

Abstract
Objectives:
To describe the clinical and electrophysiologic features of synaptotagmin II (SYT2) mutations, a novel neuromuscular syndrome characterized by foot deformities and fatigable ocular and lower limb weakness, and the response to modulators of acetylcholine release.
Methods:
We performed detailed clinical and neurophysiologic assessment in 2 multigenerational families with dominant SYT2 mutations (c.920T>G [p.Asp307Ala] and c.923G>A [p.Pro308Leu]). Serial clinical and electrophysiologic assessments were performed in members of one family treated first with pyridostigmine and then with 3,4-diaminopyridine.
Results:
Electrophysiologic testing revealed features indicative of a presynaptic deficit in neurotransmitter release with posttetanic potentiation lasting up to 60 minutes. Treatment with 3,4-diaminopyridine produced both a clinical benefit and an improvement in neuromuscular transmission.
Conclusion:
SYT2 mutations cause a novel and potentially treatable complex presynaptic congenital myasthenic syndrome characterized by motor neuropathy causing lower limb wasting and foot deformities, with reflex potentiation following exercise and a uniquely prolonged period of posttetanic potentiation.
Congenital myasthenic syndromes (CMS) are a heterogeneous group of disorders caused by abnormal signal transmission between motor axons and skeletal muscle.1 Mutations in an increasing number of presynaptic proteins, components of the synaptic basal lamina, proteins involved in endplate development and maintenance, and more recently in protein glycosylation2 have been reported, causing novel and complex phenotypes. Since most CMS are treatable, diagnosing them is of utmost importance.
Signal transmission at the neuromuscular junction is mediated via the release of acetylcholine from synaptic vesicles.3 This process is rendered calcium sensitive by members of the synaptotagmin protein family, which also have a role in vesicle priming and in reducing spontaneous transmitter release.4,5 Synaptotagmin II (SYT2) is the major isoform expressed at the neuromuscular junction, and Syt2 knockout mice show markedly reduced calcium-evoked transmitter release.6 Synaptotagmins interact with SNAP-25,5 and mutations in SNAP25B have been described in patients with myasthenia and additional CNS phenotypes.7
Herein, we describe the clinical and electrophysiologic characteristics of 2 multigenerational families displaying a novel human motor syndrome caused by dominant SYT2 mutations: c.920T>G (p.Asp307Ala) and c.923G>A (p.Pro308Leu).8 These heterozygous missense mutations alter adjacent amino acids within the C2B calcium-binding domain of Syt2. Patients present with childhood-onset foot deformities, lower limb weakness, and wasting with areflexia. However, in some cases, additional fatigability of the eye and limb muscles is observed, and strength and reflexes improve with exercise. Furthermore, neurophysiologic testing reveals features indicative of a presynaptic CMS, with a uniquely prolonged period of posttetanic potentiation.
METHODS
Patients were investigated at the peripheral neuropathy clinic at University of Rochester and at the inherited peripheral neuropathy and congenital myasthenia clinics in Newcastle upon Tyne. They were included in the study on the basis of a genetically confirmed autosomal dominant SYT2 mutation. Patients were assessed using the Charcot-Marie-Tooth Neuropathy Score, second version,9 and the CMS Scale (CMSS), which is a modified version of the myasthenia gravis score,10 before, during, and after medication discontinuation.
Standard protocol approvals, registrations, and patient consents.
The project received approval by local ethics committees. Patients gave informed consent for all clinical, electrophysiologic, and therapeutic studies.
Electrophysiologic studies.
Studies were performed on a Viking-Nicolet (US) or a Dantec Keypoint G4 (UK) EMG machine. Surface electrical stimulation was applied via either CareFusion ring electrodes or a handheld Alpine Biomed bipolar stimulating electrode. Responses were recorded using Natus Neurology disposable disk electrodes (1 cm diameter). Amplitudes were measured baseline to peak. Single-fiber EMG was performed using Natus Neurology disposable 30G concentric needles with a bandpass of 2 to 10 kHz.
