

Abstract
High-throughput, quantitative processing of N-linked glycans would facilitate large-scale studies correlating the glycome with disease and open the field to basic and applied researchers. We sought to meet these goals by coupling Filter-Aided-N-Glycan Separation (FANGS) to the individuality normalization when labeling with glycan hydrazide tags (INLIGHT™) for analysis of plasma. A quantitative comparison of this method was conducted against solid phase extraction (SPE), a ubiquitous and trusted method for glycan purification. We demonstrate that FANGS-INLIGHT purification was not significantly different from SPE in terms of glycan abundances, variability, functional classes, or molecular weight distributions. Furthermore, to increase the depth of glycome coverage, we executed a definitive screening design of experiments (DOE) to optimize the MS parameters for glycan analyses. We optimized MS parameters across five N-glycan responses using a standard glycan mixture, translated these to plasma and achieved up to a three-fold increase in ion abundances.
Introduction
Glycomics remains a nascent field, with quantification, separation, and bioinformatic methods quickly evolving. Development of high-throughput, quantitative methods is vital to attaining robust data that can provide insights into differential regulation of glycosylation from disease and the environment1–8. N-glycans decorate over 50% of proteins9 and are critical for cellular functions including protein folding10,11, enzymatic activity12–14, and cellular adhesion15,16.
Over the last decade, numerous purification methods for N-glycans have been reported in the literature, though nearly all involve clean-up post-deglycosylation by PNGase F17. Lectin columns18–20 or lectin-bound filters21 containing, for example, wheat germ agglutinin (sialic acid affinity), concanavalin A (mannose affinity), and/or Ricinus communis agglutinin (bi-antennary/galactose affinity), may be used to affinity purify N-glycans. Multiple lectins are incorporated onto the solid-phase to provide a range of carbohydrate specificities, but the capture of glycans with unusual structures can prove difficult. Furthermore, lectin affinities in vitro are considerably weaker than that measured in vivo, where the formation of clusters ensures tight and complete binding22,23.
Alternatively, solid phase extraction (SPE) columns contain functional groups that interact with glycan chemistries. The hydrophilicity of oligosaccharides facilitates differential separation from peptides and small organic molecules on graphitized carbon24, C1825, or hydrophilic interaction (HILIC) columns26. Borate27 and titanium oxide (TiO2)28 may be used to enrich the sialic acid containing subset of glycans/glycoproteins via chelation. The sialylated sub-fraction can also be purified by chemical ligation of oxidized sialic acids to hydrazide containing stationary phases or beads29. While SPE is highly effective, gravity elution methods are often associated with increased variability and can be time-consuming. These challenges can be overcome through the use of vacuum-operated 96-well SPE plates, which are compatible with hydrophobic and HILIC filters26,30.
Filter Aided N-Glycan Separation (FANGS)31 may be an attractive alternative to the SPE workflows. Filter aided sample preparation (FASP) transformed proteomics, making sample preparation facile, efficient, and cost-effective32. Likewise, in FANGS, biological samples can be cleaned on a molecular weight (MW) cutoff filter prior to glycan release, allowing the removal of complex lysis buffer or metabolic contaminants that can yield inconsistent PNGase F activity. With the exception of PNGase F and glycan derivatization reagents, FANGS utilizes the same materials and tools available in proteomics labs, is highly scalable, and reduces the needs for specialty equipment, such as vacuum manifolds and affinity plates. Published in 2014, this method was qualitatively demonstrated on permethylated N-glycans purified from standard glycoproteins and cell lysates. If shown to be unbiased and quantitative and eliminates the need for a glycoprotein enrichment step, FANGS promises to achieve similar gains for glycomics as FASP for proteomics.
Mass spectrometry (MS) is the gold standard for glycan detection and identification33–35. Compositions can be determined from high resolving power accurate mass (≤ 5 ppm), though MSn is needed to elucidate glycan linkages due to the large number of isomeric and/or isobaric species36,37. MALDI and electrospray ionization (ESI) are the two most common MS techniques used in glycan analysis. MALDI may be used to detect very large molecular weight glycans with reasonable relative quantification, but incorporation of off-line, pre-MS fractionation is difficult, time consuming, and often fails to resolve isomers. Reproducibility is stymied by the poor efficiency of laser ablation and the inhomogeneity of crystal formation38. ESI offers additional advantages in that it can be readily coupled to liquid chromatography (LC) or capillary electrophoresis (CE) separation platforms. With isomer resolution achievable35, the need for MSn for structural resolution is significantly reduced. Separations also decrease signal suppression from contaminants and co-eluting glycans, expanding the effectiveness of MS quantification. Derivatization is the preferred approach to overcome challenges with ionization. Detection of glycans in ESI is hindered by the hydrophobic bias, and labels that add non-polar-surface area reduce this effect39,40.
A limited number of derivatization options have been designed to specifically enhance MS signal and facilitate quantitation of biological samples41. Permethylation of glycans has historically been used to stabilize glycans in MALDI, though when applied to ESI, can achieve a several-fold increase in signal response42,43. Challenges of this technique include obtaining complete, reproducible reaction efficiency, which decreases the signal-to-noise ratio and can impact quantification. In the individuality normalization when labeling with glycan hydrazide tags (INLIGHT™) strategy, a hydrophobic, hydrazine tag is introduced at the glycan reducing sugar under mild conditions and yields an average 4-fold increase in abundance by nano-flow LC-MS44,45. Both labeling methods offer 13C stable isotope labeled (SIL) tags for accurate relative quantitation44,46,47. Alternatively, TMT can be used to multiplex up to six samples for relative quantitation of co-eluting, isobaric species in MS2 spectra; however, it has not been validated in complex samples48.
