

Abstract
Objective:
To determine whether cortical β-amyloid (Aβ) deposition is associated with circadian blood pressure (BP) profiles and dynamic cerebral blood flow (CBF) regulation in patients with amnestic mild cognitive impairment (aMCI).
Methods:
Forty participants with aMCI were included in this study. Cortical Aβ depositions were measured by 18F-florbetapir PET and expressed as the standardized uptake value ratio (SUVR) relative to the cerebellum. Circadian BP profiles were measured by 24-hour ambulatory monitoring during awake and sleep periods. The dipping status of sleep BP (i.e., the percent changes from the awake BP) was calculated and dichotomized into the dipper (≥10%) and nondipper (<10%) groups. Dynamic CBF regulation was assessed by a transfer function analysis between beat-to-beat changes in BP and CBF velocity measured from the middle cerebral artery during a repeated sit-stand maneuver.
Results:
Age was positively correlated with a greater Aβ deposition in the posterior cingulate, precuneus, and mean cortex. Accounting for the age effect, attenuated reductions in sleep systolic BP were associated with higher levels of posterior cingulate SUVR. Consistently, the nondippers exhibited a higher SUVR in the posterior cingulate than the dippers. Transfer function gain between changes in BP and CBF velocity was diminished in the nondippers, and moreover those individuals with a lower gain exhibited a higher SUVR in the posterior cingulate.
Conclusions:
Attenuated reductions in sleep BP are associated with a greater Aβ burden in the posterior cingulate and altered dynamic CBF regulation in patients with aMCI.
Alzheimer disease (AD) may begin decades before the manifestation of clinical symptoms, as revealed by β-amyloid (Aβ) depositions in the cerebral cortex.1 Mounting evidence suggests a link between AD and the presence of cardiovascular risk factors such as elevated blood pressure (BP).2 Physiologically, normal BP maintains a proper brain perfusion, which provides appropriate oxygen and nutrient supply, and may also play a pivotal role for Aβ clearance. Conversely, sustained elevations in BP can impose a substantial mechanical stress on the cerebrovascular beds and subsequently cause vascular remodeling and dysfunction.3
In daily life, BP fluctuates considerably and presents prominent circadian rhythms.4 Thus, a snapshot measurement of BP in the routine clinic setting is unlikely to represent the overall impact of BP on the brain.5 Indeed, elevated BP not only while awake but also during sleep has been linked to brain structural deteriorations6–8 and cognitive dysfunction.9–12 Nevertheless, the potential effects of changes in BP circadian rhythms on brain Aβ deposition are yet to be understood.
Amnestic mild cognitive impairment (aMCI), often a prodromal phase of AD, may represent a critical time window to prevent or slow AD progression.13 Therefore, it would be important to identify modifiable risk factors in these individuals. The purpose of this study was twofold: (1) to investigate whether cortical Aβ deposition is associated with circadian BP patterns, as assessed by 24-hour ambulatory monitoring (ABPM); and (2) to determine whether dynamic cerebral blood flow (CBF) regulation is altered and whether these changes are associated with cortical Aβ deposition.
METHODS
Participants.
Forty participants with aMCI were recruited through a community-based advertisement and the University of Texas Southwestern Medical Center Alzheimer's Disease Center. The diagnosis of aMCI was based on Petersen criteria,14 as modified by the Alzheimer's Disease Neuroimaging Initiative project (http://adni-info.org). Specifically, a global Clinical Dementia Rating scale of 0.5 with a score of 0.5 in the memory category, objective memory loss as indicated by education-adjusted scores on the Logical Memory subtest of the Wechsler Memory Scale–Revised, and a Mini-Mental State Examination (MMSE) score between 24 and 30 were used. Clinical evaluation was performed and based on the recommendations from the Alzheimer's Disease Cooperative Study (http://adni-info.org). Inclusion criteria were men and women aged 55–80 years with aMCI. Exclusion criteria were major neurologic, vascular, or psychiatric disorders. Participants with body mass index ≥40 kg/m2, sleep disorders including clinically diagnosed or self-reported sleep apnea, uncontrolled hypertension, diabetes, and a history of smoking were also excluded.
Standard protocol approvals, registrations, and patient consents.
All participants signed the informed consent approved by the Institutional Review Boards of the University of Texas Southwestern Medical Center and Texas Health Presbyterian Hospital of Dallas.
Study protocol and measurements.