Repetitive nerve stimulation (RNS) was performed on the abductor digiti minimus, abductor pollicis brevis (APB), and tibialis anterior (TA). Ten supramaximal stimuli were applied, with the percentage increment or decrement calculated between the first and fourth response. An amplitude increase or decrease of greater than 10% was regarded as significantly abnormal.11
To assess posttetanic potentiation, single supramaximal stimuli were applied at least 60 seconds apart to establish the baseline compound muscle action potential (CMAP) amplitude. Participants were then asked to make a 10-second isometric maximum voluntary contraction (MVC) against resistance. Single supramaximal CMAP responses were then recorded every 30 seconds for 5 to 10 minutes, with longer time intervals up to 60 minutes.
RESULTS
Family 1 (United States).
This family carried a heterozygous missense mutation c.920T>G (p.Asp307Ala) in the SYT2 gene.
The proband (US II.2), a 49-year-old woman, was referred to the University of Rochester for putative Charcot-Marie-Tooth disease. She had high-arched feet and hammer toes since childhood. At age 44, she underwent surgery to correct hammer toe deformity and had delayed recovery following general anaesthesia, describing months of muscle fatigue and weakness. She continued to experience fatigue, weakness, and exertional dyspnea that improved with rest.
Physical examination showed pes cavus (figure 1A). Strength testing showed symmetric, proximal, and distal Medical Research Council (MRC) grade 4 weakness in upper and lower extremities that improved with muscle activation. Deep tendon reflexes were unobtainable at rest but elicited at the knees and biceps following exercise. Acetylcholine receptor binding, blocking, and modulating antibodies and voltage-gated calcium channel (VGCC) antibodies were negative. These were not checked in the other patients. No pathogenic CACNA1A gene mutations were found, and none of the patients underwent a muscle or nerve biopsy.
Figure 1. Clinical presentation of dominant SYT2 mutations.

Family tree of the US and UK families showing autosomal dominant inheritance pattern. (A) US family members show foot deformity but no lower limb wasting. (B) Lower limb wasting and foot deformities in the UK family. Asymmetrical lower limb wasting in UK II.2. The patient had undergone multiple operations to correct foot deformities and a right knee replacement. Symmetrical distal wasting after bilateral foot operations in III.2. Mild weakness and atrophy in toes and feet were seen in the other affected patients.
US I.2: A 70-year-old woman.
She had high-arched feet and hammer toes since childhood. Examination showed pes cavus and hammer toes. Strength testing showed grade 4+ weakness of finger extensor, finger abductor, ankle dorsiflexion, and hip girdle muscles. Deep tendon reflexes were absent and did not facilitate after exercise. Gait was narrow-based with mild waddling. She could not tandem, toe, or heel walk.
US III.1: A 23-year-old woman.
Examination revealed slight pes cavus and hammer toes. Deep tendon reflexes were unobtainable at the ankles and in the arms and hypoactive at the knees. The biceps reflex could be elicited after brief exercise. She had slight difficulty squatting and heel walking.
US III.2: A 15-year-old male.
He experienced tripping and falls starting in elementary school. He participated in school sports but described early muscle fatigue. Examination revealed pes cavus and hammer toes (figure 1A). Deep tendon reflexes were initially unobtainable, but could be elicited following brief exercise. The remainder of the neurologic examination was normal, except for mild difficulty with tandem gait.
Family 2 (United Kingdom).
In this family, a heterozygous missense mutation of SYT2 in the adjacent amino acid residue to that in the US family was found (c.923G>A [p.Pro308Leu]).
UK III.2: The index patient is a 27-year-old woman.
She was diagnosed with congenital hip dysplasia after birth and had foot deformities since childhood. At age 12 years, she underwent bilateral foot surgery for her high arches. On general physical examination she had severe bilateral foot deformities (figure 1B). Neurologic examination revealed fatigable eye movements, with slight ptosis after exercise and distal lower extremity muscle wasting, normal power in proximal limb muscles, and grade 4+ weakness in hand intrinsic muscles and grade 3 in ankle dorsiflexion and plantar flexion. Deep tendon reflexes were absent. Vibration and pinprick sensation was diminished in the feet and hands. Casual gait was nonataxic but aided by bilateral ankle foot orthotics and orthotic shoes.