While hydrophobic tagging significantly reduces difficulties in effective ionization of glycans, MS parameter settings still contribute significantly to analyte abundance. A classical one-factor-at-a-time (OFAT) approach to optimization is time and resource intensive, and is not appropriate to yield globally optimum parameters49,50. Design of experiments (DOE) efficiently examines multiple parameters in a minimum number of runs51. The type of design and number of experiments is tailored to the depth of information desired from the system. Fractional designs are commonly used, but the required number of experiments increases exponentially with the number of parameters tested52. Alternatively, a new class of split-plot screening designs, definitive screening designs, has been recently published53. The definitive screening design more evenly samples the parameter space, allowing all secondary interactions to be examined with a low-level of aliasing, and its experimental requirements increase only linearly as parameters are added. These advantages make it particularly useful for experiments where quadratic effects are anticipated and secondary interactions are expected, yet undefined. DOE’s utility has been shown to model MS instrument responses, reveal interesting secondary interactions, and yield significant increases in the abundance of peptides from complex mixtures54,55. However, to the best of our knowledge, no optimization has been reported for glycans, which are expected to exhibit significantly different ionization characteristics than peptides.
The following “FANGS-INLIGHT” coupled method was developed to create a novel, alternative, high-throughput, quantitative glycan workflow for complex biological samples. We demonstrated that plasma N-glycans derived by this novel strategy were comparable in abundance to the SPE preparation, quantitatively reproducible, and had no apparent bias towards classes of glycans. Furthermore, a definitive screening design was constructed using standard glycans to gain insight into ESI abundance and glycan integrity as a function of twelve MS parameters. When optimized parameters were applied to complex plasma samples, we achieved up to a 3-fold increase in abundance.
Methods
Experimental materials, including the preparation of pooled hen plasma, are detailed in the supplementary methods.
Solid Phase Extraction of N-Glycans from Pooled Hen Plasma
The solid phase extraction of N-glycans was completed as described previously44 (Figure 1). N-glycans were obtained from 2.5 μL (~150 μg total protein) of pooled hen plasma by standard PNGase F digestion as described. SPE columns were freshly conditioned with the sequential addition of 16 mL of Solvent 1 (0.1% TFA in DI Water), 8 mL of Solvent 2 (80% ACN, 0.05% TFA), and 16 mL of Solvent 1. Glycans were resuspended in 1 mL of Solvent 1 and quantitatively transferred to the SPE column. The columns were washed with 40 mL of Solvent 1 and samples were eluted in four, 1 mL fractions of Solvent 3 (25% ACN, 0.1% TFA). Eluted fractions were stored at −80 °C overnight, dried by Thermo Scientific Savant™ SPD131 SpeedVac™ at 45 °C, reconstituted, and pooled. The final, pooled glycan-containing eluent was dried and stored at −20 °C until INLIGHT™ derivatization.
Figure 1.
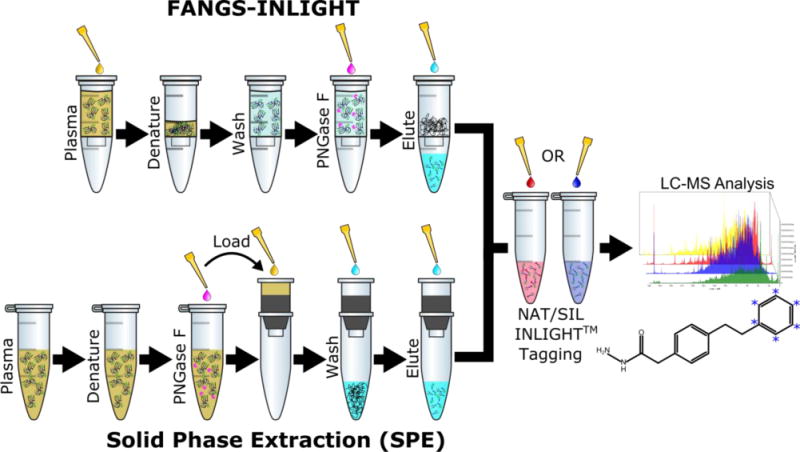
The FANGS-INLIGHT workflow steps are compared with the traditional SPE extraction method to prepare INLIGHT™ tagged N-glycans from plasma for relative quantification by UPLC-MS/MS.
FANGS-INLIGHT Coupled Strategy for N-glycans from Hen Plasma
The FANGS strategy31 was modified for a new application of processing of plasma (Figure 1). 2.5 μL of hen plasma was loaded onto a Vivacon 500, 30kDa molecular weight cutoff filter and diluted with 200 μL of 10mM DTT in digest buffer. Filters were gently vortexed, incubated at 56 °C for 30 minutes to denature protein, and concentrated onto the filter by centrifugation at 14,000 × g for 40 minutes. The samples were washed with 100 μL of digest buffer and concentrated at 14,000 × g for 20 min three times to remove small molecular weight contaminants. The filter was transferred to a fresh collection vial and 2 μL of PNGase F and 98 μL of digest buffer was added. The reaction was incubated at 37 °C for 18 hours to enzymatically cleave N-glycans. Glycans were eluted by centrifugation at 14,000 × g for 20 minutes. Two washes with 100 μL buffer were also collected to ensure complete elution of the glycans from the filter. Purified glycans were frozen at −80 °C, evaporated to dryness, and stored at −20 °C until INLIGHT™ derivatization.
INLIGHT™ Derivatization of N-linked glycans
Derivatization buffer was prepared containing 3:1 (v/v) MeOH: acetic acid. Dried N-glycans were tagged with 200 μL (50 μg) of the NAT or SIL label in derivatization buffer (250 μg/mL) for 3 hours at 56 °C. To quench the reaction, tagged glycans were dried at 55 °C in a SpeedVac. Dried, tagged species were stored at −20 °C until LC-MS analysis.
N-Glycan Standard Solution Preparation
For DOE optimization, an equimolar mixture of INLIGHT™ NAT and SIL tagged standard glycan solution was used. The mixture contained oligosaccharides representative of multiple glycan classes, including H6N5A3 glycan (trisialylated, 280 nM), H5N4F1 glycan (fucosylated, 270 nM), and MD-6 calibrated maltodextrin ladder (neutral, 260 nM). Glycan compositions are given based on their hexose (H), hexosamine (N), sialic acid (A), and fucose (F) subunits.