Amyloid PET imaging.
Image acquisition.
Participants underwent an IV bolus injection of 10 mCi 18F-florbetapir. At 30 minutes postinjection, participants were positioned on the imaging table of a Siemens (Munich, Germany) ECAT HR PET scanner. Velcro straps and foam wedges secured the participant's head and the participant was positioned using laser guides. A 2-minute scout scan was acquired to ensure that the brain was completely in the field of view without rotation in either the transverse or sagittal planes. At 50 minutes postinjection, 2 frames of 5-minute PET emission scan and a 7-minute transmission scan were acquired in 3D mode using the following parameters: matrix size = 128 × 128, resolution = 5 × 5 mm, slice thickness = 2.42 mm, and field of view = 58.3 cm. The emission images were processed by iterative reconstruction, 4 iterations and 16 subsets with a 3-mm full width at half maximum (FWHM) ramp filter. The transmission image was reconstructed using back-projection and a 6-mm FWHM Gaussian filter for attenuation correction.15,16
Image processing.
Each participant's PET image was spatially normalized to a florbetapir uptake template (2 × 2 × 2 mm3 voxels) using SPM8 (Wellcome Department of Cognitive Neurology, London, UK) and in-house MATLAB (MathWorks, Sherborn, MA) scripts, and visually inspected for registration quality. Standardized uptake value ratio (SUVR) was computed using mean cerebellar uptake as a reference. Three regions of interest relevant to AD pathology were selected a priori: posterior cingulate, precuneus, and mean cortex.17 The mean cortical SUVR was calculated by taking an average of the posterior and anterior cingulate, precuneus, temporal, dorsolateral prefrontal, orbital frontal, parietal, and occipital SUVRs.15
Ambulatory BP monitoring.
ABPM was performed for ≥24 hours using a noninvasive oscillometric BP monitor (SunTech Medical Instruments, Morrisville, NC).18 Each participant wore a brachial cuff on the nondominant arm. Measurements were made every 30 minutes during the participant's individually scheduled awake period and every 60 minutes during the scheduled sleep period. The ABPM data are reported as an average of 24-hour, awake, and sleep periods. Awake and sleep BP were calculated according to the standard clinical guideline.4 To quantify the relative magnitude of BP changes from awake to sleep period, we calculated sleep BP dip using the following formula7:
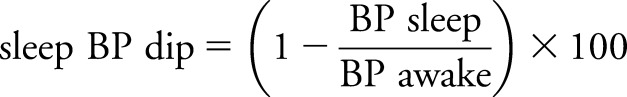 |
We analyzed the dipping status of sleep BP using continuous and categorical variables. The dippers were characterized as having falls in sleep systolic BP (SBP) ≥10%, whereas nondippers had falls in sleep SBP <10%.7,19
Dynamic CBF regulation assessment.
Dynamic CBF regulation assessment was conducted in an environmentally controlled laboratory. After collecting a 5-minute baseline, a repeated sit-stand maneuver was performed for 5 minutes with a duty cycle of a 10-second sit and a 10-second stand (i.e., 0.05 Hz). The purpose of this maneuver is to nonpharmacologically induce oscillations of BP using a movement relevant to daily life, while recording dynamic changes in CBF.20 CBF velocity (CBFV) was measured from the middle cerebral artery using a 2-MHz transcranial Doppler (TCD) probe (Multi-Dop X2; Compumedics/DWL, Singen, Germany). Beat-by-beat changes in CBFV and BP at the level of heart using finger plethymography (Finapress; Ohmeda, Boulder, CO) were continuously and simultaneously recorded and stored for offline analysis (Acqknowledge; BIOPAC Systems, Goleta, CA).
To quantify the cerebral dynamic pressure flow (CDPF) relation, transfer function (TF) metrics (i.e., gain, phase, and coherence) were calculated in the frequency domain.21 First, all hemodynamic data were averaged over each cardiac cycle to obtain beat-by-beat mean values. Second, the beat-by-beat values of mean arterial pressure (MAP) and CBFV were linearly interpolated, resampled at 2 Hz, and detrended by a third-order polynomial curve fitting. These data were subdivided into 256-point segments with 50% overlap, and a Hanning window was applied for Fourier spectral analysis.
TF gain represents a magnitude response of CBF to dynamic changes in BP and was calculated using both the absolute (cm/s) and relative (%) units.22 TF phase represents a temporal relation between changes in BP and CBF, while the coherence function reflects strength of their linear correlations (0 = no correlation and 1 = perfect correlation).