UK IV.1: A 7-year-old male.
He was born by emergency Cesarean section for failure to progress. His motor milestones were delayed with poor balance and frequent falls. He had a mild language delay, but normal academic achievement. Physical examination showed joint hyperlaxity and pes planus. Neurologic examination showed poor fine motor skills, reduced muscle tone, with normal muscle bulk and strength. All deep tendon reflexes were absent. Gait was unsteady, and he was unable to heel walk.
UK III.6: A 16-year-old male.
He was asymptomatic. General examination revealed pes cavus and clawing of his toes bilaterally and mild wasting of distal lower limb musculature. Strength was normal but deep tendon reflexes were absent.
UK III.7: A 12-year-old female.
She was born with developmental hip dysplasia and had bilateral foot deformity since early childhood. She complained of difficulty with sports participation and of muscle cramps and pain in her lower limbs with prolonged exertion. Examination showed bilateral foot deformities. Muscle tone and bulk were normal, with MRC grade 4 weakness of right ankle dorsiflexion. She had generalized areflexia. Sensation was intact. She was unable to heel walk.
UK III.3 (a 24-year-old male) and UK III.8 (a 9-year-old female).
These patients are unaffected with no neurologic symptoms or signs and no foot deformities.
UK II.2: A 44-year-old woman.
She had bilateral hip dysplasia, corrected with braces. She had a history of progressive weakness and wasting predominantly in the right lower extremity since early childhood. She had been diagnosed with a variant of localized morphea, contributing to loss of soft tissue bulk in the right thigh. She also complained of right leg pain and abnormal sensory symptoms in her right foot and hand. She required surgery for bilateral foot deformity and complex reconstructive right knee surgery. General examination showed bilateral pes cavus and reduced muscle bulk proximally and distally in the right lower extremity and distally in the left lower limb. There was a slight ptosis and diplopia triggered by exercise. She had grade 4+ weakness in intrinsic hand muscles bilaterally, grade 4 weakness in proximal and distal right lower limb muscles, and 4+ in left ankle plantar and dorsiflexion. Deep tendon reflexes were diffusely unobtainable. Sensation was mildly reduced to pinprick in the feet. She had a waddling gait, with bilateral foot drop, and was unable to heel or toe walk.
UK II.3: A 42-year-old man.
He had his first foot surgery at the age of 27 years for clawing of his toes. He described muscle cramps and pain in his lower limbs with sustained exertion. Examination demonstrated grade 4 strength in ankle dorsiflexion and plantar flexion. Upper limb strength was normal. Deep tendon reflexes were absent, but patella reflexes were obtainable after brief exercise. He was unable to heel or toe walk.
Repetitive nerve stimulation.
Because of the history of fatigable weakness, we performed RNS to assess the reliability of neuromuscular transmission. RNS places the neuromuscular junction under stress, and a decrement of greater than 10% in the motor amplitude indicates a significant failure of transmission. Low-frequency (0.5 Hz) RNS of APB produced a decrement of −12% in the US proband (US II.2) and −18% in the UK proband (UK III.2). In UK III.2 and III.6, 0.5-Hz RNS of TA produced decremental responses of −20% and −15%, respectively.
Response to brief MVC.
Brief (10 seconds) MVC produced a significant amplitude increment in all of the muscles examined (table 1, figure 2A). In the US family, the mean amplitude increase was +91.2% (range +21.3% to +200%). In the UK family, the mean amplitude increase was +87.2% (range +19.0% to +420%) with a larger increment in lower limb vs upper limb muscles (APB: mean +34%, range +21.3% to +119.5%; abductor digiti minimus: mean +35%, range +20.5% to +200%: TA: mean +149%, range +19% to +420%).
Table 1.