Definitive Screening Design of UPLC-MS/MS Parameters Using Standard Glycans
Derivatized and dried standard glycan mixture was resuspended in 200 μL of mobile phase A (98% H2O, 2% ACN, 0.1% formic acid) (MPA). Equimolar mixture (10 μL) was loaded on a 15 cm, Kinetex 2.6 μm (100 Å) C18 (Phenomenex), in-house packed 75 μm ID PicoTip® emitter column (New Objective). The column was conditioned each run with a 5 μl analytical column equilibration whose flow was pressure controlled at 500 bar. Separations were performed at 300 nL/min on an Easy-nLC 1000 (Thermo Scientific) with bi-linear gradient A that increased the percentage of mobile phase B (98% ACN, 2% H2O, 0.1% formic acid) (MPB) in the system: 0–25% (2 min), 25–45% (40 min), 45–90% (1 min), 90% (8 min), 90–0% (1 min), and 0% (10 min).
Data was collected on a Thermo Scientific Q Exactive™ High Field (HF) mass spectrometer. Twelve MS acquisition parameters were varied according to a definitive screening design (Supplementary Table 1). Data dependent acquisition (DDA) was used in Top 12 mode, with a standard inclusion list (Supplementary Table 2), a dynamic exclusion window (15.0s), and isotope exclusion “on.” Other constant parameters included: scan range (600–1900 Th), fixed first mass (125 Th), and apex trigger (OFF). For model validation experiments, MS parameters were set according to DOE optimized parameters (Table 1) and the standard mixture was injected in triplicate.
Table 1.
The initial definitive screening design parameter bounds and optimized parameters are given for each of the 12 independent factors. The optimized parameters were determined based on models across five response variables to maintain N-glycan integrity (minimize sialic acid loss), augment MS1 response, and increase MS2 information.
| Continuous Variables | Variable Code | Low Range | High Range | Initial Parameters | Optimized Parameters |
|---|---|---|---|---|---|
| S Lens (rf) | S-Lens | 45 | 65 | 55 | 65 |
| Ionization Time, MS1 (ms) | IT-1 | 50 | 225 | 250 | 64 |
| Temperature (°C) | Temp | 175 | 325 | 275 | 325 |
| Voltage (kV) | Volt | 1.75 | 3.75 | 2.25 | 1.75 |
| Underfill Ratio (%) | Underfill | 1 | 10 | 1 | 1 |
| Window (Th) | Window | 0.7 | 2 | 2.0 | 1.4 |
| Ionization Time, MS2 (ms) | IT-2 | 25 | 100 | 120 | 100 |
| Stepped Normalized Collision Energy, Midpoint | Mid-SNCE | 20 | 35 | 25 | 20 |
| Stepped Normalized Collision Energy, Step Size | Step-SNCE | 5 | 10 | 0 | 10 |
| Categorical Variables | Variable Code | Low Range | High Range | Initial Parameters | Optimized Parameters |
| Automatic Gain Control, MS1 | AGC-1 | 5E5 | 1E6 | 1E6 | 5E5 |
| Automatic Gain Control, MS2 | AGC-2 | 2E4 | 5E4 | 2E4 | 5E4 |
| Resolution, MS1 | Res-1 | 60k | 120k | 60,000 | 60,000 |
UPLC-MS/MS of Hen Plasma N-Glycans
Derivatized and dried hen plasma N-glycans were resuspended in 50 μL of MPA. For the inter-workflow comparison, equimolar mixtures of FANGS-INLIGHT and SPE glycans, tagged with NAT and SIL reagent, or vice versa, were prepared (4 biological replicates, 2 technical replicates). For intra-workflow analysis, native (NAT) and SIL glycan equimolar mixtures, purified from the same method, were combined (3 biological replicates per workflow, 2 technical replicates).
Sample (5 μL) was loaded onto a 5 cm × 100 μm ID trap packed with Kinetex 2.6 μm (100 Å) C18 (Phenomenex). Traps were prepared in-house according to Meiring et al.56. Separations were performed at 300 nl/min on a 30 cm × 75 μm ID PicoTip® emitter column (New Objective) packed similarly. The column was conditioned each run with a 2 μl pre-column and a 5 μl analytical column equilibration whose flows were pressure controlled at 500 bar. For comparison of the old and optimized MS parameters (Table 1), an equimolar mixture of NAT and SIL FANGS-INLIGHT derived glycans was analyzed in triplicate under each condition using gradient A. For workflow comparisons, the reverse-phase bi-linear gradient B was constructed to better compare the non-glycan components of the FANGS-INLIGHT and SPE purification schemes by incorporating an extended hydrophobic elution region: 5–30% (1 min), 30–40% (40 min), 40–63% (5 min), 63–90% (1 min), 90% (8 min), 90–5% (1 min), 5% (10 min). Method comparison experiments employed the optimized MS parameters determined by DOE optimization of standards (Table 1).
Data Processing and Quantitation
Glycans were identified by accurate mass (MMA ≤ 3ppm) manually in the Quan Browser of Thermo XCalibur using either a data list for the standard glycan mixture or for the hen plasma (Supplementary Table 3). The areas under each extracted ion chromatogram were integrated, corrected for SIL and NAT molecular weight overlap and individually normalized within each spectrum44.
Results and Discussion
Development of the High-Throughput FANGS-INLIGHT™ Coupled Workflow
The driving force behind the development of a filter-based separation method is the unbiased purification and extraction of glycans from complex biological matrices. The use of size exclusion as the primary method of glycan enrichment is not unprecedented in the literature31,57, but previous applications were qualitative or semi-quantitative and utilized MALDI detection. This novel FANGS-INLIGHT methodology is deeply rooted in high throughput, quantitative investigations of glycosylation, which call for the inclusion of chromatographic separation and SIL tagging via INLIGHT derivatization. The accessibility of protein samples on the filter following deglycosylation is a second motivation for the use of FANGS-INLIGHT™ over column based protocols, though at this point we have not assessed coverage of the proteome from the material remaining on the filter. In spite of its cross-platform limitations, SPE remains the most pervasive purification method for glycomics, and we sought to achieve equivalent performance by the FANGS-INLIGHT method.