Alteration of circadian BP patterns may lead to cerebrovascular remodeling, such as arteriolar hypertrophy,3 which may cause cerebral vasoconstriction accompanied by the elevated vascular resistance, and have significant impacts on the TF metrics via a Windkessel effect, as suggested in our previous studies.23 The TF metrics were computed at 0.05 Hz. In addition, cerebrovascular resistance index was calculated by dividing MAP by CBFV.
APOE genotyping.
Peripheral blood mononuclear cells (PBMC) were obtained via IV blood draw and centrifugal Ficoll-based separation. PBMCs were cryopreserved in the media containing 50% human serum on the day of collection. For genotype analysis, 1 million cells were thawed and DNA extracted using the DNeasy Blood and Tissue kit (Qiagen; Venlo, Netherlands). APOE genotype was identified using TaqMan SNP genotyping assays (Life Technologies; Carlsbad, CA). APOE genotype data were available from 35 participants.
Statistical analyses.
The nonparametric statistical analyses were used according to the result of normality check performed by the Shapiro-Wilk test and the visual inspection of histograms and Q-Q plots. The changes in BP from awake to sleep period were examined by the Wilcoxon signed-rank test. Spearman correlation examined simple correlations among continuous variables. Group differences in categorical and continuous variables were examined by the χ2 and Mann-Whitney U tests, respectively. Analysis of covariance using general linear model and partial correlation analysis were performed on rank-order variables with an adjustment for covariates. The covariates were determined by the significant correlates of cortical SUVR in this sample. All data are reported as mean ± SD. Statistical significance was set a priori at p < 0.05 for all tests. All statistical analyses were performed using SPSS 20 (SPSS Inc.; Chicago, IL).
RESULTS
Table e-1 on the Neurology® Web site at Neurology.org presents basic characteristics of all participants. All participants were physically healthy and had a normal or controlled BP. Despite the diagnosis of aMCI, MMSE scores were within the normal range, suggesting an early phase of mild cognitive impairment. The APOE4 carriers were all heterozygotes and accounted for 34% of the sample. The older participants with aMCI showed higher SUVRs in the posterior cingulate, the precuneus, and the mean value of the overall cortex, whereas the APOE4 carriers demonstrated a trend toward higher levels in the precuneus and mean cortical values than the noncarriers (figure 1).
Figure 1. Amyloid burden with age and apolipoprotein E4 status.

(A) 18F-florbetapir standardized uptake value ratio (SUVR) in relation to age. (B) Group comparisons of 18F-florbetapir SUVR between APOE4 carriers and noncarriers. All the APOE4 carriers were heterozygotes. APOE4 data were available from 35 participants. Horizontal thick lines represent group-averaged mean values.
From awake to sleep period, SBP (figure 2A) and diastolic BP decreased. Table e-2 presents simple correlations between amyloid SUVRs and BP measures. After adjustment for age, the magnitude of sleep SBP dip remained to be negatively associated with the higher levels of the posterior cingulate SUVR (figure 2B). These results were further confirmed by the group comparisons between the dippers and nondippers (table 1). Of note, the nondippers were older than the dippers but similar in the APOE4 status. Interestingly, awake BP was similar between the groups, while sleep BP was elevated in the nondippers. Figure 3 displays the group-averaged SUVR maps of the dippers and nondippers. After adjustment for age, the posterior cingulate SUVR remained to be elevated in the nondippers.
Figure 2. Sleep systolic blood pressure and amyloid burden in the posterior cingulate cortex.