Motor nerve conduction studies before and after low-frequency RNS and brief MVC
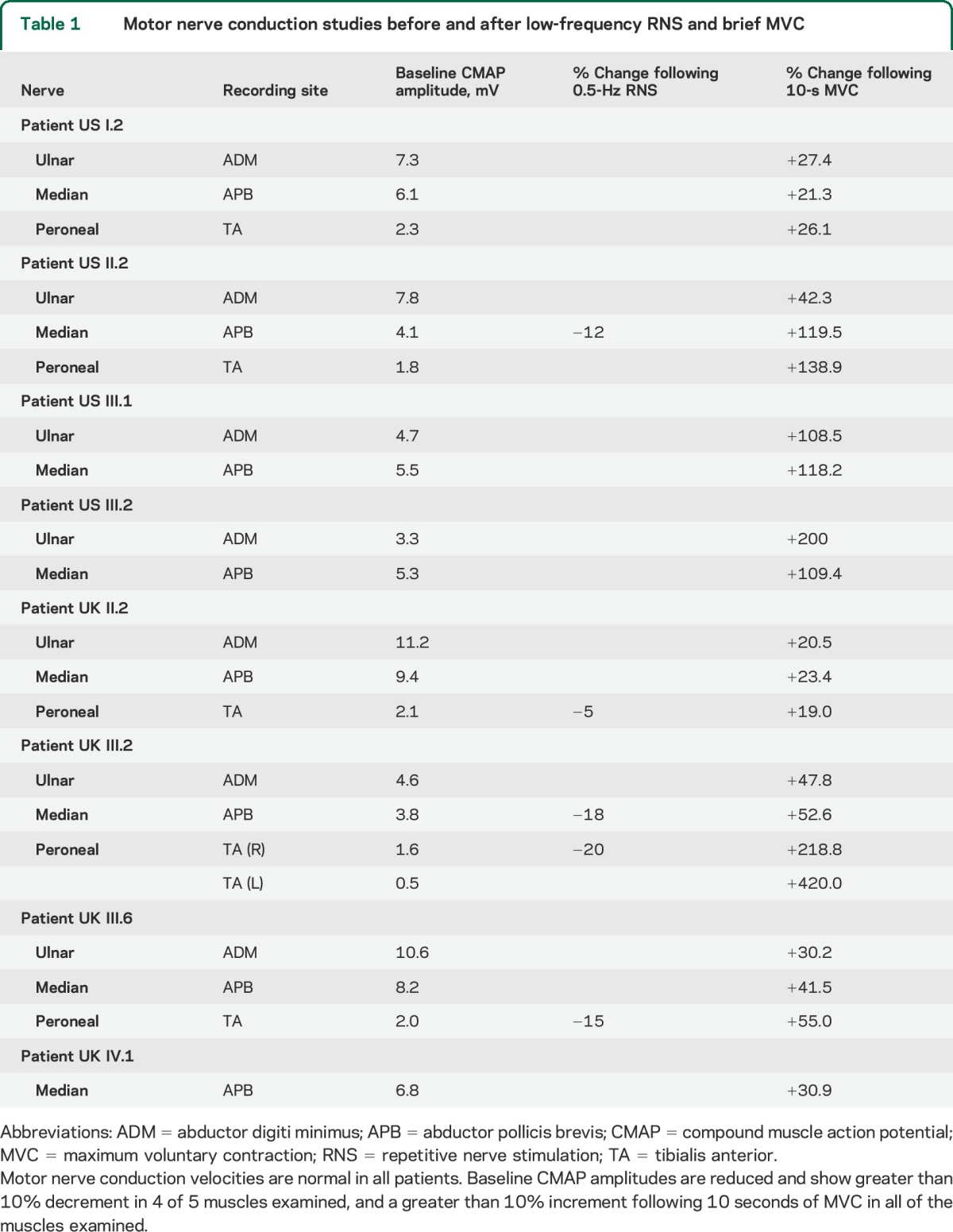
Figure 2. Neurophysiologic testing reveals features consistent with a deficit of acetylcholine release at the neuromuscular junction.