An inter-workflow comparison was made between FANGS-INLIGHT and SPE processed hen plasma. We were concerned that because the filter was cellulose based and contained traces of glycerol, it could cause inconsistent elution due to the hydrophilicity of glycans or reduce the efficiency of the INLIGHT reaction, respectively. Paired N-glycans were analyzed by UPLC-MS/MS for relative quantification, with NAT and SIL channels corresponding to different preparations. The abundances of each glycan were not statistically significantly different (p > 0.05, paired t-test) (Figure 2) between the preparatory workflows. Furthermore, the molecular weight ranges of glycans detected from FANGS-INLIGHT (1.3–3.2 kDa) and SPE (1.3–2.8 kDa) workflows were nearly identical, and all glycan classes were represented with similar probabilities. This demonstrated that despite its synthetic composition, filtration purification did not utilize physiochemical interactions, was unbiased and did not cause any unexpected non-specific interactions reducing glycan recovery.
Figure 2.
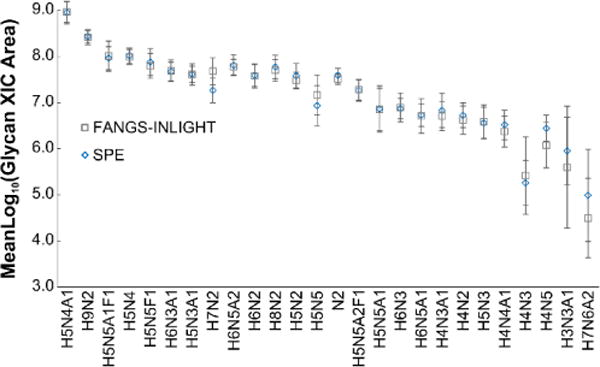
SPE and FANGS-INLIGHT derived N-glycans were analyzed as NAT/SIL INLIGHT™ pairs (N=8). The extracted ion chromatograms (XICs) of the 26 glycans common to each method were integrated, corrected for MW overlap, normalized, and log10-transformed44,45. There was no significant differences between the SPE and FANGS-INLIGHT integrated areas. The standard deviation of the mean is shown as error bars.
The intra-variability and reproducibility of each workflow was also compared; intra-workflow relative quantification of equimolar N-glycan mixtures was made on paired samples, with NAT and SIL channels derived from the same preparation method. No statistically significant difference in the distribution around the mean was determined between FANGS-INLIGHT and SPE methods (p > 0.05, paired t-test) (Figure 3). Accurate relative quantification was achieved with 80% of N-glycans falling within one log2 unit (2-fold change) of the mean. INLIGHT glycans were well resolved from contaminants by reverse phase LC and we did not see any cross-reactivity of N-glycans with other co-eluting species.
Figure 3.
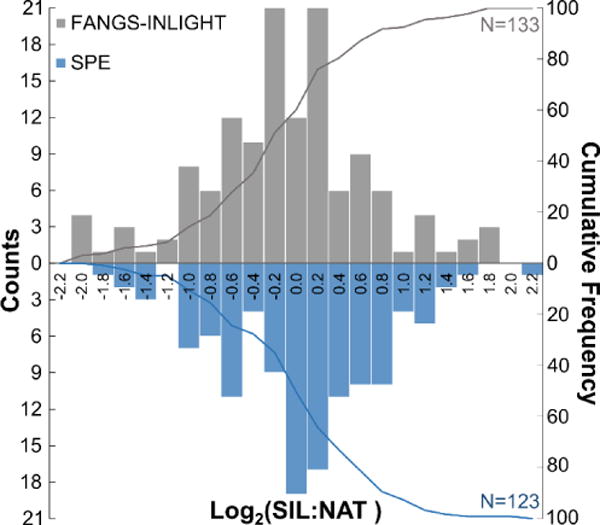
Equimolar mixtures of N-glycans (NAT:SIL) derived from the FANGS-INLIGHT (N=3) or SPE preparation (N=3) were assessed for intra-method variability. Glycans above the limit of detection (1×105) in the majority of samples were quantified by XIC area. Area measurements were corrected for MW overlap, normalized, and log2-transformed to produce a zero mean centered distribution. No statistically significant difference between the methods was detected (p = 0.92, t-test) and 80% of glycans were detected with ≤2-fold variation.
The FANGS-INLIGHT strategy offers significant benefits in terms of benchtop processing time over gravity SPE while not fundamentally impacting the quantitation of the glycome. Furthermore, FANGS-INLIGHT consumes fewer solvents, reducing per sample processing cost and waste generation compared to column based methods. Due to the similarity of the purification and LC materials required for FANGS and FASP, this enables a proteomics laboratory to immediately expand their analysis to glycomics, with the acquisition of PNGase F. FANGS also facilitates quantitative recovery of the deglycosylated proteins generated from PNGase F digestion (data not shown). This offers the potential for proteomic analysis of the retained proteins using in-line FASP procedures, including targeted glycosylation site analysis.
Definitive Screening Design Construction for MS Optimization of N-Glycans
Ionization of glycans is particularly challenging compared to other classes of molecules (i.e. peptides) due to their hydrophilicity and lability. The INLIGHT™ strategy confers 350 Å2 of non-polar-surface-area, achieving up to a 4-fold increase in nano-LC-MS abundance. However, adjustments of critical MS parameters, such as voltage and temperature, are needed to ensure stable spray and prevent gas-phase fragmentation of glycans as they travel from the emitter, through the S-lens, and into the quadrupole. While the magnitude of these effects are instrument specific, previous work on mass spectrometer peptide optimization has indicated that the basic relationships behind these parameters are to some extent universal54,58–60. Despite evidence that design of experiments can lead to improvements in protein abundance, to date, there has been no parallel investigation of MS parameters for glycans.