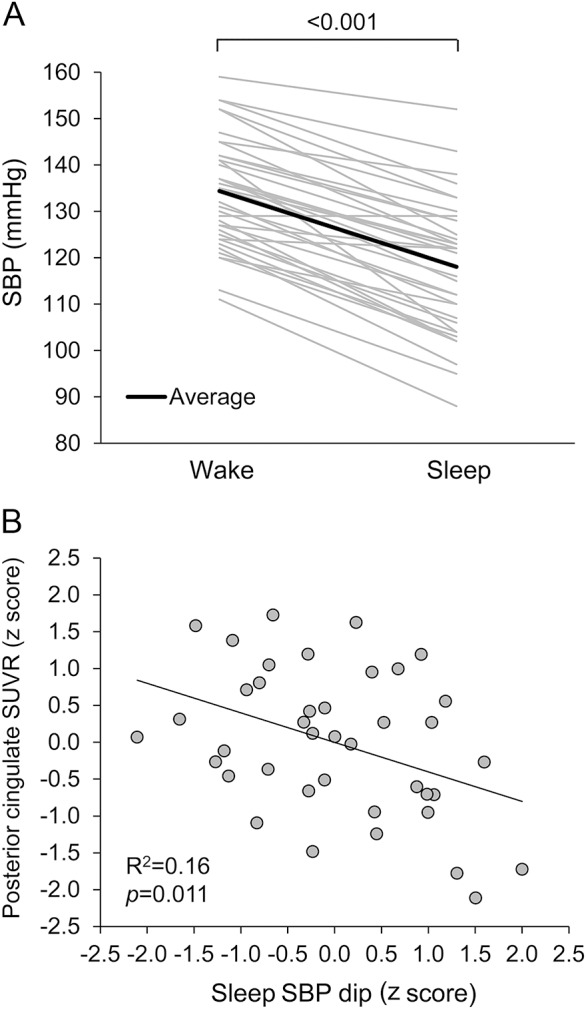
(A) Circadian changes in ambulatory systolic blood pressure (SBP) from awake to sleep period. (B) Age-adjusted partial correlation between sleep SBP dip and posterior cingulate standardized uptake value ratio (SUVR). Standardized residuals of sleep SBP dip and posterior cingulate SUVR were calculated by regressing out the effect of age.
Table 1.
Brain amyloid burden, blood pressures, and cerebral pressure–flow relation compared between dippers and nondippers
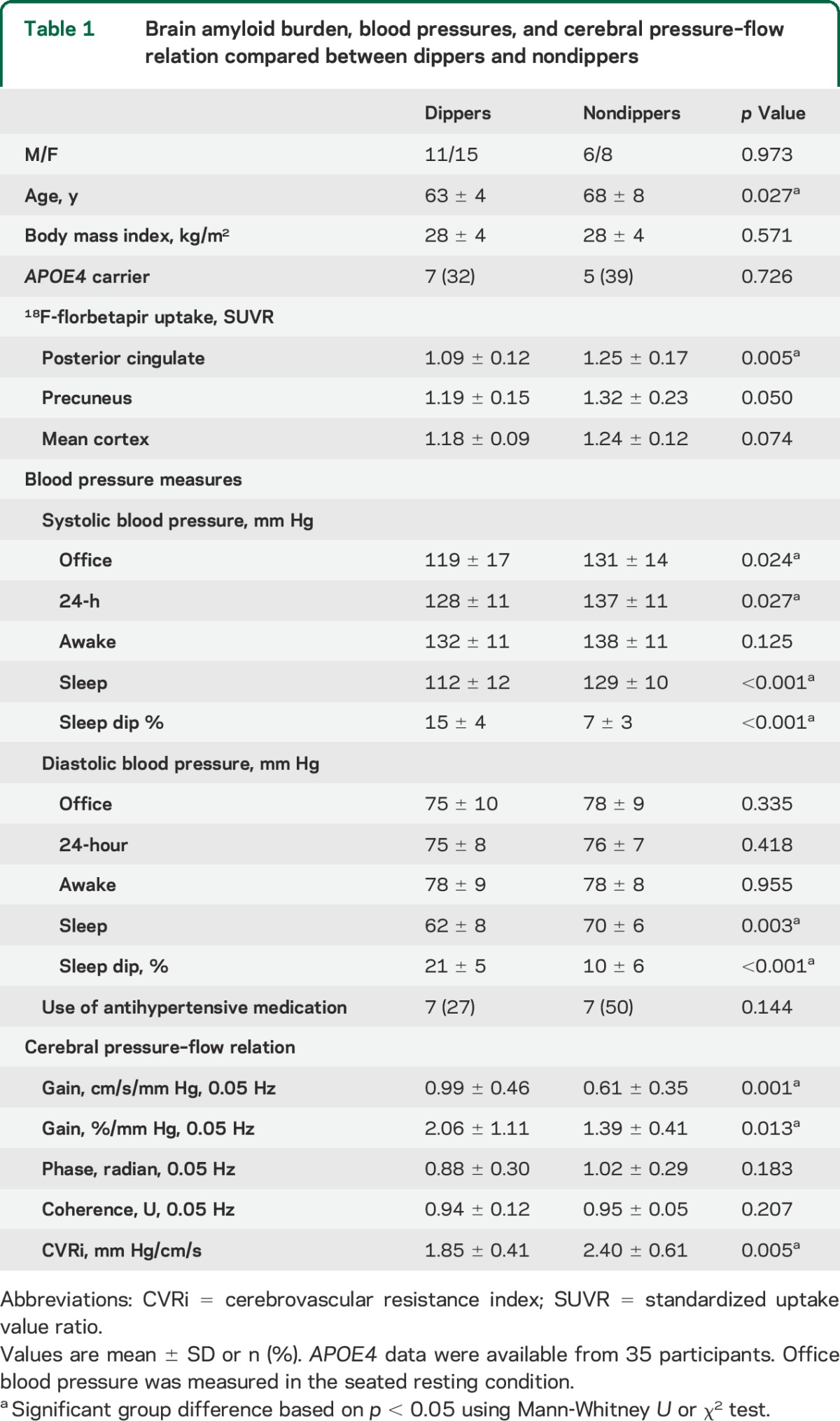
Figure 3. Amyloid burden in sleep systolic blood pressure dippers and nondippers.