(A) Recording electrodes were placed over the APB muscle and supramaximal electrical stimulation was applied to the median nerve at the wrist. Marked incremental response in CMAP amplitude was seen following brief MVC. (B) Prolonged time course in decay of the incremental response following MVC in the TA muscle of UK III.2. The period of posttetanic potentiation lasted at least 60 minutes. The incremental response was replicated by a second period of maximum contraction, indicating that the recording electrodes remained correctly positioned. Δ% = % change compared to baseline CMAP amplitude. Red bar = 10-second period of MVC. (C) Posttetanic potentiation lasting at least 10 minutes was seen in all of the UK and US family members tested. Modeling of the decay was best fitted assuming a 2-component exponential decay. ADM = abductor digiti minimus; APB = abductor pollicis brevis; CMAP = compound muscle action potential; TA = tibialis anterior.
Time course of the incremental response.
To estimate the time course of this incremental response, the study was repeated in UK III.2, UK III.6, UK II.2, and US II.2 with more frequent time points (every 30 seconds after MVC for 10 minutes) (figure 2C). The initial increment varied between +270% and +19%, and in all participants showed an initial decay over 2 to 3 minutes followed by a persistent >10% potentiation for the entire 10 minutes.
A particularly striking response (+187%) was seen in the TA muscle in patient UK III.2. This patient also underwent a prolonged study in which CMAP responses were measured at baseline, after 10-second MVC, and subsequently at intervals over the next 60 minutes (figure 2B). The potentiated response decayed over this period but remained increased compared with baseline even after 60 minutes (+53%).
The decay for both US and UK families was best modeled using a 2-component exponential fit: ae−bt + ce−dt where a and c are the weighting constants for both components and 1/b and 1/d are the corresponding time constants for decay. Using data pooled from both families, 1/b was found to be 28 seconds and 1/d equal to 24,500 seconds (approximately 408 minutes) (R2 was 0.3167).
Effects of treatment with pyridostigmine and 3,4-diaminopyridine.
Given the evidence of significant neuromuscular junction dysfunction, we treated both the UK proband (UK III.2) and her mother (UK II.2) with a trial of pyridostigmine (60 mg, 3 times daily). Neither reported any change in their muscle strength or daily activities and the CMSS showed no difference after 1 month of therapy (data not shown). Following a period of several days’ washout, they were commenced on 20 mg, 3 times daily of 3,4-diaminopyridine (3,4-DAP). Both patients experienced slight improvement in exercise tolerance and in performing their daily activities. The CMSS confirmed an improvement in several indices, particularly eye muscle fatigability (from 26 and 45 seconds to 1 minute) and improved strength in timed head-lifting (from 45 seconds and 1 minute 30 seconds to 2 minutes) and other complex motor functions (table e-1 on the Neurology® Web site at Neurology.org). Following discontinuation of 3,4-DAP for 14 days, the values returned to the original assessment before therapy.
Electrophysiologic assessment during therapy.
Neither drug had a consistent effect on the initial CMAP amplitudes (figure 3A) or the degree of increment following 10-second MVC (figure 3B). We therefore performed single-fiber EMG in the TA muscle of the UK proband (UK III.2). This technique measures both the variability in the initiation of muscle fiber action potentials (“jitter”) and failures of neuromuscular transmission (“blocking”), and is the most sensitive test of neuromuscular instability. Normal muscles show jitter and blocking in no more than 10% of fibers.12 Before treatment with either agent, an increased jitter was seen in 16 of 16 pairs (100%), with intermittent blocking in 14 of these (88%). The mean consecutive difference was 193 μs (upper limit 50 μs) indicating a significant defect of neurotransmission.
Figure 3. Effects of treatment with pyridostigmine and 3,4-DAP.

(A) Treatment with pyridostigmine or 3,4-DAP produced no consistent change in initial CMAP amplitude or (B) percentage incremental response following 10-second maximum voluntary contraction. Δ% = % change compared to baseline CMAP amplitude. (C) Treatment with pyridostigmine produced a reduction in the percentage of muscle fiber pairs showing jitter and blocking on single-fiber EMG. A greater effect was seen with 3,4-DAP. (D) Both agents also produced a reduction in the mean consecutive jitter, with a greater reduction seen for 3,4-DAP than for pyridostigmine. APB = abductor pollicis brevis; CMAP = compound muscle action potential; 3,4-DAP = 3,4-diaminopyridine; MCD = mean consecutive difference; Pre = baseline values before treatment; Pyrido = pyridostigmine; TA = tibialis anterior.