Definitive screening designs represent a new class of split-plot screening designs that can be used to produce second-order models, which include all quadratic and secondary interactions. For twelve parameters, the maximum correlation between terms (aliasing) is 0.27, allowing all effects to be reasonably estimated61. Bounds for each parameter are given in Table 1, and twenty six runs were designed and randomized in JMP Pro 11 (SAS Institute Inc., Cary, NC). Experiments were carried out on a standard glycan mix that represented the neutral (maltodextrin), sialylated (H6N5A3), and fucosylated (H5N4F1) glycan classes. Use of a standard was necessary to quantify gas-phase losses of sialic acids from H6N5A3, which can theoretically decompose into H6N5A2, H6N5A1, H6N5A1, or H6N5. Furthermore, the MS2 spectra of H5N4F1 could be examined for critical fragments, including the core fucose (N2F), without risk of co-eluting species contaminating the channel.
Models (Supplementary Equation 1) were generated for five distinct responses, including the ion abundance for glycans belonging to each class (H9, H5N4F1, H6N5A3), percent sialic acid loss, and the number of H5N4F1 MS/MS spectra collected. We chose to employ a modified stepwise Baysian Inference Criterion (BIC) approach that penalizes models that include more variables according to a natural logarithm equation. In a definitive screening design, to prevent type II errors, the final model is limited to three or fewer interactions62. A limited number, 19/66 possible combinations, of interactions (Supplementary Table 4) were tested to prevent overfitting. These interactions were selected on the basis of importance in previous peptide DOEs and predictions about possible fundamental mass spectrometry instrument component interactions. Furthermore, we included an extra main effect term, absolute ion abundance, which quantifies a known interaction between the MS2 AGC, underfill ratio, and ionization time terms (Supplementary Equation 2).
All responses calculated by an area-under-the-curve approach (H9, H5N4F1, H6N5A3 abundances and %A3 loss) showed a left-skewed distribution and were normalized through log10-transformation (Supplementary Figure 1). Fixed effects models for each of the five responses were constructed by standard least squares regression methods in JMP Pro 11. All main and quadratic effects and selected secondary interactions (Supplementary Table 4) were assessed for inclusion within the model using a forward stepwise approach and selecting for the BIC value. The five models constructed were all statistically significant (ANOVA, F-test) (Table 2, Supplementary Figure 2), with the best fit calculated for sialic acid loss. Equations describing raw glycan abundances contained a maximum of ten terms, with no significant interactions. Models for sialic acid loss and MS2 were considerably larger and included main, quadratic, and interaction effects (Supplementary Equation 1). Effects were standardized, as a function of their range, in order to compare magnitudes across parameters within a model (Supplementary Table 5).
Table 2.
Statistical parameters are reported to characterize each of the five response models for H5N4F1, H9, and H6N5A3 abundance, percent H6N5A3 sialic acid loss, and the number of MS2 spectra. These models were simultaneously optimized and their predicted response values were calculated. The experimental values (N=3) generated under the optimized parameters were not significantly different (p > 0.05) from the estimates for all models with the exception of MS2 spectra.
| Log(H5N4F1) | Log(H9) | Log(H6N5A3) | Log(%Sialic Acid Loss) | # MS2 Scans | |
|---|---|---|---|---|---|
| Model Parameters | |||||
| p-value | <0.0001 | <0.0001 | <0.0001 | 0.0016 | <0.0001 |
| R2/R2adj. | 0.90/0.84 | 0.91/0.85 | 0.76/0.70 | 0.99/0.97 | 0.99/0.97 |
| DF | 9 | 10 | 5 | 21 | 19 |
| Optimized Model Responses | |||||
| Predicted | 5.47 × 108 | 1.33 × 108 | 1.67 × 108 | 8.98 | 7.56 |
| Experimental | 1.46 × 108 | 2.83 × 108 | 3.80 × 108 | 5.27 | 19.3 |
Importantly, for optimization, a lower ESI voltage was correlated with both increased glycan abundance and decreased sialic acid loss. This relationship did not hold for temperature, where increasing temperature was significantly positively correlated with abundances but negatively correlated with sialic acid loss. These relationships suggest that the best stable spray and desolvation occurs at high temperatures and (relatively) low voltage, and that both temperature and voltage affect the mechanism for ionization-induced peeling of sialic acids.
The sialic acid loss model had the greatest predictive power and most confidence, with a negative BIC value (−58.7), an RMSE < 0.05, and a highly significant p-value (0.0016). Unique to this model, the S-lens was added as a main, quadratic, and interaction (with voltage) effect. We hypothesized that if increased voltage was responsible for sialic acid loss, then it may have also been responsible for shifting the preferred charge state of the analyte. Comparison of DOE experiments at different voltages, but similar S-lens and temperature parameters, showed a change in the distribution of charge states with a trend toward +3 at higher voltages. Ion trajectories through the S-lens are related to their size, and theory suggests that an increased RF should be used for species with larger m/z values63. A shift in charge state distributions, induced by emitter voltage effects, would affect the S-lens efficacy in transmitting the ions through the instrument and substantiate an interaction term between the main effects. This hypothesis warrants more detailed, formal investigations using model systems where charge state can be exactly controlled.
For glycan abundance, the largest scaled effects were voltage and, surprisingly, window2. It was unclear why a parameter that should primarily affect MS2 spectra quality would also affect MS1 abundance. A decreased isolation width necessarily increases the MS2 ionization time. Decreased MS1 abundance as a function of isolation window was attributed to variation in the timing of MS1 scan collection. Changes to the shape of the MS1 peak would alter integration under the curve. Interestingly, the sialic acid loss model also contained nearly all the MS2 parameters, including, absolute signal, which had the largest magnitude scaled to other estimates (Supplementary Table 5). The empirical nature of these relationships raise questions about how MS2 parameters can affect ions detected at the MS1 level, which need to be addressed outside of a DOE framework.