(A) Group-averaged maps of 18F-florbetapir standardized uptake value ratio (SUVR) in dippers and nondippers. (B) Age-adjusted group comparisons of 18F-florbetapir SUVR in the posterior cingulate, precuneus, and mean cortex. Standardized residual of 18F-florbetapir SUVR was calculated by regressing out the effect of age. Horizontal thick lines represent group-averaged mean values.
Figure 4 shows the estimated TF metrics of the CDPF relation in the dippers and nondippers. The nondippers demonstrated a diminished TF gain and elevated cerebrovascular resistance index compared with the dippers (table 1). Of note, the diminished TF gain indicates an attenuated CBF response to BP fluctuations during postural changes. When analyzed in all participants, individuals with a diminished TF gain had higher levels of posterior cingulate SUVR (absolute gain: r = −0.43, p = 0.010 and normalized gain: r = −0.35, p = 0.038). Interestingly, group differences in posterior cingulate SUVR between dippers and nondippers were attenuated after adjustment for the TF gains (absolute gain: F = 1.25, p = 0.272 and normalized gain: F = 1.75, p = 0.196).
Figure 4. Transfer function metrics of the cerebral dynamic pressure-flow relation assessed during a repeated sit-stand maneuver.

(A) Gain, (B) phase, (C) normalized gain, (D) coherence. Group-averaged waveforms were generated from dippers and nondippers. The center of the shade areas represents the sit-stand frequency (i.e., 0.05 Hz) where hemodynamic oscillations were induced.
Participants on antihypertensive medications had similar levels of ambulatory BP, cortical Aβ deposition, or TF metrics compared with those not on the medication (table e-3). Body weight or body mass index was not related to sleep BP or cortical Aβ levels.
DISCUSSION
The primary findings from this study are twofold. First, patients with aMCI with an attenuated fall in sleep SBP had greater Aβ burden in the posterior cingulate independent of age. Consistently as a group, the nondippers demonstrated higher levels of Aβ deposition in the posterior cingulate than the dippers. Second, the nondippers exhibited a diminished TF gain of the CDPF relation, and moreover those individuals with a lower TF gain had higher Aβ deposition in the posterior cingulate. Collectively, these findings suggest that brain Aβ burden is related to elevated sleep BP and altered CBF regulation in patients with aMCI. Below we discuss the potential mechanisms and clinical implications of these findings.
Brain Aβ deposition increases with the aging process, which may reflect AD pathology.15 In patients with aMCI, age was positively correlated with greater Aβ burden, consistent with previous studies.15 Physiologically, Aβ in the brain is produced by the neurons and cleared by the 4 potential mechanisms: (1) paravascular drainage of the interstitial and CSFs, (2) direct transport across the blood–brain barrier, (3) enzyme-dependent proteolysis, and (4) endogenous autoantibodies.24–26 Current evidence suggests that brain Aβ deposition associated with age and late-onset AD are mainly attributed to a reduced clearance, which facilitates the aggregation of soluble Aβ in the interstitial fluid and the formation of neurotic plaques in a concentration-dependent manner.27,28
The patients with aMCI with elevated sleep SBP were older than those with normal BP dipping; however, after accounting for age, those nondippers still exhibited greater Aβ burden in the posterior cingulate. The age difference between the dippers and nondippers observed in the present study is consistent with the literature.29 In contrast, the link between the elevated sleep BP and Aβ provide new insights into the previous observations that nondippers have a higher risk of cognitive impairment and brain structural deteriorations.6–12 Specifically, elevated sleep BP has been associated with ischemic brain injuries,6 stroke incidence,7 and brain atrophy.8 Moreover, nondippers have been shown to manifest lower performance in memory, sensorimotor, and global cognition9–11 and a higher risk of developing dementia12 than the dippers.