Following treatment with pyridostigmine, the proportion of fiber pairs showing increased jitter fell to 87%, with blocking in 25% and a mean consecutive difference of 139 μs (figure 3, C and D). Following treatment with 3,4-DAP, the proportion of fiber pairs showing increased jitter fell further to 70%, with intermittent blocking in 15% and a mean consecutive difference of 93 μs. While still abnormal, these indicate a significant improvement in neuromuscular transmission compared to baseline.
DISCUSSION
We describe a novel human neuromuscular syndrome caused by dominant mutations in SYT2, a protein known to be essential for synchronous vesicle release at the neuromuscular junction. The 2 families described have mutations in adjacent amino acids, both residing within the calcium-binding pocket of the C2B domain of Syt2. This domain has emerged as the key effector for driving synaptic vesicle fusion.13–15
Both families presented with a phenotype suggestive of a motor neuropathy. The proband of the US family was initially referred for the investigation of presumed Charcot-Marie-Tooth disease, while the UK proband had previously been diagnosed with a distal hereditary motor neuropathy. Patients with distal hereditary motor neuropathy typically present with symmetrical distal muscle wasting and weakness, and with foot deformities.16,17 Distal reflexes are absent and do not recover with exercise. The observation in our patients of reflex potentiation following brief exercise suggested an additional reversible defect of neuromuscular transmission. This clinical sign is also seen in the Lambert-Eaton myasthenic syndrome (LEMS). The classic electrophysiologic features in LEMS are reduced amplitude CMAP responses with a decrementing response to low-frequency (0.5-Hz) nerve stimulation, but a marked incremental response at high-frequency (50-Hz) stimulation or following brief MVC.18 This pattern is the hallmark of presynaptic disorders in which there is a deficit of vesicle release. The patients described here exhibit the same pattern, likewise indicating a presynaptic deficit of transmitter release. In the case of LEMS, high-frequency motor nerve depolarization is able to overcome the blockade of VGCCs. In the case of SYT2 mutations, the incremental response presumably occurs as a result of raised intracellular calcium overcoming the reduced affinity of the calcium-binding domain.
One notable feature in our study is the markedly prolonged time course over which this incremental response decays back to baseline. In one patient, this period of posttetanic potentiation lasted at least 60 minutes, and all patients showed a greater than 10% increase in CMAP amplitude after 10 minutes. Posttetanic potentiation lasts approximately 2 minutes in normal individuals,19 but can be prolonged in LEMS with a decay time constant that is increased with cooling, implying a relationship to calcium clearance from the presynaptic terminal.20 Posttetanic potentiation can also last up to 21 minutes in infantile botulism,21 another cause of reduced presynaptic transmitter release; however, our finding of potentiation lasting up to 60 minutes is unique.
As well as acting as calcium sensors for vesicle release, synaptotagmins have also been implicated in the tethering of vesicles to VGCCs.22 Opening of VGCCs in response to membrane depolarization results in a transient and highly localized microdomain of increased calcium concentration, and vesicle release occurs from a population of “primed” vesicles within a few tens of nanometers of the VGCCs.23 SYT2 mutations could affect vesicle tethering, resulting in changes in the replenishment of this population of vesicles. Whether such a redistribution of vesicles can lead to a potentiation lasting up to 60 minutes is unclear. An alternative mechanism could involve posttranslation modification (such as phosphorylation) of SYT2 or another component of the fusion machinery that leads to a long-lasting increase in release probability. Further studies are ongoing to elucidate the mechanism of the prolonged posttetanic potentiation and the molecular basis of the distal lower limb atrophy. This is of great clinical relevance since understanding this unique feature may identify a specific treatment strategy.