Optimization of MS Parameters to Increase Glycan Abundance in Plasma Samples
The five models produced for the standard glycan mixture were simultaneously optimized, with H5N4F1 abundance, H6N5A3 abundance, H9 abundance, and sialic acid loss equally weighted and a 50% weighting factor inflicted on the MS2 parameter to reduce its influence. The number of MS2 spectra was the only parameter for which maximization was not needed; instead, a minimal number of good quality spectra was required for structure identification. Qualitatively, we were able to determine that all types of fragments (individual glycan units and large glycan chains) were produced in the range of 10–30% NCE in the higher-energy collision dissociation (HCD) cell. Even at 10% NCE, the core fucose fragment was only sometimes observed; analysis of glycans at a NCE of 3–5% (data not shown) produced the fragment consistently, but this setting was not accessible in the Q Exactive™ XCalibur software, only directly through the Tune window. The optimized parameters are reported in Table 2 and the standard mixture was tested under these parameters to validate the model. The experimental values were not significantly different from those predicted with the exception of the number of MS2 spectra (Table 2).
The ultimate goal of this DOE was to achieve real gains in the integrity and ion abundances of N-glycans in complex mixtures. Biological samples are analyzed significantly differently from the standard mixture in that separations are more difficult, there is more ion suppression from co-eluting glycans and contaminants, and the diversity of glycans is greater. When N-glycan abundances from hen plasma, derived via the FANGS-INLIGHT™ method, were compared between the initial and optimized parameters (Table 1), there was a significant increase in 12 glycans (p < 0.05) and a probable increase in 5 additional glycans (p < 0.07) observed (Figure 4). Of these glycans, 70% contained sialic acids, suggesting that some of the increases in abundances resulted from the prevention of sialic acid peeling. Importantly, gains were also observed for neutral and fucose-containing species, with an average 1.9-fold increase across all glycans. Interestingly, N2, the core glycan unit, was detected under the initial conditions but not under the optimized conditions. This suggests that other, non-sialic acid, oligosaccharide linkages are susceptible to fragmentation and that optimization is very important for consistent results.
Figure 4.
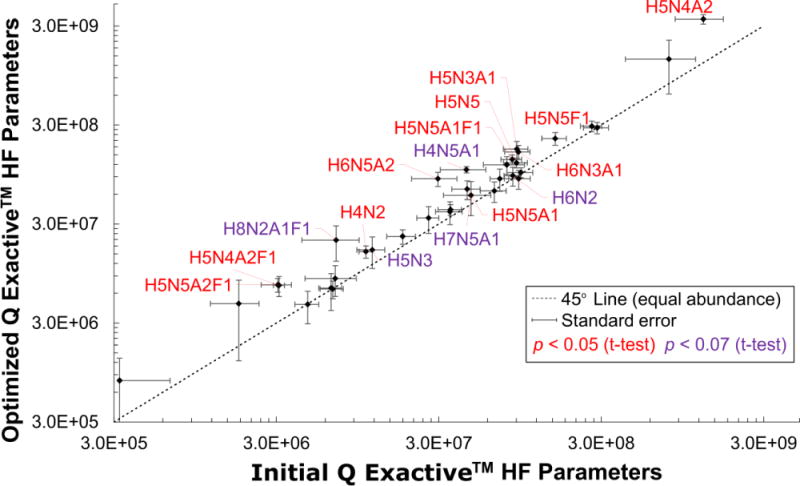
Plasma glycan abundances, derived from the FANGS-INLIGHT method, were compared between the DOE optimized MS parameters against the initial parameters on a logarithmic plot. Confidence intervals are shown for each species as error bars in the x-direction for the initial parameters (N=12) and in the y-direction for the optimized parameters (N=8). N-glycans falling to the left of the 45° line (dashed black line) were more abundant under the new conditions. N2 was not observed above the LOD for the optimized parameters and was omitted. Of the detected glycans, 12 had statistically significant (p < 0.05, one-sided t-test, red labels) and 5 had borderline (p < 0.07, purple labels) increases in abundance, with an average 1.90-fold change, and maximum 3-fold gain.
The advantages and efficiency of DOE for mass spectrometry applications are supported by the validation of the models’ predicted response values in the standard mixture and the gains achieved in glycan signal in plasma. These optimized parameters may be directly adopted by groups derivatizing glycans with tags of similar hydrophobicity. For groups analyzing glycans with different chemistries (e.g. permethylation) by LC-MS, the design table may be employed on these samples, though new models would need to be generated for optimization. Importantly, because the sialic acid loss model was so dependent on ESI voltage, we propose that for un-derivatized sialic acids analyzed by ESI, that a maximum threshold of 2.5 kV is needed to minimize losses to less than 5%. Despite the limitations of DOE in providing a theoretical framework for the empirical relationships it describes, it spurred the development of new hypotheses that can be formally tested in future research. The decreased LOD and increased signal-to-noise were achieved entirely through manipulation of MS parameters, orthogonal to improvements in sample preparation. These significant gains in MS response, coupled to the accurate relative quantification achieved by FANGS-INLIGHT, will increase the depth of glycome coverage and provide new opportunities for the comparison of diseased and healthy plasma.
Conclusions
We have quantitatively validated a new high-throughput processing method for N-glycans from plasma that offers flexibility in sample handling and has the potential for in-line discovery proteomics. Through DOE, for MS/MS analysis, we achieved up to three-fold increases in glycan abundance, which comes on top of the four-fold enhancement from the INLIGHT™ derivatization. With the parameters established in the study, confidence is gained in the integrity of glycans analyzed and increased sensitivity is achieved, positioning these methods for use in biomarker discovery projects.
Supplementary Material
Acknowledgments
This research was generously funded by the NIH NCI IMAT Program Grant R33 (CA147988–02), the NIH NIGMS Graduate Training in Molecular Biotechnology at NC State Grant (T32GM008776), the US Dept. of Education GAANN Fellowship Program in Molecular Biotechnology at NC State Grant (P200A140020), the W.M. Keck Foundation, and North Carolina State University. Hen plasma was obtained with the assistance of Dr. James N. Petitte and Rebecca Wysocky in the NC State University Dept. of Poultry Science.
