravens U, Dobrev D. Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:472–480. doi: 10.1161/CIRCEP.110.954636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Wagoner DR, Pond AL, McCarthy PM, Trimmer JS, Nerbonne JM. Outward K+ current densities and Kv1. 5 expression are reduced in chronic human atrial fibrillation. Circ Res. 1997;80:772–781. doi: 10.1161/01.res.80.6.772. [DOI] [PubMed] [Google Scholar]
- 40.Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U. The G protein-gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706. doi: 10.1161/CIRCULATIONAHA.105.575332. [DOI] [PubMed] [Google Scholar]
- 41.Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol. 2008;1:93–102. doi: 10.1161/CIRCEP.107.754788. [DOI] [PubMed] [Google Scholar]
- 42.Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. doi: 10.1152/ajpheart.00390.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schotten U, Ausma J, Stellbrink C, Sabatschus I, Vogel M, Frechen D, Schoendube F, Hanrath P, Allessie MA. Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation. 2001;103:691–698. doi: 10.1161/01.cir.103.5.691. [DOI] [PubMed] [Google Scholar]
- 44.Wettwer E, Hala O, Christ T, Heubach JF, Dobrev D, Knaut M, Varro A, Ravens U. Role of IKur in controlling action potential shape and contractility in the human atrium: influence of chronic atrial fibrillation. Circulation. 2004;110:2299–2306. doi: 10.1161/01.CIR.0000145155.60288.71. [DOI] [PubMed] [Google Scholar]
- 45.Belus A, Piroddi N, Ferrantini C, Tesi C, Cazorla O, Toniolo L, Drost M, Mearini G, Carrier L, Rossi A, Mugelli A, Cerbai E, van der Velden J, Poggesi C. Effects of chronic atrial fibrillation on active and passive force generation in human atrial myofibrils. Circ Res. 2010;107:144–152. doi: 10.1161/CIRCRESAHA.110.220699. [DOI] [PubMed] [Google Scholar]
- 46.Hilgemann DW, Collins A, Matsuoka S. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol. 1992;100:933–961. doi: 10.1085/jgp.100.6.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llach A, Molina CE, Prat-Vidal C, Fernandes J, Casado V, Ciruela F, Lluis C, Franco R, Cinca J, Hove-Madsen L. Abnormal calcium handling in atrial fibrillation is linked to up-regulation of adenosine A2A receptors. Eur Heart J. 2011;32:721–729. doi: 10.1093/eurheartj/ehq464. [DOI] [PubMed] [Google Scholar]
- 48.Hove-Madsen L, Prat-Vidal C, Llach A, Ciruela F, Casado V, Lluis C, Bayes-Genis A, Cinca J, Franco R. Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc Res. 2006;72:292–302. doi: 10.1016/j.cardiores.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 49.Rivet-Bastide M, Vandecasteele G, Hatem S, Verde I, Benardeau A, Mercadier JJ, Fischmeister R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J Clin Invest. 1997;99:2710–2718. doi: 10.1172/JCI119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vandecasteele G, Verde I, Rucker-Martin C, Donzeau-Gouge P, Fischmeister R. Cyclic GMP regulation of the L-type Ca2+ channel current in human atrial myocytes. J Physiol. 2001;533:329–340. doi: 10.1111/j.1469-7793.2001.0329a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.