There are a few mechanisms that may explain the relation between elevated sleep BP and brain Aβ burden. First, cerebrovascular disease or dysfunction may contribute to Aβ deposition in the brain. We found a diminished TF gain of the CDPF relation in the nondippers, which in turn was correlated with higher Aβ burden in the posterior cingulate. Physiologically, these observations indicate the presence of elevated cerebrovascular resistance in the nondippers, which dampened the oscillations of CBF in the face of BP fluctuations.23 Consistently, we found a higher level of cerebrovascular resistance index in the nondippers, suggesting BP-induced cerebrovascular remodeling and constriction.3 The elevated cerebrovascular resistance may increase the risk of hypoperfusion, which has been shown to accelerate Aβ deposition.30 Moreover, significant dampening of CBF dynamics may also impair the paravascular clearance of Aβ from the interstitial fluid.31
Second, Aβ clearance may be impaired by an enhanced sympathetic neural activity during sleep associated with attenuated reductions in BP dip.32 A recent study in rodents demonstrated that sleep expands the volume of brain interstitial space, which facilitates Aβ clearance via the paravascular pathway.32 Furthermore, such sleep-induced clearance of Aβ from the brain may be mediated by a suppression of adrenergic signaling.32 In healthy individuals, sympathetic neural activity is reduced during sleep and decreases BP, which may promote Aβ clearance from the brain.33 In contrast, with advanced age or presence of cardiovascular disease, elevated sympathetic neural activity may not only increase BP but also impair Aβ clearance from the brain during sleep.32,34
Third, AD and Aβ deposition in the brain are influenced by the presence of hypertension.35 This relation may be mediated by an Aβ-induced activation of α1-adrenergic receptors, leading to cerebral vasoconstriction, endothelial dysfunction, and alterations in chronotropic control of the heart rate or cardiac output, which is related to brain perfusion.36
The mechanism leading to the observed regional vulnerability of the posterior cingulate to Aβ deposition in the nondippers remains unclear. However, it is well-known that this area of the brain functions as the neuronal hub of the default mode network and exhibits a higher metabolic rate than the cortical average.37,38 Therefore, the posterior cingulate has a higher rate of Aβ production and manifests an earlier sign of Aβ deposition.17 In the context of the present study, Aβ homeostasis may be disturbed by the disrupted neurovascular coupling due to elevated sleep BP and altered CBF regulation (e.g., hypoperfusion).30 Consequently, the posterior cingulate may express an accelerated Aβ deposition, which can subsequently increase the risk of neurodegeneration and cognitive impairment associated with AD.39,40
This study is strengthened by the use of a multimodality method to investigate the effects of ambulatory BP profile and dynamic CBF regulation on cortical Aβ deposition in patients with aMCI. First, 18F-florbetapir PET was used to quantify the degree of Aβ burden in the cortical areas relevant to AD pathology.16 Second, the use of 24-hour ABPM allowed us to measure circadian rhythms of BP, which reflects the overall impact of changes in BP in daily life on the brain and also increases the reliability of BP measurement.4 Third, to better understand the mechanism of the BP-Aβ link, we assessed the CDPF relation, which revealed the potential role of altered CBF regulation in cortical Aβ deposition.21
There are several limitations that need to be discussed. First, this is a cross-sectional study, which limits the understanding of causality. Second, our sample size was limited, and only highly educated individuals with aMCI were enrolled. Thus, the generalizability of our findings awaits confirmation in larger samples. The relatively small sample size also posed a limitation in detecting the effect of APOE4 on the cortical SUVR in this study (i.e., only 12 participants were APOE4 carriers). Third, although TCD has a high temporal resolution to assess dynamic changes in CBF, it does not measure volumetric flow and is limited by the spatial resolution. Therefore, we cannot determine whether the altered cerebral hemodynamics observed at a relatively global level reflects regional differences.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the study participants for their time and effort.