Footnotes
Supporting Information Available
Supporting information is as indicated throughout the text. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Oefner CM, Winkler A, Hess C, Lorenz AK, Holecska V, Huxdorf M, Schommartz T, Petzold D, Bitterling J, Schoen AL, Stoehr AD, Van DV, Darcan-Nikolaisen Y, Blanchard V, Schmudde I, Laumonnier Y, Strover HA, Hegazy AN, Eiglmeier S, Schoen CT, Mertes MMM, Loddenkemper C, Lohning M, Konig P, Petersen A, Luger EO, Collin M, Kohl J, Hutloff A, Hamelmann E, Berger M, Wardemann H, Ehlers M. J Allergy Clin Immunol. 2012;129:1647–1655. doi: 10.1016/j.jaci.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 2.Rekedal LR, Massarotti E, Garg R, Bhatia R, Gleeson T, Lu B, Solomon DH. Arthritis Rheum. 2010;62:3569–3573. doi: 10.1002/art.27703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogers KM, Heise M. J Innate Immun. 2009;1:405–412. doi: 10.1159/000226422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reis CA, Osorio H, Silva L, Gomes C, David L. J Clin Pathol. 2010;63:322–329. doi: 10.1136/jcp.2009.071035. [DOI] [PubMed] [Google Scholar]
- 5.Preston RJS, Rawley O, Gleeson EM, O’Donnell JS. Blood. 2013;121:3801–3810. doi: 10.1182/blood-2012-10-415000. [DOI] [PubMed] [Google Scholar]
- 6.Goetz JA, Mechref Y, Kang P, Jeng MH, Novotny MV. Glycoconjugate J. 2009;26:117–131. doi: 10.1007/s10719-008-9170-4. [DOI] [PubMed] [Google Scholar]
- 7.Gardinassi LG, Dotz V, Hipgrave Ederveen A, de Almeida RP, Nery Costa CH, Costa DL, de Jesus AR, Mayboroda OA, Garcia GR, Wuhrer M, de Miranda Santos IKF. mBio. 2014;5:1–12. doi: 10.1128/mBio.01844-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Proteomics. 2014;14:525–546. doi: 10.1002/pmic.201300387. [DOI] [PubMed] [Google Scholar]
- 9.Apweiler R, Hermjakob H, Sharon N. Biochim Biophys Acta Gen Subj. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 10.Branza-Nichita N, Petrescu AJ, Negroiu G, Dwek RA, Petrescu SM. Chem Rev. 2000;100:4697–4711. doi: 10.1021/cr990291y. [DOI] [PubMed] [Google Scholar]
- 11.Live DH, Kumar RA, Beebe X, Danishefsky SJ. P Natl Acad Sci USA. 1996;93:12759–12761. doi: 10.1073/pnas.93.23.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bloem K, Vuist IM, van der Plas AJ, Knippels LMJ, Garssen J, Garcia-Vallejo JJ, van Vliet SJ, van Kooyk Y. Plos One. 2013;8:e66266. doi: 10.1371/journal.pone.0066266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen VP, Choi RCY, Chan WKB, Leung KW, Guo AJY, Chan GKL, Luk WKW, Tsim KWK. J Biol Chem. 2011;286:32948–32961. doi: 10.1074/jbc.M111.261248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryšlavá H, Doubnerová V, Kavan D, Vaněk O. J Proteomics. 2013;92:80–109. doi: 10.1016/j.jprot.2013.03.025. [DOI] [PubMed] [Google Scholar]
- 15.Fan H, Meng W, Kilian C, Grams S, Reutter W. Eur J Biochem. 1997;246:243–251. doi: 10.1111/j.1432-1033.1997.00243.x. [DOI] [PubMed] [Google Scholar]
- 16.Gu J, Isaji T, Xu Q, Kariya Y, Gu W, Fukuda T, Du Y. Glycoconjugate J. 2012;29:599–607. doi: 10.1007/s10719-012-9386-1. [DOI] [PubMed] [Google Scholar]
- 17.Tarentino AL, Gomez CM, Plummer TH. Biochemistry. 1985;24:4665–4671. doi: 10.1021/bi00338a028. [DOI] [PubMed] [Google Scholar]
- 18.Hirabayashi J. Glycoconjugate J. 2004;21:35–40. doi: 10.1023/B:GLYC.0000043745.18988.a1. [DOI] [PubMed] [Google Scholar]
- 19.Turner GA. Clin Chim Acta. 1992;208:149–171. doi: 10.1016/0009-8981(92)90073-y. [DOI] [PubMed] [Google Scholar]
- 20.Faye L, Salier J-P. Electrophoresis. 1989;10:841–847. doi: 10.1002/elps.1150101208. [DOI] [PubMed] [Google Scholar]
- 21.Zielinska DF, Gnad F, Wiśniewski JR, Mann M. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 22.Drickamer K. Nat Struct Mol Biol. 1995;2:437–439. doi: 10.1038/nsb0695-437. [DOI] [PubMed] [Google Scholar]
- 23.Haab BB. Proteom Clin Appl. 2012;6:346–350. doi: 10.1002/prca.201200014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redmond JW, Packer NH. Carbohyd Res. 1999;319:74–79. [Google Scholar]
- 25.Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H. Nat Protoc. 2007;2:334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruhaak LR, Huhn C, Waterreus W-J, de Boer AR, Neusüss C, Hokke CH, Deelder AM, Wuhrer M. Anal Chem. 2008;80:6119–6126. doi: 10.1021/ac800630x. [DOI] [PubMed] [Google Scholar]
- 27.Siegel D. Analyst. 2012;137:5457–5482. doi: 10.1039/c2an35697j. [DOI] [PubMed] [Google Scholar]