Given the evidence of significant neuromuscular junction dysfunction, we treated 2 of the UK family members with pyridostigmine and subsequently 3,4-DAP. Pyridostigmine potentiates the effect of acetylcholine by inhibiting acetylcholinesterase in the synaptic cleft, and has previously been used in the treatment of LEMS,24 albeit with limited efficacy.25 3,4-DAP increases calcium entry to the presynaptic terminal via blockade of potassium channels, and reduces weakness in patients with LEMS26 and other neuromuscular disorders.27 Both of our patients reported a reduction in fatigable weakness, and several objective measures on the CMSS improved during 3,4-DAP treatment. Furthermore, neurophysiologic testing revealed an improvement in synaptic transmission. These preliminary results provide an impetus for clinical trials and emphasize the potential treatability of this disease. Of note, a patient with SNAP25 mutation also benefited from 3,4-DAP treatment but not from pyridostigmine, emphasizing the similarity in presynaptic pathophysiology.7
We describe the clinical and neurophysiologic features of a novel human neuromuscular syndrome caused by autosomal-dominant mutations in the synaptic vesicle calcium sensor SYT2. The phenotype of this mutation is suggestive of a distal hereditary neuropathy. However, in some patients, fatigable ptosis and reflex facilitation following exercise are also found. Furthermore, neurophysiologic testing demonstrates a decrementing response at low-frequency RNS and an incrementing response following brief MVC, features indicative of presynaptic CMS. Prolonged testing reveals a uniquely prolonged period of posttetanic potentiation. Crucially, patients with pharmacologic intervention experienced improvement in neuromuscular transmission but it remains to be seen whether it also improves motor nerve function.
Supplementary Material
ACKNOWLEDGMENT
The authors thank two of the patients (UK III.2 and II.2) for their helpful comments on the manuscript.
GLOSSARY
- APB
abductor pollicis brevis
- CMAP
compound muscle action potential
- CMS
congenital myasthenic syndrome
- CMSS
Congenital Myasthenic Syndrome Scale
- 3,4-DAP
3,4-diaminopyridine
- LEMS
Lambert-Eaton myasthenic syndrome
- MRC
Medical Research Council
- MVC
maximum voluntary contraction
- RNS
repetitive nerve stimulation
- SYT2
synaptotagmin II
- TA
tibialis anterior
- VGCC
voltage-gated calcium channel
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Whittaker: study design, acquisition of data, analysis of data, writing of manuscript. Dr. Herrmann: study design, acquisition of data, analysis of data, writing of manuscript. Dr. Bansagi: acquisition of data, analysis of data. Dr. Hasan: analysis of data. Dr. Lofra: acquisition of data. Dr. Logigian: acquisition of data, critical revision of the manuscript for important intellectual content. Dr. Sowden: acquisition of data, analysis of data. Dr. Almodovar: acquisition of data, analysis of data. Dr. Littleton: critical revision of the manuscript for important intellectual content. Dr. Zuchner: critical revision of the manuscript for important intellectual content. Dr. Horvath: study design, acquisition of data, analysis of data, writing of manuscript. Dr. Lochmüller: study design, acquisition of data, analysis of data, writing of manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
R. Whittaker receives funding from the EPSRC (EP/K028421/1) and the Wellcome Trust (G102037). D. Herrmann is supported by the Inherited Neuropathies Consortium Rare Disease Clinical Research Network, National Institute of Neurological Disorders and Stroke (U54NS065712). B. Bansagi is supported by the MRC Centre for Neuromuscular Diseases. B. Hasan, R. Lofra, and E. Logigian report no disclosures relevant to the manuscript. J. Sowden is supported by the Inherited Neuropathies Consortium Rare Disease Clinical Research Network, National Institute of Neurological Disorders and Stroke (U54NS065712). J. Almodovar reports no disclosures relevant to the manuscript. J. Littleton is funded by NIH grant NS40296 and the JPB Foundation. S. Zuchner is supported by NIH grants U54NS065712, R01NS075764, and R01NS072248, the MDA, and the CMT Association. R. Horvath is supported by the Medical Research Council (UK) (G1000848) and the European Research Council (309548). H. Lochmüller is supported by a grant from the Medical Research Council UK (reference G1002274, grant 98482). H.L. receives funding from the European Union Seventh Framework Programme (FP7/2007–2013) under grant agreement 305444 (RD-Connect) and 305121 (Neuromics). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 2015;14:420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Senderek J, Müller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet 2011;88:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Acuna C, Guo Q, Burré J, et al. Microsecond dissection of neurotransmitter release: SNARE-complex assembly dictates speed and Ca2+ sensitivity. Neuron 2014;82:1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JS, Kim MH, Ho WK, Lee SH. Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci 2008;28:7945–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohrmann R, de Wit H, Connell E, et al. Synaptotagmin interaction with SNAP-25 governs vesicle docking, priming, and fusion triggering. J Neurosci 2013;33:14417–14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang ZP, Melicoff E, Padgett D, et al. Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 2006;26:13493–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 2014;83:2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrmann DN, Horvath R, Sowden JE, et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert-Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet 2014;95:332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2011;16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan W. Reliability testing of the quantitative myasthenia gravis score. Ann NY Acad Sci 1998;841:769–772. [DOI] [PubMed] [Google Scholar]
- 11.Jablecki CK. AAEM case report #3: myasthenia gravis. Muscle Nerve 1991;14:391–397. [DOI] [PubMed] [Google Scholar]
- 12.Stålberg E, Ekstedt J, Broman A. The electromyographic jitter in normal human muscles. Electroencephalogr Clin Neurophysiol 1971;31:429–438. [DOI] [PubMed] [Google Scholar]
- 13.Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)B calcium binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 2002;418:340–344. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Guan Z, Akbergenova Y, Littleton JT. Genetic analysis of C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J Neurosci 2013;33:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshihara M, Guan Z, Littleton JT. Differential regulation of synchronous versus asynchronous neurotransmitter release by the C2 domains of synaptotagmin 1. Proc Natl Acad Sci USA 2010;1707:14869–14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103:259–280. [DOI] [PubMed] [Google Scholar]
- 17.Irobi J, De Jonghe P, Timmerman V. Molecular genetics of distal hereditary motor neuropathies. Hum Mol Genet 2004;13:R195–R202. [DOI] [PubMed] [Google Scholar]
- 18.Oh SJ, Kurokawa K, Claussen GC, Ryan HF., Jr Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve 2005;32:515–520. [DOI] [PubMed] [Google Scholar]
- 19.Grob D, Harvey AM, Johns RJ. Studies in neuromuscular function: II: effects of nerve stimulation in normal subjects and in patients with myasthenia gravis. Bull Johns Hopkins Hosp 1956;99:125–135. [PubMed] [Google Scholar]
- 20.Maddison P, Newsom-Davis J, Mills KR. Decay of postexercise augmentation in the Lambert-Eaton myasthenic syndrome: effect of cooling. Neurology 1998;50:1083–1087. [DOI] [PubMed] [Google Scholar]
- 21.Fakadej AV, Gutmann L. Prolongation of post-tetanic facilitation in infant botulism. Muscle Nerve 1982;5:727–729. [Google Scholar]
- 22.Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron 2009;63:482–496. [DOI] [PubMed] [Google Scholar]
- 23.Meinrenken CJ, Borst JG, Sakmann B. Calcium secretion coupling at calyx of held governed by nonuniform channel-vesicle topography. J Neurosci 2002;22:1648–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verschuuren JJ, Wirtz PW, Titulaer MJ, Willems LN, van Gerven J. Available treatment options for the management of Lambert-Eaton myasthenic syndrome. Expert Opin Pharmacother 2006;7:1323–1336. [DOI] [PubMed] [Google Scholar]
- 25.Wirtz PW, Verschuuren JJ, van Dijk JG, et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert–Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther 2009;86:44–48. [DOI] [PubMed] [Google Scholar]
- 26.McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med 1989;321:1567–1571. [DOI] [PubMed] [Google Scholar]
- 27.Flet L, Polard E, Guillard O, et al. 3,4-Diaminopyridine safety in clinical practice: an observational, retrospective cohort study. J Neurol 2010;257:937–946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.