GLOSSARY
- Aβ
β-amyloid
- ABPM
ambulatory blood pressure monitoring
- AD
Alzheimer disease
- aMCI
amnestic mild cognitive impairment
- BP
blood pressure
- CBF
cerebral blood flow
- CBFV
cerebral blood flow velocity
- CDPF
cerebral dynamic pressure flow
- FWHM
full width at half maximum
- MAP
mean arterial pressure
- MMSE
Mini-Mental State Examination
- PBMC
peripheral blood mononuclear cells
- SBP
systolic blood pressure
- SUVR
standardized uptake value ratio
- TCD
transcranial Doppler
- TF
transfer function
Footnotes
Supplemental data at Neurology.org
Editorial, page 1918
AUTHOR CONTRIBUTIONS
Study concept/design: Dr. Tarumi and Dr. Zhang. Data analysis/interpretation: Dr. Tarumi, Dr. Zhang, Dr. Cullum, T. Harris, J. Riley, M. Turner, C. Hill, Z. German, Dr. Monson, Dr. Stowe, Dr. Mathews, and Dr. Kerwin. Drafting/revising of the manuscript: Dr. Tarumi, Dr. Zhang, Dr. Cullum, and J. Riley. Statistical analysis: Dr. Tarumi. Obtained funding: Dr. Zhang, Dr. Cullum, Dr. Womack, Dr. Stowe, Dr. Monson, and Dr. Kerwin.
STUDY FUNDING
National Institute of Health (R01AG033106, R01HL102457, and P30AG012300) and American Heart Association (14POST20140013). The 18F-florbetapir PET radiotracer was provided to the study by Avid Radiopharmaceuticals.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kivipelto M, Helkala E-L, Laakso MP, et al. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ 2001;322:1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension 1989;13:968–972. [DOI] [PubMed] [Google Scholar]
- 4.O'Brien E, Coats A, Owens P, et al. Use and interpretation of ambulatory blood pressure monitoring: recommendations of the British Hypertension Society. BMJ 2000;320:1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piper MA, Evans CV, Burda BU, Margolis KL, O'Connor E, Whitlock EP. Diagnostic and predictive accuracy of blood pressure screening methods with consideration of rescreening intervals: an updated systematic review for the US Preventive Services Task Force. Ann Intern Med 2015;162:192–204. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz GL, Bailey KR, Mosley T, et al. Association of ambulatory blood pressure with ischemic brain injury. Hypertension 2007;49:1228–1234. [DOI] [PubMed] [Google Scholar]
- 7.Kario K, Pickering TG, Matsuo T, Hoshide S, Schwartz JE, Shimada K. Stroke prognosis and abnormal nocturnal blood pressure falls in older hypertensives. Hypertension 2001;38:852–857. [DOI] [PubMed] [Google Scholar]
- 8.Nagai M, Hoshide S, Ishikawa J, Shimada K, Kario K. Ambulatory blood pressure as an independent determinant of brain atrophy and cognitive function in elderly hypertension. J Hypertension 2008;26:1636–1641. [DOI] [PubMed] [Google Scholar]
- 9.Conway KS, Forbang N, Beben T, Criqui MH, Ix JH, Rifkin DE. Relationship between 24-Hour ambulatory blood pressure and cognitive function in community-Living older Adults: the UCSD ambulatory blood pressure study. Am J Hypertens Epub 2015 Apr 19. [DOI] [PMC free article] [PubMed]
- 10.van Boxtel MP, Gaillard C, Houx PJ, Buntinx F, de Leeuw PW, Jolles J. Is nondipping in 24 h ambulatory blood pressure related to cognitive dysfunction? J Hypertens 1998;16:1425–1432. [DOI] [PubMed] [Google Scholar]
- 11.Bellelli G, Frisoni GB, Lucchi E, et al. Blunted reduction in night-time blood pressure is associated with cognitive deterioration in subjects with long-standing hypertension. Blood Press Monit 2004;9:71–76. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto Y, Akiguchi I, Oiwa K, Hayashi M, Kasai T, Ozasa K. Twenty-four–Hour blood pressure and MRI as predictive factors for Different Outcomes in patients with Lacunar Infarct. Stroke 2002;33:297–305. [PubMed] [Google Scholar]
- 13.Selkoe DJ. Preventing Alzheimer's disease. Science 2012;337:1488–1492. [DOI] [PubMed] [Google Scholar]
- 14.Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Arch Neurol 2001;58:1985–1992. [DOI] [PubMed] [Google Scholar]
- 15.Rodrigue K, Kennedy K, Devous M, et al. β-Amyloid burden in healthy aging Regional distribution and cognitive consequences. Neurology 2012;78:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging β-amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buckner RL, Snyder AZ, Shannon BJ, et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci 2005;25:7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodwin J, Bilous M, Winship S, Finn P, Jones SC. Validation of the Oscar 2 oscillometric 24-h ambulatory blood pressure monitor according to the British Hypertension Society protocol. Blood Press Monit 2007;12:113–117. [DOI] [PubMed] [Google Scholar]