- 28.Palmisano G, Lendal SE, Engholm-Keller K, Leth-Larsen R, Parker BL, Larsen MR. Nat Protoc. 2010;5:1974–1982. doi: 10.1038/nprot.2010.167. [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Li X-j, Martin DB, Aebersold R. Nat Biotech. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 30.Royle L, Campbell MP, Radcliffe CM, White DM, Harvey DJ, Abrahams JL, Kim Y-G, Henry GW, Shadick NA, Weinblatt ME, Lee DM, Rudd PM, Dwek RA. Anal Biochem. 2008;376:1–12. doi: 10.1016/j.ab.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 31.Abdul Rahman S, Bergström E, Watson CJ, Wilson KM, Ashford DA, Thomas JR, Ungar D, Thomas-Oates JE. J Proteome Res. 2014;13:1167–1176. doi: 10.1021/pr401043r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nat Meth. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 33.Rakus JF, Mahal LK. Annu Rev Anal Chem. 2011;4:367–392. doi: 10.1146/annurev-anchem-061010-113951. [DOI] [PubMed] [Google Scholar]
- 34.Roseman S. J Biol Chem. 2001;276:41527–41542. doi: 10.1074/jbc.R100053200. [DOI] [PubMed] [Google Scholar]
- 35.Zaia J. Chem Biol. 2008;15:881–892. doi: 10.1016/j.chembiol.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laine RA. Glycobiology. 1994;4:759–767. doi: 10.1093/glycob/4.6.759. [DOI] [PubMed] [Google Scholar]
- 37.Werz DB, Ranzinger R, Herget S, Adibekian A, von der Lieth C-W, Seeberger PH. ACS Chem Biol. 2007;2:685–691. doi: 10.1021/cb700178s. [DOI] [PubMed] [Google Scholar]
- 38.Harvey DJ. Int J Mass spectrom. 2003;226:1–35. [Google Scholar]
- 39.Shuford CM, Muddiman DC. Expert Rev Proteomics. 2011;8:317–323. doi: 10.1586/epr.11.24. [DOI] [PubMed] [Google Scholar]
- 40.Walker SH, Lilley LM, Enamorado MF, Comins DL, Muddiman DC. J Am Soc Mass Spectr. 2011;22:1309–1317. doi: 10.1007/s13361-011-0140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mechref Y, Hu Y, Desantos-Garcia JL, Hussein A, Tang H. Mol Cell Proteomics. 2013;12:874–884. doi: 10.1074/mcp.R112.026310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baldwin MA, Stahl N, Reinders LG, Gibson BW, Prusiner SB, Burlingame AL. Anal Biochem. 1990;191:174–182. doi: 10.1016/0003-2697(90)90405-x. [DOI] [PubMed] [Google Scholar]
- 43.Harvey DJ. J Chromatogr B. 2011;879:1196–1225. doi: 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Walker SH, Taylor AD, Muddiman DC. J Am Soc Mass Spectr. 2013;24:1376–1384. doi: 10.1007/s13361-013-0681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bendiak B, Salyan ME, Pantoja M. J Org Chem. 1995;60:8245–8256. [Google Scholar]
- 46.Walker SH, Budhathoki-Uprety J, Novak BM, Muddiman DC. Anal Chem. 2011;83:6738–6745. doi: 10.1021/ac201376q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvarez-Manilla G, Warren NL, Abney T, Atwood J, Azadi P, York WS, Pierce M, Orlando R. Glycobiology. 2007;17:677–687. doi: 10.1093/glycob/cwm033. [DOI] [PubMed] [Google Scholar]
- 48.Hahne H, Neubert P, Kuhn K, Etienne C, Bomgarden R, Rogers JC, Kuster B. Anal Chem. 2012;84:3716–3724. doi: 10.1021/ac300197c. [DOI] [PubMed] [Google Scholar]
- 49.Czitrom V. Am Stat. 1999;53:126–131. [Google Scholar]
- 50.Yates F. Biometrika. 1939;30:440–466. [Google Scholar]
- 51.Fisher RA. J Min Agric. 1926;33:503–513. [Google Scholar]
- 52.Myers RH, Montgomery DC, Vining GG, Borror CM, Kowalski SM. J Qual Technol. 2004;36:53–77. [Google Scholar]
- 53.Jones B, Nachtsheim CJ. J Qual Technol. 2013;45:121–129. [Google Scholar]
- 54.Randall SM, Cardasis HL, Muddiman DC. J Am Soc Mass Spectr. 2013;24:1501–1512. doi: 10.1007/s13361-013-0693-y. [DOI] [PubMed] [Google Scholar]
- 55.Oberg AL, Vitek O. J Proteome Res. 2009;8:2144–2156. doi: 10.1021/pr8010099. [DOI] [PubMed] [Google Scholar]
- 56.Meiring HD, van der Heeft E, ten Hove GJ, de Jong APJM. J Sep Sci. 2002;25:557–568. [Google Scholar]
- 57.Sun N, Deng C, Li Y, Zhang X. Anal Chem. 2014;86:2246–2250. doi: 10.1021/ac404103r. [DOI] [PubMed] [Google Scholar]
- 58.Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M. Mol Cell Proteomics. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zachariadis GA, Rosenberg E. Rapid Commun Mass Sp. 2013;27:489–499. doi: 10.1002/rcm.6475. [DOI] [PubMed] [Google Scholar]
- 60.Houbart V, Cobraiville G, Lecomte F, Debrus B, Hubert P, Fillet M. J Chromatogr A. 2011;1218:9046–9054. doi: 10.1016/j.chroma.2011.10.030. [DOI] [PubMed] [Google Scholar]
- 61.Jones B, Nachtsheim CJ. J Qual Technol. 2011;43:1–15. [Google Scholar]
- 62.Li W, Nachtsheim CJ. Technometrics. 2000;42:345–352. [Google Scholar]
- 63.Beu SC, Hendrickson CL, Marshall AG. J Am Soc Mass Spectrom. 2011;22:591–601. doi: 10.1007/s13361-010-0057-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.