- 19.O'Brien E, Sheridan J, O'Malley K. Dippers and non-dippers. Lancet 1988;332:397. [DOI] [PubMed] [Google Scholar]
- 20.Claassen JA, Levine BD, Zhang R. Dynamic cerebral autoregulation during repeated squat-stand maneuvers. J Appl Physiol 2009;106:153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang R, Zuckerman JH, Giller CA, Levine BD. Transfer function analysis of dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol 1998;274:H233–H241. [DOI] [PubMed] [Google Scholar]
- 22.Bishop C, Powell S, Rutt D. Transcranial Doppler measurement of middle cerebral artery blood flow velocity: a validation study. Stroke 1986;17:913–915. [DOI] [PubMed] [Google Scholar]
- 23.Zhang R, Behbehani K, Levine BD. Dynamic pressure–flow relationship of the cerebral circulation during acute increase in arterial pressure. J Physiol 2009;587:2567–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Trans Med 2012;4:147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weller R, Subash M, Preston S, Mazanti I, Carare R. Perivascular drainage of amyloid-b peptides from the brain and its Failure in cerebral amyloid angiopathy and Alzheimer's disease. Brain Pathol 2008;18:253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y-J, Zhou H-D, Zhou X-F. Clearance of amyloid-beta in Alzheimer's disease: progress, problems and perspectives. Drug Discov Today 2006;11:931–938. [DOI] [PubMed] [Google Scholar]
- 27.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS β-amyloid in Alzheimer's disease. Science 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kress BT, Iliff JJ, Xia M, et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 2014;76:845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de la Sierra A, Redon J, Banegas JR, et al. Prevalence and factors associated with circadian blood pressure patterns in hypertensive patients. Hypertension 2009;53:466–472. [DOI] [PubMed] [Google Scholar]
- 30.Okamoto Y, Yamamoto T, Kalaria RN, et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 2012;123:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iliff JJ, Wang M, Zeppenfeld DM, et al. Cerebral arterial pulsation drives paravascular csf–interstitial fluid exchange in the murine brain. J Neurosci 2013;33:18190–18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science 2013;342:373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Somers VK, Dyken ME, Mark AL, Abboud FM. Sympathetic-nerve activity during sleep in normal subjects. N Engl J Med 1993;328:303–307. [DOI] [PubMed] [Google Scholar]
- 34.Yamada Y, Miyajima E, Tochikubo O, Matsukawa T, Ishii M. Age-related changes in muscle sympathetic nerve activity in essential hypertension. Hypertension 1989;13:870–877. [DOI] [PubMed] [Google Scholar]
- 35.Elias MF, Davey A. Midlife blood pressure, amyloid-β, and risk for Alzheimer disease one More Reason to Treat hypertension. Hypertension 2012;59:771–772. [DOI] [PubMed] [Google Scholar]
- 36.Haase N, Herse F, Spallek B, et al. Amyloid-β peptides activate α1-adrenergic cardiovascular receptors. Hypertension 2013;62:966–972. [DOI] [PubMed] [Google Scholar]
- 37.Leech R, Sharp DJ. The role of the posterior cingulate cortex in cognition and disease. Brain 2014;137:12–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfefferbaum A, Chanraud S, Pitel A-L, et al. Cerebral blood flow in posterior cortical nodes of the default mode network decreases with task engagement but remains higher than in most brain regions. Cereb Cortex 2011;21:233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang X, Zou Q, He Y, Yang Y. Coupling of functional connectivity and regional cerebral blood flow reveals a physiological basis for network hubs of the human brain. Proc Natl Acad Sci USA 2013;110:1929–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crossley NA, Mechelli A, Scott J, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain 2014;137:2382–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.